

Abstract
A general theory has been developed for proton-coupled electron transfer (PCET), which is vital to a wide range of chemical and biological processes. This theory describes PCET reactions in terms of nonadiabatic transitions between reactant and product electron-proton vibronic states and includes the effects of thermal fluctuations of the solvent or protein environment, as well as the proton donor-acceptor motion. Within the framework of this general PCET theory, a series of analytical rate constant expressions has been derived for PCET reactions in well-defined regimes. Herein the application of this theory to PCET in the enzyme soybean lipoxygenase illustrates the regimes of validity for the various rate constant expressions and elucidates the fundamental physical principles dictating PCET reactions. Such theoretical studies provide significant physical insights that guide the interpretation of experimental data and lead to experimentally testable predictions. A combination of theoretical treatments with atomic-level simulations is essential to understanding PCET.
1. Introduction
Proton-coupled electron transfer (PCET) plays a vital role in key steps of photosynthesis, respiration, and many enzymatic reactions.2–8 A variety of chemical and energy conversion processes, including those occurring in solar energy devices, also rely on PCET. In general, a PCET reaction can occur by a sequential or a concerted mechanism.5 Although the distinction between these two mechanisms is not always clear, a PCET reaction is definitely sequential if a stable intermediate corresponding to either electron transfer or proton transfer is observed experimentally. In the absence of such an observed intermediate, however, the mechanism cannot be rigorously determined, although it can be deduced to be concerted if the single electron and single proton transfer reactions are shown to be significantly less thermodynamically favorable than the overall PCET reaction. In this case, the reaction is expected to be concerted to avoid the intermediates with relatively high free energy. Concerted PCET reactions can be broken down further into hydrogen atom transfer (HAT), in which the electron and proton transfer between the same donor and acceptor, or electron-proton transfer (EPT), in which the electron and proton transfer between different donors and acceptors.9 Again, the distinction between HAT and EPT reactions is not always clear, especially because the donors and acceptors can be defined as atoms, orbitals, or groups, and the electron and proton are often delocalized.
Concerted PCET reactions can be described in terms of quantum transitions between a set of reactant and product diabatic electron-proton vibronic states.10, 11 Typically, concerted PCET reactions are vibronically nonadiabatic in that the vibronic coupling, which is the Hamiltonian matrix element between the diabatic reactant and product electron-proton vibronic states, is much less than the thermal energy associated with the solvent or protein environment. As a result, nonadiabatic rate constants based on the Golden Rule formulation, in which the rate constant is proportional to the square of the vibronic coupling, are valid for most PCET reactions. The specific form of the vibronic coupling is determined by the electron-proton nonadiabaticity (i.e., the electronic nonadiabaticity of proton transfer), which reflects the changes in the electronic wavefunction as the proton transfers from its donor to its acceptor. A general form of the vibronic coupling, as well as expressions in the electronically adiabatic and nonadiabatic regimes, is available in the literature.12 The degree of electron-proton nonadiabaticity can also be used to distinguish the HAT and EPT mechanisms.9, 13 Specifically, HAT reactions are electronically adiabatic, corresponding to negligible electronic charge redistribution, whereas EPT reactions are electronically nonadiabatic, corresponding to significant electronic charge redistribution.
Previously nonadiabatic rate constant expressions for PCET reactions in various well-defined regimes have been derived.4, 11, 14, 15 In this formulation, a PCET reaction is described in terms of nonadiabatic transitions between reactant and product electron-proton vibronic states that become degenerate as a result of thermal fluctuations of the environment. The probability of these nonadiabatic transitions is proportional to the square of the vibronic coupling between the reactant and product vibronic states. This vibronic coupling is assumed to be only weakly dependent on the configuration of the environment but has been shown to depend strongly on the proton donor-acceptor distance coordinate R. As a result, the effects of the proton donor-acceptor motion are included explicitly in theoretical treatments of PCET,14, 15 analogous to previous treatments of proton transfer.16–21 To incorporate these effects, the natural logarithm of the coupling is expanded around the equilibrium proton donor-acceptor distance. In the original derivations of PT and PCET rate constants, only terms up to first order in these fluctuations were included in this expansion, and the coupling was assumed to decrease exponentially with R. Recently, these rate constants were derived again with the inclusion of terms up to second order in these fluctuations;15 in this case, the exponential decay of the coupling involved both linear and quadratic terms in R. Studies of model systems indicated that the quadratic terms in the coupling significantly impact the rate constants at high temperatures for proton transfer interfaces with soft proton donor-acceptor modes that are associated with small force constants and weak hydrogen bonds.15
On the basis of these model studies, the present paper applies the recently derived rate constant expressions to the enzyme soybean lipoxygenase (SLO), which catalyzes the oxidation of unsaturated fatty acids. Specifically, SLO catalyzes the net hydrogen atom transfer from the linoleic acid substrate to the iron cofactor. As depicted in Figure 1, this reaction occurs by a concerted PCET mechanism, in which the electron transfers from the π backbone of the linoleic acid to the iron, and the proton transfers from C11 of the linoleic acid to the oxygen of the iron-bound hydroxide. The evidence of this concerted mechanism is that the single electron transfer and single proton transfer reactions are highly endoergic, whereas the concerted PCET reaction is somewhat exoergic by ~5 kcal/mol, as determined from the reduction potentials and pKa’s of the species involved.22, 23 Moreover, analysis of the Kohn-Sham orbitals in DFT calculations have illustrated that the electron is transferred from the π backbone of the linoleic acid substrate to the iron, confirming distinct donors and acceptors for the electron and proton.24, 25 Experimental measurements have indicated that the kinetic isotope effect (KIE), which is the ratio of the rate constants for hydrogen and deuterium transfer, is ~80 at room temperature.22 Moreover, the temperature dependences of the rate constants and KIE are relatively weak, and distal mutations of the enzyme significantly influence the magnitude and temperature dependence of the KIE.1 Recently a KIE of ~700 was measured experimentally at room temperature for the double mutant L546A/L754A.26 This enormous KIE is unprecedented for such systems and is a prime example of the significance of hydrogen tunneling in biology.
Figure 1.
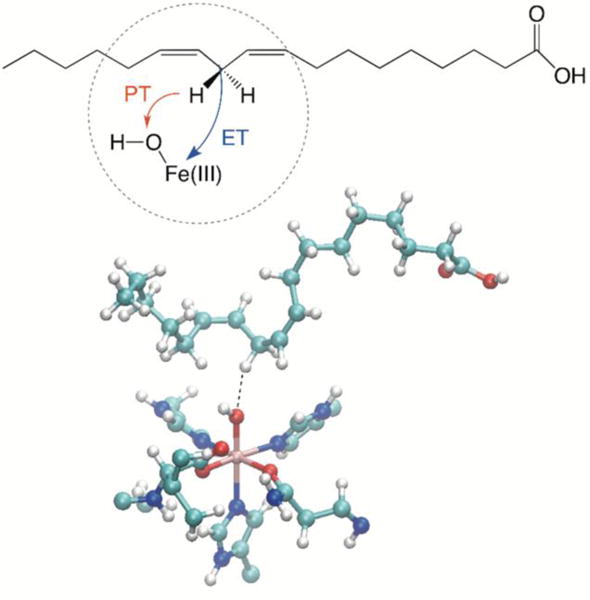
Schematic depiction of the PCET reaction in the active site of SLO. The electron transfers from the π backbone of the linoleic acid to Fe(III), and the proton transfers from C11 of the linoleic acid to the oxygen of the iron-bound hydroxide to generate water bound to Fe(II). The substrate linoleic acid and the iron cofactor are shown in the molecular representation.
Given the wealth of experimental data and the large KIEs, the SLO system has become the prototype for hydrogen tunneling in enzymes. As a result, the experimental observations have inspired numerous theoretical studies of SLO.23, 26–33 In particular, we applied the original PCET nonadiabatic rate constant expressions to wild-type SLO,23, 32 as well as to a series of I553 mutants and to the double mutant L546A/L754A.26, 33 In the present paper, we apply the recently derived PCET nonadiabatic rate constant expression15 to all of these SLO systems. From a fundamental theoretical perspective, our goal is to determine the significance of the quadratic terms in the coupling for this type of PCET reaction. Our previous study of model systems included only the ground vibronic states in the calculations and focused on only the rate constants.15 In the present study, we extend this treatment to include the excited vibronic states and to investigate the magnitudes and temperature dependences of both the rate constants and the KIEs. To analyze the significance of the quadratic terms, the results from the analytical rate constant expressions are compared to the results from a two-dimensional full quantum approach, which treats the proton coordinate and the proton donor-acceptor distance coordinate quantum mechanically on the same level, and an explicit thermal averaging approach, which does not assume a specific form of the vibronic coupling and thus is more generally applicable.
An outline of the paper is as follows. Section 2 presents the nonadiabatic rate constant expressions derived with the cumulant expansion approach, the thermal averaging approach, and the two-dimensional full quantum approach, as well as approximate expressions for the KIE. Section 3 presents expressions for the vibronic coupling in the general case and in the electronically adiabatic and nonadiabatic regimes. The application to SLO is described in Section 4, and conclusions are provided in Section 5.
2. Rate constant expressions
The nonadiabatic theory of PT and PCET is based on the general Golden Rule formulation in the framework of a set of reactant and product electron-proton vibronic states.10, 11, 17, 20, 21, 34–36 Typically the reactant and product vibronic states are defined in terms of reactant and product diabatic electronic states characterized by the transferring electron localized on the electron donor or acceptor species for PCET reactions. In this formulation, thermal fluctuations of the environment lead to the degeneracy of a pair of reactant and product vibronic states, thereby enabling a nonadiabatic transition with a probability that is proportional to the square of the coupling between these two vibronic states. The electron-proton vibronic wavefunctions and energies depend strongly on the proton donor-acceptor distance R, requiring a formalism that includes fluctuations of R as well as the solvent energy gap.14
A. Cumulant expansion approach
Within the framework of the Golden Rule formulation, the rate constant is expressed as a time integral of the reaction flux correlation function. In the cumulant expansion approach, the energy gap is expanded in a Taylor series in the coordinate R, and an approximate form of the vibronic coupling is invoked.14, 17 Specifically, the natural logarithm of the vibronic coupling can be expanded to second order in , where is the equilibrium proton donor-acceptor distance, which is often assumed to be the same for all reactant and product vibronic states. This expansion can be written as
| (1) |
with , for the vibronic coupling between reactant and product vibronic states μ and ν, respectively. The majority of previous treatments included only the linear term in the exponential (i.e., assumed that ), although recently the effects of quadratic terms have been investigated.15, 37 Note that the parameters and may be different for different pairs of reactant and product vibronic states and must be determined by fitting the vibronic coupling for each pair of vibronic states to Eq. (1).
Within this formalism, a series of rate constant expressions have been derived for well-defined regimes by making a series of approximations.14, 15 First, the expression for the reaction flux correlation function is simplified by assuming Gaussian processes for the energy gap and the R coordinate and invoking the high-temperature approximation for the solvent fluctuations with a simple Debye model of solvent relaxation. These approximations lead to an expression for the rate constant in terms of a time integral of the reaction flux correlation function (Eq. (23) of Ref.15) that can be evaluated numerically. In this paper, this rate constant is denoted the “numerical full cumulant expansion approach.” Further simplification occurs by neglecting the equilibrium dynamics of the R coordinate (i.e., assuming that the characteristic time scale of the R coordinate correlation function is much longer than the time scale associated with solvent damping) and then assuming that the reactant and product equilibrium proton donor-acceptor distances are equal. Invoking all of these approximations leads to the following closed-form analytical rate constant expression:
| (2) |
where the summations are over reactant and product electron-proton vibronic states, Pμ is the Boltzmann population of reactant state μ, ν is the total reorganization energy, ΔGμν is the reaction free energy for states μ and ν, and M and Ω are the effective mass and frequency, respectively, associated with the R-mode, which is assumed to behave as an undamped quantum harmonic oscillator. Moreover, and are the coupling reorganization energies corresponding to the linear and quadratic terms, respectively, in the coupling, , and .
This expression can be further simplified in the low-frequency ( ) and high-frequency ( ) limits for the R-mode by substituting the corresponding limits for . The low-frequency expression is
| (3) |
and the high-frequency expression is
| (4) |
These expressions were originally derived under the assumption that 14 and were subsequently derived for nonzero values of .15
We emphasize that additional rate constant expressions have been derived for cases that do not satisfy the assumptions leading to Eq. (2). For example, the rate constant expression associated with the numerical full cumulant expansion approach includes the effects from the dynamical interference between the proton donor-acceptor motion and the fluctuations of the energy gap but requires numerical integration for nonzero values of .15 Furthermore, inclusion of the contributions arising from differences between the reactant and product equilibrium proton donor-acceptor distances is also straightforward.14, 15
B. Thermal averaging approach
An alternative to the cumulant expansion approach is the thermal averaging approach,14, 38, 39 which is based on the assumptions that the proton donor-acceptor mode remains in thermal equilibrium during the reaction, the nonadiabatic transitions occur independently at all values of R, and the relaxation along R is fast compared to the rate of nonadiabatic transitions. Given these assumptions, the rate constant can be expressed as a thermal average, namely the product of the rate constant as a function of R and the equilibrium probability distribution function P(R) integrated over R. Neglecting the weak dependence of the reorganization energy and the reaction free energy on R, this rate constant is
| (5) |
where is the thermal average of the square of the vibronic coupling. For the exponential form of the coupling given in Eq.(1), this thermal average is
| (6) |
In the low-frequency ( ) and high-frequency ( ) limits of a harmonic oscillator representation of , the integral in Eq. (6) can be evaluated analytically, resulting in the same rate constants as given in Eqs. (3) and (4) for the cumulant expansion approach.15, 39
The general thermal averaging approach does not require the assumption of a specific form of the vibronic coupling or . In particular, the vibronic coupling can be calculated analytically or numerically at each R coordinate along a grid, and the probability distribution function can include anharmonic effects or can be evaluated numerically at each R coordinate.40 In this explicit thermal averaging approach, the first integral given in Eq. (6) is evaluated numerically. Note that the thermal averaging approach neglects the dynamical interference effects between the R-mode and the solvent fluctuations, which can be included in the cumulant expansion approach. From a theoretical perspective, the cumulant expansion is viewed as more rigorous because the equilibrium dynamics along the R coordinate is treated explicitly on the same level as the dynamics of the energy gap in the most general form.
C. Two-dimensional full quantum approach
The two-dimensional (2D) quantum approach treats the coordinate on the same level as the electron and proton coordinates. In this case, the reactant and product vibronic states are defined as solutions of the vibrational Schrödinger equation for the proton and its donor and acceptor moving on 2D diabatic electronic potentials that depend on the proton coordinate and R. In the case of 2D harmonic potentials, the solutions of the vibrational Schrödinger equation can be obtained analytically.37 For more realistic anharmonic 2D potentials, the 2D vibrational wavefunctions can be calculated numerically using, for example, Fourier Grid Hamiltonian methods.41, 42 In some cases, the calculation of these 2D vibrational wavefunctions can be simplified by invoking the adiabatic separation between the proton coordinate and the R coordinate.
In the models studied in this paper, the R coordinate is uncoupled from the proton coordinate in the 2D diabatic potentials corresponding to the reactant and product states in the PCET reaction. In this case, the initial and final electron-proton vibronic states are simple products of the proton vibrational wavefunctions and harmonic wavefunctions for the R coordinate. The overlap integrals between these vibronic wavefunctions can be calculated numerically, and the nonadiabatic rate constant can be calculated by direct application of the Golden Rule expression with averaging over the reactant states and summing over the product states.14, 15 The resulting rate constant converged with respect to the number of included vibronic states is considered to be “exact” in the framework of the Golden Rule approximation and can be used to validate the approximate analytical rate constant expressions.
D. Approximate expressions for kinetic isotope effect
As defined in the Introduction, the KIE is the ratio of the rate constants for hydrogen and deuterium. Previously expressions were derived for the KIE under the assumptions that the reorganization energy and driving force are independent of isotope, , and only the ground reactant and product vibronic states contribute to the rate constants. In this case, the KIE can be approximated as
| (7) |
where and are the couplings for hydrogen and deuterium, respectively, at and and are the linear attenuation parameters for hydrogen and deuterium respectively, for the ground vibronic states. The expressions for are more complicated, even when the other assumptions are still invoked. In the low-frequency limit of the R-mode, the KIE can be approximated as
| (8) |
where and are the quadratic attenuation parameters for hydrogen and deuterium respectively, for the ground vibronic states. The analogous expression in the high-frequency limit of the R-mode can easily be obtained from Eq. (4), and the more general expression can be obtained from Eq. (2). The temperature dependences of these KIE expressions can be determined by taking the derivative of ln(KIE) with respect to temperature.
3. Vibronic couplings
The vibronic coupling Vμν(R) appearing in the rate constant expressions for PT and PCET can be calculated in the general case and in the electronically adiabatic and nonadiabatic regimes using previously derived expressions. In the general case, which spans the electronically adiabatic and nonadiabatic regimes, the following semiclassical expression12 for the coupling between two vibronic states can be utilized:
| (9) |
Here is the vibronic coupling in the electronically adiabatic limit and
| (10) |
where is the gamma function and is the electron-proton adiabaticity parameter. This adiabaticity parameter is defined in terms of the effective electronic transition time
| (11) |
and the effective proton tunneling time
| (12) |
where is the electronic coupling between the reactant and product diabatic electronic states43 and |ΔF| is the difference between the slopes of the electronically diabatic potentials at the crossing point. Moreover, the tunneling velocity is
| (13) |
where is the energy at which the potential energy curves cross, E is the tunneling energy associated with the unperturbed degenerate proton vibrational levels in the reactant and product diabatic potentials, and m is the mass of the proton.
The adiabaticity parameter can be used to determine if a system is in the electronically adiabatic or nonadiabatic regime. When , the system is in the electronically adiabatic regime, in which the electrons respond instantaneously to the proton motion, resulting in the reaction proceeding entirely on the ground adiabatic electronic state. When , the system is in the electronically nonadiabatic regime, in which the electrons do not respond instantaneously to the proton motion, resulting in involvement of the excited electronic state. In addition to the adiabaticity parameter, which reflects the relative effective timescales of the electronic transition and proton tunneling, other diagnostics for this type of electron-proton nonadiabaticity have been devised.44, 45 Specifically, the reaction is electronically nonadiabatic when the component of the nonadiabatic coupling vector between the ground and first excited adiabatic electronic states along the proton coordinate is large and when a significant amount of electronic charge redistribution occurs in these two electronic states during proton transfer. Most PT and HAT reactions are electronically adiabatic, whereas EPT reactions, which are concerted PCET reactions involving electron and proton transfer between different donors and acceptors, are typically electronically nonadiabatic.9, 13
Approximate expressions for the vibronic coupling in the electronically adiabatic and nonadiabatic regimes have been derived previously.12 In the electronically adiabatic limit, the semiclassical vibronic coupling simplifies to , which can be computed as half the tunneling splitting between the proton vibrational levels in the ground state adiabatic electronic potential. Several semiclassical expressions have been derived for in terms of parameters associated with proton double well potentials20, 35, 37, 46, 47 and can also be extended to excited proton vibrational states. In the electronically nonadiabatic limit, the semiclassical vibronic coupling reduces to
| (14) |
where and are the proton vibrational wavefunctions associated with vibrational states μ and ν in the reactant and product diabatic potentials, respectively. This expression is typically used for EPT reactions and will be used in the application to SLO discussed below.
4. Case study: Soybean lipoxygenase
A. Determination of appropriate rate constant expression
As mentioned in the Introduction, SLO catalyzes the PCET reaction involving an electron transfer from the π backbone of the linoleic acid substrate to the iron of the cofactor, as well as a proton transfer from C11 of the substrate to the oxygen of the iron-bound hydroxide. In our previous work,26, 32, 33 we modeled the SLO reaction for both the wild-type enzyme and various mutants using the analytical rate constant expressions above, neglecting the quadratic terms in Eq.(1) for the vibronic coupling. In other words, we assumed that for all of our previous calculations on this system. In the present work, we perform these calculations with the more exact expressions provided above to determine the significance of the quadratic terms in the coupling for this system. In the process, we identify the regimes in which these quadratic terms become important and the regimes in which other approximations underlying some of the commonly used rate constant expressions break down. Such insights will guide studies of other PCET systems.
The diagnostics described in Section 3 have been used to determine whether SLO is in the electronically adiabatic or nonadiabatic regime.25 For this purpose, constrained DFT calculations were performed on the model system depicted in Figure 2, and the diabatic proton potentials were calculated along the transferring hydrogen coordinate. According to the semiclassical formalism, the electronic transition time is much larger than the effective proton tunneling time: . Moreover, the dipole moment of the system was observed to change suddenly and significantly during the hydrogen transfer reaction, as depicted in Figure 2. Both of these diagnostics strongly indicate that the SLO reaction is electronically nonadiabatic. Moreover, the overall vibronic coupling is much less than the thermal energy , thereby justifying the use of the Golden Rule formalism for this system. Thus, the appropriate rate constant expression is Eq.(2) with the vibronic coupling from Eq. (14).
Figure 2.
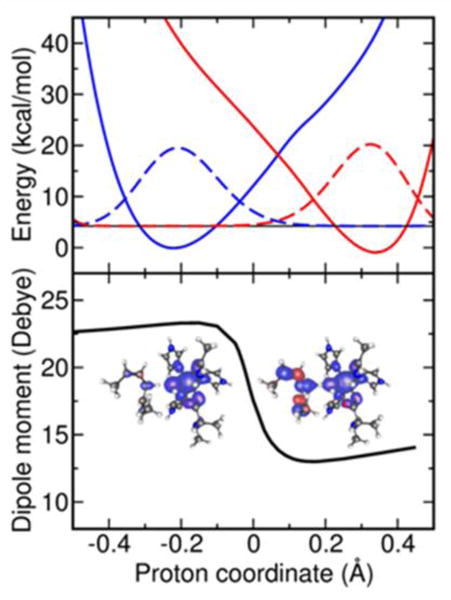
Diabatic electronic potentials along the proton coordinate obtained with CDFT-CI/ωB97X/6-31G** with the corresponding ground state proton vibrational wavefunctions (top panel) and the total dipole moment along the proton coordinate obtained with DFT/ωB97X/6-31G** (bottom panel) at the transition state geometry for the SLO model system studied in Ref.24. The insets in the bottom panel depict the spin densities of the reactant and product diabatic electronic states calculated at the minimum of each diabatic curve in the top panel. The spin densities show that the unpaired spin density is localized near the iron center for both diabatic states and is also delocalized along the π-backbone for the product state.
For comparison, we note that a purely adiabatic treatment has also been used to study the wild-type SLO reaction.48, 49 This adiabatic approach is based on transition state theory with semiclassical tunneling corrections obtained from a simple truncated inverted parabola model. Such an approach was able to reproduce the magnitude and temperature dependence for wild-type SLO using physically reasonable parameters for the simple parabolic model. However, an adiabatic treatment is unlikely to be able to reproduce the KIE of 500 – 700 measured experimentally for the double mutant SLO.26 Moreover, as discussed above, several independent diagnostics have been used to determine that the SLO reaction is both electronically and vibronically nonadiabatic.25 A comparison of adiabatic and nonadiabatic rate constant expressions, as well as a more comprehensive discussion of quantitative diagnostics to determine which expression is valid for a specific system, is provided elsewhere.50
B. Determination of input parameters for rate constant expression
The input parameters to this rate constant expression were determined in previous work. The reactant and product diabatic potentials along the proton coordinate were modeled by standard Morse potentials given by
| (15) |
where XY corresponds to the C–H and O–H bonds for the reactant and product diabatic potentials, respectively. The minima of the two Morse potentials associated with the reactant and product are separated by the difference between , the equilibrium proton donor–acceptor distance, and the C–H and O–H equilibrium bond lengths, which are chosen to be = 1.09 Å and = 0.96 Å, respectively. The values for the remaining Morse parameters for the C–H and O–H bonds in SLO are as follows:32 the bond dissociation energies are DCH = 77 kcal/mol and DOH = 82 kcal/mol, and the width parameters are βCH = 2.068 Å−1 and βOH = 2.442 Å−1.
The hydrogen and deuterium vibrational wavefunctions for the Morse potentials, as well as the overlap integrals between the reactant and product vibrational wavefunctions, were calculated analytically.51 The coupling attenuation parameters and were calculated by numerical evaluation of the first and second derivatives, respectively, of the vibronic coupling (i.e., the overlap integral between proton vibrational states μ and ν in the electronically nonadiabatic regime) at R0. Note that these numerical derivatives may be problematic for excited proton vibrational states, particularly at relatively small values of , because of the nodes in the distance dependence of the overlap integrals. Thus, the explicit thermal averaging approach is more suitable than the analytical expressions for systems in which highly excited vibrational states contribute significantly. For SLO, only the lowest three reactant and product proton vibrational states contribute, and the dominant contribution arises from the ground vibrational states. As a result, the logarithmic expansion of the coupling and numerical evaluation of the coupling attenuation parameters are reliable for this system.
The other parameters were determined utilizing available experimental data. The reaction free energies were calculated as , where and correspond to the proton vibrational state energy levels in the reactant and product diabatic potentials, respectively, and was estimated to be 5.4 kcal/mol on the basis of experimental data.32 The reorganization energy, λ, was chosen to be 13.4 kcal/mol to reproduce the temperature dependence of the absolute rate constant for wild-type SLO.26 The effective mass M associated with the proton donor-acceptor mode was assumed to be 100 amu in the previous studies of SLO.26, 33 Recent studies of molecular systems suggest that the effective mass M is typically smaller, even for relatively delocalized vibrational modes.40 Thus, we explore the results with both M = 10 and M = 100 amu in the present work, adjusting the frequency Ω accordingly. The electronic coupling, Vel, was chosen to be 1.7 cm−1 for M = 100 amu and 5.7 cm−1 for M = 10 amu to reproduce the magnitude of the absolute rate constant for wild-type SLO at 303 K with each of these effective masses. Note that the electronic coupling does not impact the KIE because it cancels out in the ratio of rate constants. The equilibrium proton donor-acceptor distance and the associated frequency Ω were chosen to reproduce the magnitude and temperature dependence of the KIE for wild-type SLO.
These parameters were retained for the calculations of the mutants with the following exceptions. The equilibrium proton donor-acceptor distance and the associated frequency Ω were varied to reproduce the magnitude and temperature dependence of the KIE for the series of I553 mutants, while keeping all other parameters fixed. We found that these two parameters are independent of the reorganization energy within the physically reasonable regime and therefore kept λ = 13.4 kcal/mol for these mutants.26 For the double mutant L546A/L754A, however, the reorganization energy was increased to λ = 45.6 kcal/mol to reproduce the temperature dependence of the absolute rate constant for this double mutant. The enormous KIE of 500 – 700 was reproduced by covariance of and Ω, although the specific values could not be determined definitively in the absence of the experimental temperature dependence of the KIE.
C. Results on wild-type and mutant SLO
All of our previous calculations included only the linear term in the vibronic coupling exponential. To determine the impact of the quadratic term in the vibronic coupling, we compared the rate constant and the KIE as a function of the inverse temperature using five different methods: the 2D full quantum approach, the numerical full cumulant expansion approach with inclusion of the cumulants up to 10-th order,15 the explicit thermal averaging approach, and the analytical expression in Eq. (2) with and with (i.e., without quadratic terms and with quadratic terms). The rate constants and KIEs for M = 10 amu and for M = 100 amu are depicted in Figure 3. The parameters and Ω were fit to reproduce the experimental magnitude and temperature dependence of the KIE for each value of the effective mass M using the analytical expression in Eq. (2) with (solid black lines in Figure 3). The parameters and Ω were the same for all five methods for each mass. For the effective mass M = 100 amu, the rate constants and KIES obtained with the 2D full quantum approach, the numerical full cumulant expansion approach, the explicit thermal averaging approach, and the analytical expression with the quadratic terms are similar (dashed red, green, blue, and solid black lines in Figure 3). This agreement provides validation for the approximations used to derive the analytical expression in Eq. (2). For the effective mass M = 10 amu, however, greater discrepancies are observed. Due to the higher frequency Ω = 368 cm−1 associated with the smaller mass, the interference between the equilibrium dynamics of the R coordinate and the fluctuations of the energy gap becomes more important because of the shorter time scale of the R coordinate time correlation function. As a result, we observe more significant deviations of the analytical rate constants calculated using Eq. (2) from the rate constant obtained with the 2D full quantum approach (solid black and dashed red lines). Note that the rate constant obtained using the numerical full cumulant expansion approach, which includes these interference effects, agrees well with the rate constant obtained with the 2D full quantum approach (dashed green and red lines in Figure 3). In principle, the numerical full cumulant expansion approach could be used to fit the parameters and Ω to the experimental data.
Figure 3.
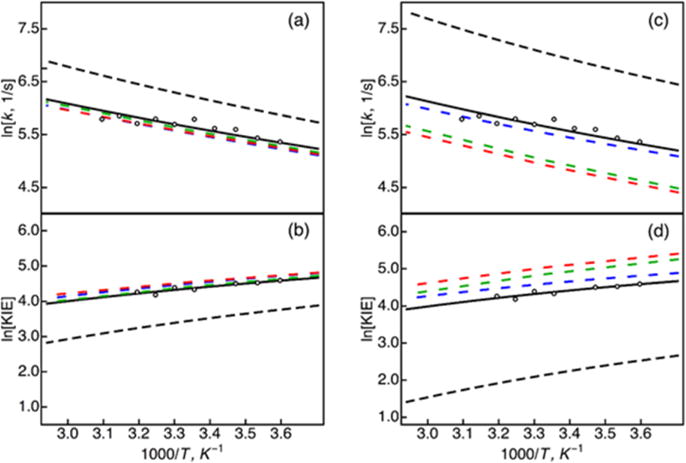
PCET rate constants, (a) and (c), and KIEs, (b) and (d), as a function of temperature, calculated using the 2D full quantum approach (dashed red lines), the numerical full cumulant expansion approach with cumulants up to 10-th order (dashed green lines), the explicit thermal averaging approach (dashed blue lines), and the analytical expression in Eq. (2) with quadratic terms, (solid black lines), and without quadratic terms, (dashed black lines). The rate constants and KIEs in panels (a) and (b) were calculated with the effective proton donor-acceptor mode mass M = 100 amu, = 2.77 Å, and Ω = 132.8 cm−1 for panels (a) and (b) and M = 10 amu, R0 = 2.88 Å, and Ω = 368.2 cm−1 for panels (c) and (d). The open circles represent the experimental data for wild-type SLO, as provided in Ref.21.
The rate constants and KIEs for the analytical expression without the quadratic terms are not in good agreement with the results obtained with the other methods (dashed black lines in Figure 3). Specifically, the rate constants are too high, and the KIEs are too low. Note that and Ω could be varied to obtain excellent agreement between the analytical expression without the quadratic terms and the experimental result, as in our previous work. Thus, the simpler expressions including only the linear term in the coupling contain the essential physics of PCET, but including the quadratic terms provides a more quantitatively accurate description of PCET rate constants. We also found that the low-frequency expression given by Eq. (3) is in good agreement with the more general expression given by Eq. (2) for M = 100 amu but deviates significantly from the more general expression for M = 10 amu because the frequency is much higher for the smaller mass. Thus, as expected, the low-frequency approximation also breaks down for lower values of the effective mass.
The calculated and experimental KIEs as a function of temperature for wild-type SLO and the series of I553 mutants are depicted in Figure 4, and the values of and Ω determined by fitting to the experimental data are provided in Table 1. In addition, the calculated KIEs for the double mutant are depicted in Figure 5 to illustrate the range of reasonable and Ω values that can reproduce the experimental KIE of 500 – 700, indicated by the shaded horizontal strip in the figure. The calculated KIEs in both of these figures were obtained using the analytical expression in Eq. (2) including the quadratic terms for M = 100 amu. These data are qualitatively similar to our previous results that were based on Eq. (3),26, 33 which is the low-frequency limit of Eq. (2), without the quadratic terms (i.e., ). The analogous results for M = 10 amu are also qualitatively similar after reparameterization of and Ω. Thus, the trends and conclusions based on this analysis are not sensitive to the specific analytical rate constant expression as long as the parameters are modified accordingly. On the other hand, because inclusion of the quadratic terms in the coupling is more mathematically rigorous, the values of the parameters are more meaningful when these terms are included in the general expression in Eq. (2).
Figure 4.
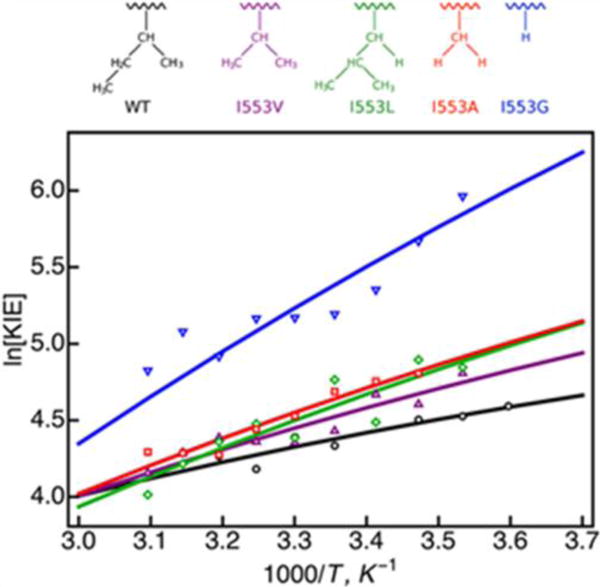
KIE versus inverse temperature for WT SLO and the series of I553 mutants. The lines were obtained from calculations using the analytical expression with quadratic terms, as given in Eq. (2), in conjunction with the parameters given in Table 1. Open symbols are the experimental data from Ref.1. The color scheme corresponds to the I553 mutants with residues depicted above the plot: WT (black), I553V (purple), I553L (green), I553A (red), and I553G (blue).
Table 1.
Proton donor-acceptor equilibrium distances R0 and frequencies Ω determined for WT SLO and a series of I553 mutants by fitting the experimental KIE magnitudes and temperature dependences using the analytical rate constant expression with quadratic terms, as given in Eq. (2).
| M = 100 amu | M = 10 amu | |||
|---|---|---|---|---|
| R0, Å | Ω, cm−1 | R0, Å | Ω, cm−1 | |
| WT | 2.77 | 132.8 | 2.88 | 368.2 |
| I553V | 2.89 | 105.5 | 3.00 | 316.3 |
| I553A | 2.97 | 96.3 | 3.08 | 295.1 |
| I553L | 3.00 | 92.8 | 3.10 | 266.4 |
| I553G | 3.24 | 82.0 | 3.35 | 257.5 |
Figure 5.
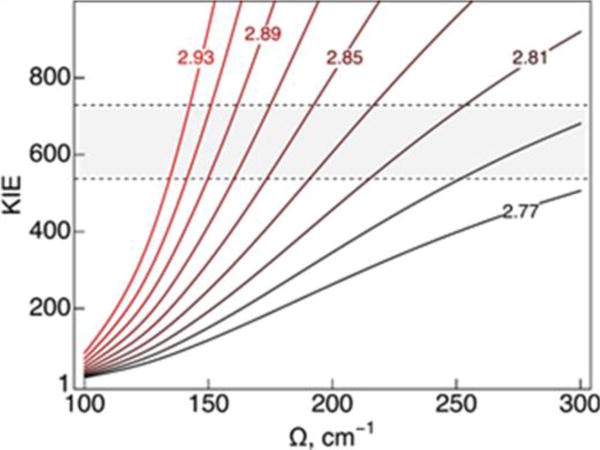
Calculated KIE for SLO double mutant L546A/L754A as a function of the proton donor-acceptor equilibrium distance and associated frequency Ω. The curves are labeled according to R0 given in Å. In this model, the effective mass of the proton donor-acceptor mode is M = 100 amu, the reorganization energy is λ = 45.6 kcal/mol, and the reaction free energy is ΔG° = −5.4 kcal/mol. The shaded area of the plot corresponds to the experimental range of the observed KIE between 30 and 35°C
These calculations provide physical understanding of the experimental data for the wild-type and mutant SLO. The analysis of the KIEs is simplified using the approximate KIE expression given in Eq. (7), which contains the essential physics of PCET but is not as quantitatively accurate as calculating the ratio of hydrogen and deuterium rate constants with the full expression. The magnitude of the KIE is strongly influenced by the prefactor, which is the ratio of the square of the vibronic coupling for hydrogen and deuterium. For the electronically nonadiabatic regime, where the coupling is given by Eq. (14), this prefactor is simply the ratio of the square of the overlap integral for hydrogen and deuterium. Both the hydrogen and deuterium overlaps decrease as the proton donor-acceptor distance increases, but the deuterium overlap decreases faster because of its larger mass. Consequently, the ratio of overlaps increases as the proton donor-acceptor distance increases or, more generally, as the overlap decreases.52 In SLO, the proton is transferred from carbon to oxygen through a relatively weak CH—O hydrogen bond. Thus, the equilibrium proton donor-acceptor distance is relatively large, and the overlap is relatively small, leading to a large prefactor in the KIE expression in Eq. (7).23, 32 In addition, the ground vibronic states are the dominant contributors to the overall rate constant for SLO. When excited states contribute significantly, the KIE tends to be moderated by the larger overlaps between excited proton vibrational wavefunctions.
The changes in the magnitude and temperature dependence of the KIE observed experimentally for the series of I553 mutants can also be explained physically based on the approximate KIE expression given in Eq. (7). The calculations indicate that the equilibrium proton donor-acceptor distance increases and the associated frequency decreases as residue I553 becomes less bulky (Table 1).33 As the equilibrium proton donor-acceptor distance increases, the ratio of the square of the overlap for hydrogen and deuterium increases, and therefore the magnitude of the KIE increases, as illustrated across the series in Figure 4. As the frequency of the proton donor-acceptor mode decreases, the temperature dependence of the KIE increases because of the exponential term in Eq. (7). The increase in temperature dependence across the series illustrated in Figure 4 reflects this decrease in the frequency. Thus, these analytical rate constant expressions provide physical explanations for the experimentally observed trends in the KIE as residue 553 becomes less bulky.
Figure 5 illustrates that the enormous KIE of 500 – 700 observed experimentally for the double mutant can be reproduced using physically reasonable parameters.26 Specifically, the enormous KIE can be reproduced using equilibrium proton donor-acceptor distances of 2.8 – 2.9 Å, in conjunction with frequencies of 150 – 250 cm−1. For the I553 mutant series, the proton donor-acceptor mode frequency decreased as the equilibrium distance increased, thereby allowing the mutants to effectively sample the shorter distances associated with a similar rate constant and KIE as observed for wild-type SLO. In other words, the I553 mutants can recover from the longer equilibrium distance by sampling the shorter distances with a low energy penalty. In contrast, for the double mutant, the frequency remains similar to the value determined for wild-type SLO, even though the equilibrium distance increases. Thus, the double mutant is not able to sample the shorter distances effectively and cannot recover from the longer equilibrium distance. The result is that the double mutant has a very slow rate constant and an enormous KIE compared to wild-type SLO. The temperature dependence of the KIE for the double mutant is predicted to be similar to that for wild-type SLO (i.e., relatively weak) on the basis of the similar proton donor-acceptor mode frequency. In this case, the analytical rate constant expressions provide a physical explanation for this unprecedented colossal KIE in the double mutant. Understanding the underlying physical principles dictating the KIE also enables predictions that can be tested experimentally. For example, an unusually large KIE is predicted to occur for systems with proton transfer interfaces that are constrained to non-optimal distances or orientations without the flexibility to recover.
The sensitivity of the rate constants and KIEs to the various parameters in the rate constant expressions has been examined previously.4, 50, 52, 53 For the specific case of SLO, previous work has shown that the KIE is not sensitive to the choice of the reorganization energy within a physically reasonable regime (i.e., 10 – 50 kcal/mol).23, 26, 33 The KIE will not be sensitive to the reaction free energy as long as the ground vibronic states are dominant in the rate constant expressions but could change significantly when contributions from excited vibronic states become dominant. As discussed above through analysis of Eq. (7), the KIE is determined mainly by the equilibrium proton donor-acceptor distance and its associated frequency. The sensitivity of the magnitude and temperature dependence of the KIE to these parameters is illustrated by analysis of the series of I553 mutants through Table 1 and Figure 4 and the double mutant through Figure 5.
Although this application to SLO required fitting of parameters to experimental data, in other cases the input parameters to the analytical rate constant expressions can be determined from electronic structure calculations and molecular dynamics simulations, thereby entirely avoiding this empirical approach. The solvent reorganization energy can be calculated using dielectric continuum theory23 or molecular dynamics simulations,32 and the reaction free energy can be determined by calculating the relevant pKa’s and reduction potentials. The equilibrium proton donor-acceptor distance can be obtained from a geometry optimization using electronic structure methods. Moreover, the effective mass and frequency of the proton donor-acceptor mode can be calculated by projecting all of the normal modes onto the proton donor-acceptor axis and combining the frequencies and masses of the normal modes with appropriate weightings.40 The proton potentials can be calculated with electronic structure methods for each proton donor-acceptor distance,40 and the proton vibrational wavefunctions and overlaps can be determined with Fourier Grid Hamiltonian methods.41 All of this information can be used as input to the analytical rate constant expressions or can be used in conjunction with the explicit thermal averaging approach.40, 54 In this manner, PCET rate constants can be calculated without the assistance of any empirical data, although comparison to experimental data is useful for validating the results.
5. Conclusions
This paper presents a series of analytical rate constant expressions for PCET reactions in well-defined regimes. A comparison among the theoretical approaches indicates that these theories should be used with an understanding of the underlying approximations and knowledge of the appropriate regimes of validity for each rate constant expression. The case study of SLO illustrates that these expressions are useful for modeling chemical and biological reactions and can provide significant physical insights that assist in the interpretation of experimental data and provide experimentally testable predictions. Understanding the fundamental physical principles dictating PCET reactions and elucidating the overarching concepts in the field is critical for designing more effective catalysts for applications in biomedical and energy science.
In addition to these types of theoretical treatments, molecular dynamics simulations of PCET reactions can provide further information about these systems.55–58 Nonadiabatic surface hopping methods59, 60 are particularly conducive to describing PCET processes. Such methods are critical for studying the nonequilibrium dynamics of photoinduced PCET because the analytical expressions described above assume that the solvent is in equilibrium and therefore cannot describe the nonequilibrium dynamics. Recent nonadiabatic dynamics simulations of photoinduced PCET in solution have elucidated the role of solvent dynamics and proton vibrational relaxation in these types of processes.56–58 In general, a full understanding of complex PCET processes will require a combination of analytical theories and molecular dynamics simulations.
Acknowledgments
We are grateful for support from National Institutes of Health Grant GM056207 (applications to enzymes) and the National Science Foundation Grant CHE-13-61293 (fundamental PCET theory).
Notes and references
- 1.Meyer MP, Tomchick DR, Klinman JP. Proc Natl Acad Sci USA. 2008;105:1146–1151. doi: 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 3.Huynh MHV, Meyer TJ. Chem Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammes-Schiffer S, Soudackov AV. J Phys Chem B. 2008;112:14108–14123. doi: 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammes-Schiffer S, Stuchebrukhov AA. Chem Rev. 2010;110:6939–6960. doi: 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Chem Rev. 2012;112:4016–4093. doi: 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- 8.Hammes-Schiffer S. J Am Chem Soc. 2015;137:8860–8871. doi: 10.1021/jacs.5b04087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammes-Schiffer S. Energy Environ Sci. 2012;5:7696–7703. [Google Scholar]
- 10.Cukier RI. J Phys Chem. 1994;98:2377–2381. [Google Scholar]
- 11.Soudackov AV, Hammes-Schiffer S. J Chem Phys. 2000;113:2385–2396. [Google Scholar]
- 12.Georgievskii Y, Stuchebrukhov AA. J Chem Phys. 2000;113:10438–10450. [Google Scholar]
- 13.Skone JH, Soudackov AV, Hammes-Schiffer S. J Am Chem Soc. 2006;128:16655–16663. doi: 10.1021/ja0656548. [DOI] [PubMed] [Google Scholar]
- 14.Soudackov AV, Hatcher E, Hammes-Schiffer S. J Chem Phys. 2005;122:014505. doi: 10.1063/1.1814635. [DOI] [PubMed] [Google Scholar]
- 15.Soudackov AV, Hammes-Schiffer S. J Chem Phys. 2015;143:194101. doi: 10.1063/1.4935045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trakhtenberg LI, Klochikhin VL, Pshezhetsky SY. Chem Phys. 1982;69:121–134. [Google Scholar]
- 17.Borgis D, Lee S, Hynes JT. Chem Phys Lett. 1989;162:19–26. [Google Scholar]
- 18.Suarez A, Silbey RJ. J Chem Phys. 1991;94:4809–4816. [Google Scholar]
- 19.Smedarchina ZK. Chem Phys. 1991;150:47–56. [Google Scholar]
- 20.Borgis D, Hynes JT. Chem Phys. 1993;170:315–346. [Google Scholar]
- 21.Borgis D, Hynes JT. J Phys Chem. 1996;100:1118–1128. [Google Scholar]
- 22.Knapp MJ, Rickert K, Klinman JP. J Am Chem Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 23.Hatcher E, Soudackov AV, Hammes-Schiffer S. J Am Chem Soc. 2004;126:5763–5775. doi: 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- 24.Lehnert N, Solomon EI. J Biol Inorg Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
- 25.Soudackov AV, Hammes-Schiffer S. J Phys Chem Lett. 2014;5:3274–3278. doi: 10.1021/jz501655v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu S, Sharma SC, Scouras AD, Soudackov AV, Carr CAM, Hammes-Schiffer S, Alber T, Klinman JP. J Am Chem Soc. 2014;136:8157–8160. doi: 10.1021/ja502726s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mincer JS, Schwartz SD. J Chem Phys. 2004;120:7755–7760. doi: 10.1063/1.1690239. [DOI] [PubMed] [Google Scholar]
- 28.Olsson MHM, Siegbahn PEM, Warshel A. J Am Chem Soc. 2004;126:2820–2828. doi: 10.1021/ja037233l. [DOI] [PubMed] [Google Scholar]
- 29.Siebrand W, Smedarchina Z. J Phys Chem B. 2004;108:4185–4195. [Google Scholar]
- 30.Tejero I, Garcia-Viloca M, Gonzalez-Lafont A, Lluch JM, York DM. J Phys Chem B. 2006;110:24708–24719. doi: 10.1021/jp066263i. [DOI] [PubMed] [Google Scholar]
- 31.Mavri J, Liu H, Olsson MHM, Warshel A. J Phys Chem B. 2008;112:5950–5954. doi: 10.1021/jp0758420. [DOI] [PubMed] [Google Scholar]
- 32.Hatcher E, Soudackov AV, Hammes-Schiffer S. J Am Chem Soc. 2007;129:187–196. doi: 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- 33.Edwards SJ, Soudackov AV, Hammes-Schiffer S. J Phys Chem B. 2010;114:6653–6660. doi: 10.1021/jp100133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trakhtenberg LI, Klochikhin VL, Pshezhetsky SY. Chem Phys. 1981;59:191–198. [Google Scholar]
- 35.Lavtchieva L, Smedarchina Z. Chem Phys Lett. 1991;184:545–552. [Google Scholar]
- 36.Cukier RI. J Phys Chem. 1996;100:15428–15443. [Google Scholar]
- 37.Benabbas A, Salna B, Sage JT, Champion PM. J Chem Phys. 2015;142:114101. doi: 10.1063/1.4913591. [DOI] [PubMed] [Google Scholar]
- 38.Kuznetsov AM, Ulstrup J. Can J Chem. 1999;77:1085–1096. [Google Scholar]
- 39.Hammes-Schiffer S, Hatcher E, Ishikita H, Skone JH, Soudackov AV. Coord Chem Rev. 2008;252:384–394. doi: 10.1016/j.ccr.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auer B, Fernandez LE, Hammes-Schiffer S. J Am Chem Soc. 2011;133:8282–8292. doi: 10.1021/ja201560v. [DOI] [PubMed] [Google Scholar]
- 41.Marston CC, Balint-Kurti GG. J Chem Phys. 1989;91:3571–3576. [Google Scholar]
- 42.Balint-Kurti GG, Ward CL. Comput Phys Commun. 1991;67:285–292. [Google Scholar]
- 43.Newton MD. Chem Rev. 1991;91:767–792. [Google Scholar]
- 44.Sirjoosingh A, Hammes-Schiffer S. J Phys Chem A. 2011;115:2367–2377. doi: 10.1021/jp111210c. [DOI] [PubMed] [Google Scholar]
- 45.Sirjoosingh A, Hammes-Schiffer S. J Chem Theor Comput. 2011;7:2831–2841. doi: 10.1021/ct200356b. [DOI] [PubMed] [Google Scholar]
- 46.Child MS. Molecular Collision Theory. Academic Press; New York: 1974. [Google Scholar]
- 47.Fain B. Theory of Rate Processes in Condensed Media. Springer; New York: 1980. [Google Scholar]
- 48.Truhlar DG. J Phys Org Chem. 2010;23:660–676. [Google Scholar]
- 49.Glowacki DR, Harvey JN, Mulholland AJ. Nature Chemistry. 2012;4:169–176. doi: 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- 50.Layfield JP, Hammes-Schiffer S. Chem Rev. 2014;114:3466–3494. doi: 10.1021/cr400400p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahl JP, Springborg M. J Chem Phys. 1988;88:4535–4547. [Google Scholar]
- 52.Edwards SJ, Soudackov AV, Hammes-Schiffer S. J Phys Chem A. 2009;113:2117–2126. doi: 10.1021/jp809122y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Decornez H, Hammes-Schiffer S. J Phys Chem A. 2000;104:9370–9384. [Google Scholar]
- 54.Horvath S, Fernandez LE, Soudackov AV, Hammes-Schiffer S. Proc Natl Acad Sci USA. 2012;109:15663–15668. doi: 10.1073/pnas.1118333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kretchmer JS, Miller TF., III J Chem Phys. 2013;138:134109. doi: 10.1063/1.4797462. [DOI] [PubMed] [Google Scholar]
- 56.Goyal P, Hammes-Schiffer S. J Phys Chem Lett. 2015;6:3515–3520. doi: 10.1021/acs.jpclett.5b01475. [DOI] [PubMed] [Google Scholar]
- 57.Goyal P, Schwerdtfeger CA, Soudackov AV, Hammes-Schiffer S. J Phys Chem B. 2015;119:2758–2768. doi: 10.1021/jp5126969. [DOI] [PubMed] [Google Scholar]
- 58.Goyal P, Schwerdtfeger CA, Soudackov AV, Hammes-Schiffer S. J Phys Chem B. 2016;120:2407. doi: 10.1021/acs.jpcb.5b12015. [DOI] [PubMed] [Google Scholar]
- 59.Tully JC. J Chem Phys. 1990;93:1061–1071. [Google Scholar]
- 60.Hammes-Schiffer S, Tully JC. J Chem Phys. 1994;101:4657–4667. [Google Scholar]