

Abstract
Proteolysis is a vital mechanism to regulate the cellular proteome in all kingdoms of life, and ATP-dependent proteases play a crucial role within this process. In Escherichia coli, ClpYQ is one of the primary ATP-dependent proteases. In addition to function with removals of abnormal peptides in the cells, ClpYQ degrades regulatory proteins if necessary and thus let cells adjust to various environmental conditions. In E. coli, SulA, RcsA, RpoH and TraJ as well as RNase R, have been identified as natural protein substrates of ClpYQ. ClpYQ contains ClpY and ClpQ. The ATPase ClpY is responsible for protein recognition, unfolding, and translocation into the catalytic core of ClpQ. In this study, we use an indirect identification strategy to screen possible ClpY targets with E. coli K12 proteome chips. The chip assay results showed that YbaB strongly bound to ClpY. We used yeast two-hybrid assay to confirm the interactions between ClpY and YbaB protein and determined the Kd between ClpY and YbaB by quartz crystal microbalance. Furthermore, we validated that YbaB was successfully degraded by ClpYQ protease activity using ClpYQ in vitro and in vivo degradation assay. These findings demonstrated the YbaB is a novel substrate of ClpYQ protease. This work also successfully demonstrated that with the use of recognition element of a protease can successfully screen its substrates by indirect proteome chip screening assay.
Protease is responsible for protein degradation that can digest long protein chains into shorter fragments by splitting the peptide bonds (1). Protease exists in a wide range of organisms. One of the most important functions for protease is to degrade the misfolding or functionless proteins. Misfolding or functionless proteins usually lead to severe diseases, such as Alzheimer's disease, bovine spongiform encephalopathy, cardiovascular disease, cancer, osteoporosis, neurological disorders, and type II diabetes (2–4). In addition, the protease-induced protein regulation is very important in the wide-range organism. Protease-induced protein regulation in bacterial cells is related to the antibiotic resistance, environmental resilience, and infectivity of bacteria (5). Altering the growth environments of bacteria (e.g. adjusting temperature, osmotic pressure, nutrient deficiency, ultraviolet irradiation, addition of antibiotics or other chemicals) can induce abnormal protein synthesis and accumulation in bacterial cells. Under such conditions, external environmental stimuli can activate the response systems of bacteria, including heat-shock and SOS responses, which can trigger the biosynthesis of the corresponding proteases, potentially aiding bacteria in overcoming unfavorable growth conditions (6, 7). The ATP-dependent protease of E. coli provides us an excellent example to understand the importance of protein degradation role in cell physiology. The homologs of these energy (ATP or GTP)-dependent proteases were also found in eukaryotes indicating the similar regulation cascades between prokaryotes and eukaryotes (5). There are four important ATP-dependent proteases in prokaryotes, Lon, FtsH, ClpAP/XP, and ClpYQ (8). Lon protease is a major cytosol protease in E. coli, which is a DNA heat shock protein responsible for degradation of misfolding proteins (9, 10). FtsH is a membrane-bound essential protease, which degrades the heat-shock transcription factor sigma 32 (11). The ClpAP/XP and ClpYQ belong to the Clp protease family. The clp stands for caseinolytic protease, which can degrade casein in vitro (12, 13). ATP-dependent protease ClpYQ is a type of protease complex. The translational products of clpQ+Y+ operon are ClpQ (HslV), the peptidase responsible for proteolysis, and ClpY (HslU), an ATPase with unfolding activity (14). ClpYQ was suggested as an alternative protease to partially restore the function of Lon protease (15, 16). Bochtler et al. (17) reported the protein crystal structure of ClpYQ, which is composed of ClpY and ClpQ hexamers (ClpY6 and ClpQ6). The centers and both ends of two ClpQ hexamers are each connected to a ClpY hexamer, thereby forming a ClpYQ complex (18–20). Previous studies (17, 20) have verified that ClpY can only form a ring complex under the presence of nucleotides such as of ATP, ADP, and nonhydrolysable ATP analogs (e.g. AMP-PNP or ATP-γ-S). By contrast, ClpQ can spontaneously form a six-membered ring. Therefore, synthesis of a complete ClpYQ complex must involve nucleotides including ATP (21). A previous study (17) divided the structure of ClpY into three domains: the N-terminal domain (N domain, Ser2-Lys109, Ile244-Leu332), intermediate domain (I domain, Met110-Ala243), and C-terminal domain (C domain, Gln333-Leu443). The N domain comprises ATP binding sites. The I domain is comparatively further away from ClpQ than the N domain is and contains a double-loop structure for recognizing and tethering substrates. The C domain involves aggregating the atoms in the ClpY and ClpQ complexes (22–24). Previous studies on how ClpY recognizes substrates have revealed that the G90Y91V92G93 pore motif (pore 1 site) located at the center pore of ClpY is conservative and essential for unfolding and transporting substrates (25–27).
Currently, few substrates have been identified for the ClpYQ protease in comparison with other ATP-dependent proteases (25–27). Because the substrate of a protease can reflect its role in a bacterial cell, identifying potential enzyme substrates is essential in protease research. Using substrate-enzyme interactions as indicators for screening enzyme substrates in large batches has become a prevalent approach in recent studies on ATP-dependent protease substrates (28, 29). Because ClpY is responsible for substrate recognition, in this study, we used E. coli proteome microarrays to identify ClpY recognition proteins and the results showed that the ClpY exhibited strong interactions with the YbaB. We also used a yeast two-hybrid assay system to validate the interactions between ClpY and YbaB. Additionally, a quartz crystal microbalance (QCM)1 was used to measure the Kd values of YbaB- ClpY interactions. The in vitro and in vivo degradation tests were conducted to confirm the degradation of YabB by ClpYQ. These results proposed the YabB is a novel target of ATP-dependent protease ClpYQ.
EXPERIMENTAL PROCEDURES
Fabrication of the E. coli K12 Proteome Chips
For the studies of the bacterial proteome, we have constructed the E. coli K12 proteome microarray (30). In short, the E. coli K12 ASKA library (31) was first incubated with 2 × Luria Broth (LB) medium containing 30 μg/ml chloramphenicol at 37 °C overnight. Then, the overnight cultures were diluted with 2 × LB to an OD595 value of 0.1. When the OD595 value reached ∼0.8, isopropyl β-d-thiogalactoside was added to the culture and incubated at 37 °C for ∼2 h. The culture was harvested by centrifugation, and the pellets were stored at −80 °C before purification.
To purify the proteins, the frozen cell pellets were thawed on ice and re-suspended in lysis buffer consisting of 50 mm NaH2PO4, 300 mm NaCl, 30 mm imidazole, CelLyticB, 1 mg/ml lysozyme, 50 units/ml benzonase, proteinase inhibitor mixture, 1 mm phenylmethanesulfonyl fluoride (Sigma-Aldrich, St. Louis, MA), and Ni-NTA Superflow resins (Qiagen, Hilden, Germany) at 4 °C. After 2.5 h incubation on the plate shaker at 4 °C, the mixtures were transferred into 96-well filter plates (Nunc). The plates were then washed with wash buffer I (50 mm NaH2PO4, 300 mm NaCl, 20% glycerol, 20 mm imidazole, and 0.1% Tween 20) and with wash buffer II (50 mm NaH2PO4, 150 mm NaCl, 30% glycerol, 30 mm imidazole, and 0.1% Tween 20, pH 8, Sigma-Aldrich). Finally, the proteins were eluted with elution buffer (50 mm NaH2PO4, 150 mm NaCl, 30% glycerol, 300 mm imidazole, and 0.1% Tween 20, pH 7.5). After the purified proteins were prepared, they were printed in duplicate on aldehyde slides by ChipWriter Pro (Bio-Rad, Hercules, CA) with 48 pins in a cold room. The chips were then stored at −80 °C before utilization.
The E. coli K12 Proteome Chip Assays
To reduce nonspecific binding, the chip was blocked with 1% bovine serum albumin. 0.5 μm ClpY (27) or ClpY (ΔI+Gly) (22, 27) in TBS-T (Tris-buffered saline contains 0.05% Tween 20) contains 5 mm ATP and 1% bovine serum albumin were used to probe the chips in a hybridization chamber with shaking at room temperature for 1 h. After incubation, the chips were washed with TBS-T, and incubated with anti-ClpY antibody (22) for another 1 h. After washed with TBS-T again, DyLight 649 (Thermo Scientific, Waltham, MA) labeled goat anti-rabbit antibodies (Abcam, Cambridge, England) were used to probe the chips at room temperature for 30 min. Finally, the chips were washed several times with TBS-T and distilled water. After the final wash, the chips were dried by centrifugation at 201 × g and then scanned with a microarray scanner (Axon GenePix 4000B).
Bioinformatics Analysis of the Chip Assays
To process the images of the chip assay results, GenePix Pro 6.0 was used to align each protein spot and export all of the images to text files. For data preprocessing, ProCAT (32) was applied to normalize the signals and hit selection. Positive hits were selected based on a local cutoff, which was defined as triple standard deviations above the signal mean for each spot.
Construction of YbaB and ClpY Protein Expression Plasmids for Yeast Two-Hybrid, In Vitro and In Vivo Assay
The primers used for YbaB gene amplification, restriction site generation and his-tag fusion for expression plasmid construction are listed in supplemental Table S1. pGilda and pB42AD (Clontech®, Mountain View, CA) vector plasmids were chosen to construct pB42AD-ybaB and pGilda-ybaB for later yeast two hybridization assay. The YbaB expression plasmid (pB42AD-ybaB or pGilda-ybaB) was constructed using the restrictive enzymes EcoRI and XhoI sites in the two primers and the template from the chromosomal DNA of MG1665; subsequently, the ybaB gene fragment were amplified using polymerase chain reaction (PCR) and then ligated to pB42AD or pGilda vectors, which were prepared using the same restrictive enzymes. Next, target gene constructed in the pB42AD and pGilda plasmid were transformed into the XL-1 Blue E. coli strain, and a selection medium with ampicillin was then used to screen for the transformed bacterial colonies contained the ybaB gene. The clpY gene expression plasmids pB42AD-clpY and pGilda-clpY have already been constructed by Lee et al. (23).
The restrictive enzymes EcoRI and HindIII were used to cleavage the gene from pB42AD-ybaB plasmid, and the gene was then ligated to pMal-c2x (NEB®) and pTH18kr-mbp vectors (35), which were prepared using the same restrictive enzymes, to construct pMal-c2x-ybaB, and pTH18kr-mbp-ybaB. pET21a-(6x his tag)-clpY and pET21a-clpQ-(6x his-tag), the expression vectors used for protein purification of His(6x)-ClpY and ClpQ-His(6x) were constructed by Hsieh et, al (27). The vectors were transformed into E. coli and then screened using a growth medium containing ampicillin (pTH18kr-mbp was screened by kanamycin). Those strains with different plasmids will be stored until required for later validation assay.
Yeast Two-hybrid Assay
Two plasmid sets, pGilda-ybaB and pB42AD-clpY or pGilda-clpY and pB42AD-ybaB, were co-transformed into EGY48 yeast strain to conduct the yeast two-hybrid assays. Interactions between the proteins expressed by the two plasmids were verified by observing whether the lacZ reporter gene in the transformed strain was activated. The EGY48 yeast strain was transformed by first culturing the yeasts in a 5-ml SD/Ura medium at 30 °C for 12–14 h until the OD600 reached to 0.4–0.6. Then yeast cells were centrifuged and washed with sterile water and 100 mm lithium acetate (LiOAc) and resuspended in 240 μl of 50% (v/v) polyethylene glycol (PEG). Mixing 36 μl of 1 m LiOAc, 25 μl of salmon sperm DNA (before use, this reagent was boiled at 100 °C and placed in an ice bath for 10 min), with 50 μl of each plasmid and added to the yeast cell suspension. The mixture was vigorously shaken for 1 min, incubated at 30 °C for 30 min, followed by heat-shock treatment in a 42 °C water bath for 20–25 mins. After heat-shock, the solution was centrifuged at 14,000 rpm for 5 s, and the supernatant was removed. The yeast cells were suspended in 100 μl of sterile water and then smeared on growth plates containing selective growth media (S.D./-Ura/-His/-Trp). After incubated for 2–3 days at 30 °C, a transformed yeast colony was transferred to a new S.D.-Ura/-His/-Trp growth plates at 30 °C for 1–2 days incubation to activate the cells. The activated cells were spread on 80-mg/L X-gal (Gal/Raf/-Ura/-His/-Trp) growth plates and incubated at 30 °C for 2 days to observe the cell growth.
Measurement of Kd Between MBP-YbaB and ClpY
A QCM (Affinix QN μ) was employed to measure the Kd values of the YbaB-ClpY protein interactions, which were expressed in the transformed E. coli cells through pMal-ybaB and pET21a (6x his tag)-clpY (27). The MBP-YbaB and His-ClpY were purified using amylose resin (New England Biolabs, NEB, Ipswich, MA) and Nickel resin (Talon resin, Takara, Kusatsu, Shiga, Japan), respectively. In the beginning, the sensor cell of the QCM was cleaned in a 500-μl sodium dodecyl sulfate (SDS, 1%) solution and then dried with compressed nitrogen gas. Subsequently, the sensor cell was merged with 300 mm HEPES buffer containing 30 mm MgCl2, 3 mm DTT, and 3 mm EDTA, and the 8 μl of MBP-YbaB (1 mg/ml) was immobilized on electrodes. After the frequency of the sensor was decreased and remained stable, 5 μl of MBP-YbaB was added again to confirm the frequency remain stable, indicating that MBP-YbaB already saturated on sensor cell. Then 2 μl of bovine serum albumin (BSA) solution was added to reduce nonspecific binding. The sensor cell was washed twice with 450 μl of HEPES buffer to remove the unbound MBP-YbaB and BSA. Before adding the next sample, the sensor cell was filled with 450 μl of HEPES buffer and stand for 3 min to stabilize the frequency. Finally, 1, 3, 5, 8, and 10 μl samples of ClpY protein (10 ng/μl) were separately loaded into the sensor cell to measure the decrease in frequency. AQUA analysis software (Initium Co., Chigasaki, Kanagawa, Japan) was employed to calculate the Kd values.
MBP-YbaB In Vitro Degradation Test
Two μm ClpY, 2 μm ClpQ, and 1 μm MBP-YbaB were mixed with 15 μl of 4X degradation solution to create a 60-μl reaction solution. The reaction solution was incubated at 37 °C. After incubation started, each 20 μl of the reaction solution was collected at 0, and 4 h, respectively. An equal volume of 2× SDS protein sample buffer solution was added to each reaction solution and mix by vortex. The mixture was boiled at 100 °C for 5 min to denature the protein and inactive protease activity. The mixture containing ClpY, ClpQ, and MBP-YabB was analyzed by SDS-PAGE and then stained with Coomassie blue R-250. The image was scanned by DigiGel® scanner. The degradation of MBP-YabB proteins was also detected by Western blotting with anti-MBP antibodies. The Western blot membrane was exposed to autoradiography film (Classic Blue, MIDSCI). The signal intensity was used to calculate YbaB protein degradation by using Image J software. Means and standard deviations were taken from three independent analyses and the significant differences were calculated by Student's t test (p < 0.05).
MPB-YbaB In Vivo Degradation Assay
The pTH18kr-mbp-ybaB plasmids were transformed into E. coli SG22623 (+ClpYQ) and AC3112 (-ClpYQ) strains. The transformed bacterial cells were cultured in LB broth until the OD600 reached to 0.3–0.4, and 1 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) was then added to induce protein expression for 30 min. Subsequently, spectinomycin (at final concentration, 100 μg/ml) was added to terminate the protein expression. Next, the OD600 values of the culture medium were recorded at 0, and 240 mins. Bacterial cells were also harvested at these time points. The harvested cells were resuspended and centrifuged in sterilized water to remove the growth media solution. An equal volume of 2× SDS protein sample buffer was added to the samples and boiled at 100 °C for 5 mins. The degradation of MBP-YabB proteins was detected by Western blotting with anti-MBP antibodies. The Western blot membrane was exposed to autoradiography film (Classic Blue, MIDSCI) and the image signal intensities were used to calculate YbaB protein degradation by using Image J software. Means and standard deviations were taken from three independent analyses and the significant differences were calculated by Student's t test (p < 0.05).
RESULTS
The E. coli Proteome Chip Assays and Bioinformatics Analysis
We employed a high-throughput E. coli proteome microarray to systematically identify the recognition proteins of ClpY. The E. coli proteome microarrays were composed of ∼4300 nonredundant proteins, which were printed in duplicated on aldehyde-coated glass slides as previously reported (30). In this study, the E. coli proteome chips were probed with ClpY and ClpY(ΔI+7Gly) proteins. ClpY(ΔI+7Gly) is an I-domain deletion mutant protein from ClpY but maintains the configuration, which lost its binding ability to substrate (22, 33). In this study, we use ClpY(ΔI+7Gly) as a control for identifying specific ClpY recognition proteins. Then the rabbit anti-ClpY detection antibodies were used to recognize ClpY on the chip (Fig. 1A). At last, DyLight™ 649 labeled anti-rabbit antibodies were used to detect anti-ClpY and show fluorescent signals on the chips. After the washing steps, the chip was scanned and analyzed. A representative chip image comparison between ClpY and ClpY(ΔI+7Gly) was shown in Fig. 1B. The ClpY and ClpY(ΔI+7Gly) chip images indicated different binding affinity between particular proteins. The representative proteins YfbV, RhlB, and YbaB showed stronger signal in ClpY than ClpY(ΔI+7Gly), respectively (Fig. 1B). This indicated ClpY recognize with specific targets on E. coli chips. After conducting chip analysis, the signal was output as a text file for signal normalization and positive hit selection. Each signal was normalized with ProCAT (32). The distribution of the signal was fit to a normal distribution, and the positive hits were selected based on a local cut-off, which was defined as triple standard deviations above the signal mean for each spot. After the initial selection, we chose the possible candidates only bind to ClpY, but not ClpY(ΔI+7Gly). After eyeballing, we remove 3 of 9 hits because of not obvious difference between control and experiment. Thus, 6 proteins were identified and summarized in Table I.
Fig. 1.

A schematic representation of ClpY recognition protein identification using an E. coli proteome chip. A, The ClpY proteins were probed to the E. coli proteins on the chips and detected by anti-ClpY antibodies. The anti-rabbit antibodies were labeled with DyLight™ 649. B, Representative images of the E. coli proteome chips probed with ClpY and control ClpY(ΔI+7Gly). The representative proteins of YfbV, RhlB and YbaB on the chips were enlarged from sample images of ClpY and control ClpY(ΔI+7Gly), respectively.
Table I. Six ClpY-interacting proteins selected by E. coli K12 proteome chips.
| Accession number | Protein name | Annotation | Signal intensity |
|---|---|---|---|
| EG11100 | YbaB | DNA-binding protein | 667.1 ± 34.3 |
| EG14106 | YfbV | Ter macrodomain insulation protein | 218.4 ± 31.5 |
| EG10817 | RbsD | d-ribose pyranase | 140.6 ± 20.5 |
| EG13443 | CobB | Protein lysine deacetylase, chemotaxis regulator | 114.8 ± 44.6 |
| EG10844 | RhlB | ATP-dependent RNA helicase | 100.8 ± 71.0 |
| EG13114 | YgbT | Multifunctional endonuclease Cas1, CRISPR adaptation protein | 82.7 ± 7.7 |
Yeast Two-hybrid Assay Analysis
Because YbaB signal is much higher than other 5 proteins, we focused on YbaB for further validations and studies. First, we used yeast two-hybrid system to validate the interaction between YbaB and ClpY. The yeast two-hybrid assay system involved in transforming the target protein gene segments into pB42AD and pGilda plasmids, which can express the target active domain (AD) and binding domain (BD) fusion proteins, respectively. We cloned ybaB into pGilda plasmids, which were co-transformed along with pB42AD-clpY into EGY48 yeast cells. To account for the effect of AD or BD fusion on the target proteins, ybaB genes was cloned into pB42AD plasmids and cotransformed along with pGilda-clpY into EGY48 yeast cells. pGilda/pB42AD plasmids with and without the cloned genes were cotransformed into yeast cells. Concurrently, we verified whether the target proteins interacted with B42 peptide (the product of pB42AD) or LexA (the product of pGilda). Cotransformation of the pGilda and pB42AD plasmids without the cloned genes yielded the negative control group. To perform the X-gal test, the yeast two-hybrid assay analysis was conducted using the reporter plasmid p8op-lacZ carried by the EGY48 yeast cells. The X-gal test results revealed obvious YbaB-ClpY reactions (Fig. 2). The interaction between expressed ClpY and YbaB caused β-galactosidase expression and further degraded the X-gal compounds, forming deep blue bacterial colonies. By contrast, the control group was colorless.
Fig. 2.

Interactions between ClpY and YbaB in the yeast two-hybrid assay. The vector-only pB42AD and pGilda were cotransformed as a negative control. Interactions between ClpY and YbaB protein were tested by streaking the yeast colony on X-Gal containing minimal medium with an addition of raffinose and galactose. Turning blue of the colony indicates interactions.
Measurement of Kd Between MBP-YbaB and ClpY
QCM was applied by fixing MBP-YbaB on the sensor electrodes, and various volumes of ClpY protein were then loaded to measure the changes in the frequencies of the sensor cell. The average Kd of triplicate assays were 1.37 × 10−9 ± 3.12 × 10−10 m. Fig. 3 showed one of the representative image of ClpY dose response curve. This Kd was smaller than the Kd (3 × 10−9 m) between ClpY and MBP-SulA that reported by Azim et al. (34). Therefore, the interaction between ClpY and MBP-YbaB was stronger than the interaction between ClpY and MBP-SulA.
Fig. 3.
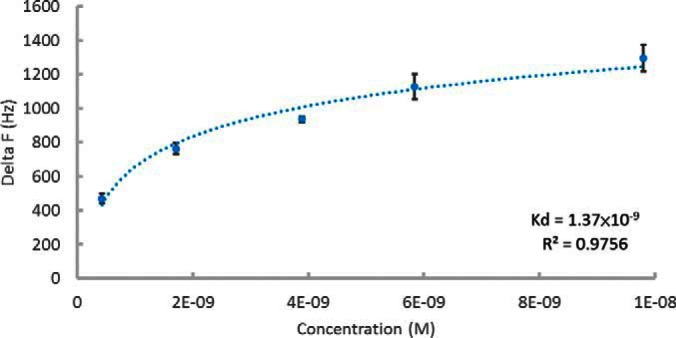
Determination of the Kd between ClpY and YbaB by QCM assay. The assay was conducted by Affinix QN μ and performed in triplicate. The representative figure was chosen for showing curve fitting and Kd calculation.
MBP-YbaB In Vitro Degradation Test
After confirming the interactions between MBP-YbaB and ClpY proteins, we purified the ClpY, ClpQ, and YbaB proteins to investigate whether ClpYQ proteases can degrade YbaB. The His tag was used to purify the ClpY and ClpQ through a nickel column. The 6X His tag of ClpY was located at the N terminals, whereas that of ClpQ was located at the C terminals. MBP was fused to YbaB to purify the MBP-YbaB itself. Consequently, the molecular weight of the purified product was increased by ∼40 kDa. The SDS-PAGE with Coomassie staining showed clear single band to ClpY, ClpQ, and YbaB (Fig. 4A).
Fig. 4.

In vitro degradation assay of MBP-YbaB. Degradations of MBP-YbaB by ClpYQ at 37 °C were determined by anti-MBP antibody and analyzed by Image J. A, Coomassie blue staining of purified MBP-YbaB, ClpY and ClpQ. B, SDS-PAGE of YbaB degradation by ClpYQ. (C) YbaB degradation by ClpYQ determined by Anti-MBP Western blotting. Means and standard deviations (error bars) were taken from three independent analyses. The asterisk indicates the significant difference by Student's t test (p < 0.05).
The in vitro degradation test was configured by adding 2 μm ClpY, 2 μm ClpQ, 1 μm MBP-YbaB, and 5 mm ATP to the HEPES degradation buffer. The resulting solution was reacted at 37 °C for 4 h, and samples were collected at 0, and 4 h after the reaction was initiated. The SDS-PAGE was used to observe the protein content. A reaction solution containing MBP-YbaB and ATP only was used as a control to determine whether the proteins can self-degrade within 4 h. The test results revealed the experimental group containing ClpYQ exhibited much higher degradation rate compared with that of the control without the proteases (Fig. 4B). Western blotting results also showed the degradation of YbaB after 4 h of reaction (Fig. 4C). This indicates that YbaB not only binds to ClpY but also is the substrate of ClpYQ.
MBP-YbaB In Vivo Degradation Test
In addition to exploring the in vitro degradation effect of ClpYQ, this study also applied lon− E. coli strain (SG22623) and lon− clpQ− clpY− E. coli strain (AC3112). The SG22623 was constructed from wild type, which was knocking out lon gene. The AC3112 was constructed from SG22623 by further knocking out the clpQ and clpY gene (35). These 2 KO strains will help us to investigate the in vivo degradation of YbaB. E. coli has two major ATP-dependent proteases, which are Lon and Clp proteases. Lon is responsible for the degradation of abnormal or unstable regulatory proteins in many bacteria. Moreover, the Lon protease shares same substrates with ClpYQ protease. It suggested the substrate of ClpYQ protease may be degraded by the Lon protease when E. coli suffering from ClpYQ functionless mutation (36). Thus, both strains SG22623 and AC3112 were considered as ATP-dependent protease deficient strains without the Lon protease, and the difference in the proteomic profiles between these two strains would be derived by the expression of ClpYQ protease in SG22623 (35). Samples of ybaB were cloned into pTH18kr-mbp plasmids and then transformed into SG22623 and AC3112 strains. IPTG was applied to express MBP-YbaB fusion proteins for 30 mins, and spectinomycin was then used to terminate the biosynthesis of the proteins in the bacterial cells. Therefore, the amounts of protein in the bacterial cells no longer increased. If the YbaB protein level decreased in SG22623 more than that in AC3112, this would confirm that YbaB proteins were degraded by ClpYQ in vivo. Western blotting with anti-MBP antibodies was used to measure the protein levels. The quantitative analysis results were standardized based on the bacterial growth OD600 values.
In Fig. 5, the protease deficient strain AC3112 shows no significant degradation of MBP-YbaB after 4 h. On the contrast, the Western blotting shows MBP-YbaB signal decreasing significantly after 4 h with ClpYQ coxpression strain SG22623 in vivo. This suggested the YbaB was degraded by ClpYQ proteolytic activity and the results are consistent with YbaB being a substrate of ClpYQ in vitro.
Fig. 5.
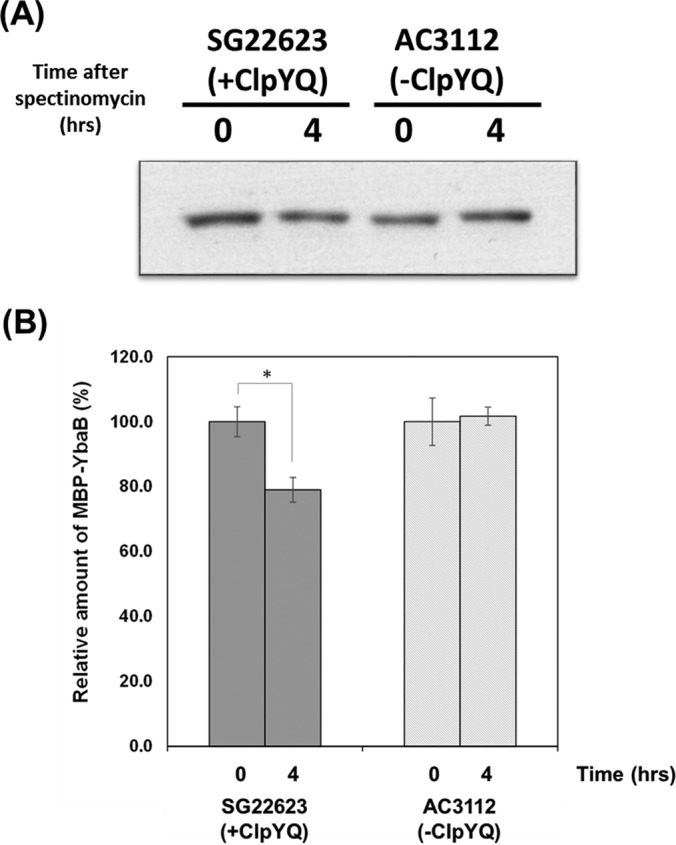
In vivo degradation assay of MBP-YbaB. Determination of the degradation of MBP-YbaB in SG22623 and AC3112 by anti-MBP antibody. The bar chart was the quantification of degradation rate by Image J. Means and standard deviations (error bars) were taken from three independent analyses. The asterisk indicates the significant difference by Student's t test (p < 0.05).
DISCUSSION
This is the first study to globally screen protease substrates using protein microarrays. Because the protein substrates on a protein chip would be degraded by the protease probe, it is very challenging to identify a protease substrate by using a regular proteome microarray assay directly. In this study, we chose an indirect strategy to first identity the protease-binding proteins. We used ClpY only, which is the recognition unit for ClpYQ protease, to screen for ClpY-binding proteins with E. coli proteome microarrays. After identifying the protein recognized by ClpY, we validated the protease activity by in vitro and in vivo degradation assay. By using this indirect strategy, we identified that the YbaB is the substrate of ClpYQ.
When E. coli suffers in heat, the misfolding proteins will accumulate. Thus, the heat shock proteins are induced (37). However; in the normal physiological state, overexpression of heat shock proteins will be toxic to the cell. Some of antibiotics, such as puromycin, induced heat shock regulation protein expression. Missiaka et al. (19) founds ClpYQ is able to inhibit the constitutive heat shock regulation induced by puromycin. They reported the ClpYQ can degrade abnormal “puromycyl polypeptides”. Thus, it is helping cell to lower the toxic effect of the abnormal heat shock response. The SOS response protein SulA (38) is a known substrate of ClpYQ (15). SulA inhibited the cell division when SOS response activated (38). The other knowing targets of ClpYQ are RcsA, RpoH, TraJ and RNaseR (15, 16, 39–41). RpoH is a heat shock transcription factor (16). RcsA is a capsule synthesis activation protein and also a substrate of Lon (42). TraJ is responsible for F plasmid conjugation activation (39). RNaseR is an important exoribonuclease responsible for degradation of structured RNAs in E. coli (41). These findings suggested the ClpYQ participate in important cell physiological functions. In this study, we showed that YbaB is a novel target of ClpYQ. YbaB is a DNA binding protein with a probable histone-like activity in bacterial cells that facilitates packing the genetic materials of prokaryotes by forming nucleoids (43, 44). However, the actual function of YbaB remains unclear (43). Because YbaB is a histone-like protein, it may mediate gene-silencing and antisilencing activities through regulating the nucleoid structure (44). This suggested that ClpYQ protease participated in the YbaB's regulations functions and other gene regulation cascade. Also, YbaB shares homology with many pathogens, such as Haemophilus influenzae, E. coli, Vibrio cholerae, Pseudomonas putida, Rickettsia rickettsiae, Neisseria gonorrhoeae, Bdellovibrio bacteriovorus, Clostridium perfringens, Bacillus subtilis, Enterococcus faecalis, Streptococcus pneumoniae, Mycobacterium tuberculosis, Bacteroides capillosus, and Borrelia burgdorferi (45). Their hosts include human, plants, and animals. This suggests that the regulation of YbaB function through digestion by ClpYQ is a very important mechanism in bacteria.
Supplementary Material
Footnotes
Author contributions: W.W. and C.C. designed research; C.T., Y.H., and T.S. performed research; Y.H., T.S., and C.C. analyzed data; T.S., W.W., and C.C. wrote the paper.
* This work was supported by Ministry of Science and Technology, Taiwan grants NSC-101-2313-B-002-065-MY3, MOST 103-2627-M-008-001, MOST 104-2320-B-002-038 and MOST 104-2320-B-008-002-MY3, The Aim for the Top University Project, National Central University and Landseed Hospital Join Research Program (NCU-LSH-103-A-001), National Central University and Cathay General Hospital Join Research Program (102NCU-CGH-02, 102NCU-CGH-07, 103CGH-NCU-A3), and National Health Research Institutes Career Development Grant (NHRI-EX103-10233SC).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- QCM
- quartz crystal microbalance.
REFERENCES
- 1. Neurath H., and Walsh K. A. (1976) Role of proteolytic enzymes in biological regulation (a review). Proc. Natl. Acad. Sci. U.S.A. 73, 3825–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harper J. D., and Lansbury P. T. (1997) Models of amyloid seeding in Alzheimer's disease and scrapie:mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407 [DOI] [PubMed] [Google Scholar]
- 3. Kahn S. E., Andrikopoulos S., and Verchere C. B. (1999) Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 48, 241–253 [DOI] [PubMed] [Google Scholar]
- 4. Turk B. (2006) Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 5, 785–799 [DOI] [PubMed] [Google Scholar]
- 5. Gottesman S. (1996) Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 30, 465–506 [DOI] [PubMed] [Google Scholar]
- 6. Wickner S., Maurizi M. R., and Gottesman S. (1999) Posttranslational quality control: folding, refolding, and degrading proteins. Science 286, 1888–1893 [DOI] [PubMed] [Google Scholar]
- 7. Zwickl P., Baumeister W., and Steven A. (2000) Dis-assembly lines: the proteasome and related ATPase-assisted proteases. Curr. Opin. Struc. Biol. 10, 242–250 [DOI] [PubMed] [Google Scholar]
- 8. Gottesman S. (2003) Proteolysis in bacterial regulatory circuits. Annu. Rev. Cell Dev. Biol. 19, 565–587 [DOI] [PubMed] [Google Scholar]
- 9. Parsell D. A., and Sauer R. T. (1989) Induction of a heat shock-like response by unfolded protein in Escherichia coli: dependence on protein level not protein degradation. Genes Dev. 3, 1226–1232 [DOI] [PubMed] [Google Scholar]
- 10. Goff S. A., and Goldberg A. L. (1985) Production of abnormal proteins in E. coli stimulates transcription of lon and other heat shock genes. Cell 41, 587–595 [DOI] [PubMed] [Google Scholar]
- 11. Tomoyasu T., Gamer J., Bukau B., Kanemori M., Mori H., Rutman A. J., Oppenheim A. B., Yura T., Yamanaka K., and Niki H. (1995) Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor sigma 32. EMBO J. 14, 2551–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katayama-Fujimura Y., Gottesman S., and Maurizi M. R. (1987) A multiple-component, ATP-dependent protease from Escherichia coli. J. Biol. Chem. 262, 4477–4485 [PubMed] [Google Scholar]
- 13. Gottesman S., Roche E., Zhou Y., and Sauer R. T. (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 12, 1338–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chuang S. E., Burland V., Plunkett G. 3rd, Daniels D. L., and Blattner F. R. (1993) Sequence analysis of four new heat-shock genes constituting the hslTS/ibpAB and hslVU operons in Escherichia coli. Gene 134, 1–6 [DOI] [PubMed] [Google Scholar]
- 15. Wu W. F., Zhou Y., and Gottesman S. (1999) Redundant in vivo proteolytic activities of Escherichia coli Lon and the ClpYQ (HslUV) protease. J. Bacteriol. 181, 3681–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuo M. S., Chen K. P., and Wu W. F. (2004) Regulation of RcsA by the ClpYQ (HslUV) protease in Escherichia coli. Microbiology 150, 437–446 [DOI] [PubMed] [Google Scholar]
- 17. Bochtler M., Hartmann C., Song H. K., Bourenkov G. P., Bartunik H. D., and Huber R. (2000) The structures of HslU and the ATP-dependent protease HslU-HslV. Nature 403, 800–805 [DOI] [PubMed] [Google Scholar]
- 18. Kessel M., Wu W. F., Gottesman S., Kocsis E., Steven A. C., and Maurizi M. R. (1996) Six-fold rotational symmetry of ClpQ, the E. coli homolog of the 20S proteasome, and its ATP-dependent activator, ClpY. FEBS Lett. 398, 274–278 [DOI] [PubMed] [Google Scholar]
- 19. Missiakas D., Schwager F., Betton J. M., Georgopoulos C., and Raina S. (1996) Identification and characterization of HsIV HsIU (ClpQ ClpY) proteins involved in overall proteolysis of misfolded proteins in Escherichia coli. EMBO J. 15, 6899–6909 [PMC free article] [PubMed] [Google Scholar]
- 20. Rohrwild M., Coux O., Huang H. C., Moerschell R. P., Yoo S. J., Seol J. H., Chung C. H., and Goldberg A. L. (1996) HslV-HslU: A novel ATP-dependent protease complex in Escherichia coli related to the eukaryotic proteasome. Proc. Natl. Acad. Sci. U.S.A. 93, 5808–5813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rohrwild M., Pfeifer G., Santarius U., Muller S. A., Huang H. C., Engel A., Baumeister W., and Goldberg A. L. (1997) The ATP-dependent HslVU protease from Escherichia coli is a four-ring structure resembling the proteasome. Nat. Struct. Mol. Biol. 4, 133–139 [DOI] [PubMed] [Google Scholar]
- 22. Lien H. Y., Shy R. S., Peng S. S., Wu Y. L., Weng Y. T., Chen H. H., Su P. C., Ng W. F., Chen Y. C., Chang P. Y., and Wu W. F. (2009) Characterization of the Escherichia coli ClpY (HslU) substrate recognition site in the ClpYQ (HslUV) protease using the yeast two-hybrid system. J. Bacteriol. 191, 4218–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee Y. Y., Chang C. F., Kuo C. L., Chen M. C., Yu C. H., Lin P. I., and Wu W. F. (2003) Subunit oligomerization and substrate recognition of the Escherichia coli ClpYQ (HslUV) protease implicated by in vivo protein-protein interactions in the yeast two-hybrid system. J. Bacteriol. 185, 2393–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song H. K., Hartmann C., Ramachandran R., Bochtler M., Behrendt R., Moroder L., and Huber R. (2000) Mutational studies on HslU and its docking mode with HslV. Proc. Natl. Acad. Sci. U.S.A. 97, 14103–14108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sundar S., Baker T. A., and Sauer R. T. (2012) The I domain of the AAA+ HslUV protease coordinates substrate binding, ATP hydrolysis, and protein degradation. Protein Sci. 21, 188–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park E., Rho Y. M., Koh O. J., Ahn S.W., Seong I. S., Song J. J., Bang O., Seol J. H., Wang J., Eom S. H., and Chung C. H. (2005) Role of the GYVG pore motif of HslU ATPase in protein unfolding and translocation for degradation by HslV peptidase. J. Biol. Chem. 280, 22892–22898 [DOI] [PubMed] [Google Scholar]
- 27. Hsieh F. C., Chen C. T., Weng Y. T., Peng S. S., Chen Y. C., Huang L.-Y., Hu H. T., Wu Y. L., Lin N. C., and Wu W. F. (2011) Stepwise activity of ClpY (HslU) mutants in the processive degradation of Escherichia coli ClpYQ (HslUV) protease substrates. J. Bacteriol. 193, 5465–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Westphal K., Langklotz S., Thomanek N., and Narberhaus F. (2012) A Trapping approach reveals novel substrates and physiological functions of the essential protease FtsH in Escherichia coli. J. Biol. Chem. 287, 42962–42971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flynn J. M., Neher S. B., Kim Y.-I., Sauer R. T., and Baker T. A.. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11, 671–683 [DOI] [PubMed] [Google Scholar]
- 30. Chen C. S., Korobkova E., Chen H., Zhu J., Jian X., Tao S. C., He C., and Zhu H. (2008) A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli. Nat. Methods 5, 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., and Mori H. (2005) Complete set of ORF clones of Escherichia coli ASKA library (A complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299 [DOI] [PubMed] [Google Scholar]
- 32. Zhu X., Gerstein M., and Snyder M. (2006) ProCAT: a data analysis approach for protein microarrays. Genome Biol. 7, R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sousa M. C., Trame C. B., Tsuruta H., Wilbanks S. M., Reddy V. S., and McKay D. B. (2000) Crystal and solution structures of an HslUV protease–chaperone complex. Cell 103, 633–643 [DOI] [PubMed] [Google Scholar]
- 34. Azim M. K., Goehring W., Song H. K., Ramachandran R., Bochtler M., and Goettig P. (2005) Characterization of the HslU chaperone affinity for HslV protease. Protein Sci. 14, 1357–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang C.-Y., Hu H.-T., Tsai C.-H., and Wu W.-F. (2016) The degradation of RcsA by ClpYQ(HslUV) protease in Escherichia coli. Microbiol. Res. 184, 42–50 [DOI] [PubMed] [Google Scholar]
- 36. Tsilibaris V., Maenhaut-Michel G., and Van Melderen L. (2006) Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 157, 701–713 [DOI] [PubMed] [Google Scholar]
- 37. Rosen R., Biran D., Gur E., Becher D., Hecker M., and Ron E. Z. (2002) Protein aggregation in Escherichia coli: role of proteases. FEMS Microbiol. Lett. 207, 9–12 [DOI] [PubMed] [Google Scholar]
- 38. Bi E., and Lutkenhaus J. (1990) Analysis of ftsZ mutations that confer resistance to the cell division inhibitor SulA (SfiA). J. Bacteriol. 172, 5602–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lau-Wong I. C., Locke T., Ellison M. J., Raivio T. L., and Frost L. S. (2008) Activation of the Cpx regulon destabilizes the F plasmid transfer activator, TraJ, via the HslVU protease in Escherichia coli. Mol. Microbiol. 67, 516–527 [DOI] [PubMed] [Google Scholar]
- 40. Kanemori M., Nishihara K., Yanagi H., and Yura T. (1997) Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of sigma32 and abnormal proteins in Escherichia coli. J. Bacteriol. 179, 7219–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liang W., and Deutscher M. P. (2012) Transfer-messenger RNA-SmpB protein regulates ribonuclease R turnover by promoting binding of HslUV and Lon proteases. J. Biol. Chem. 287, 33472–33479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gottesman S., Trisler P., and Torres-Cabassa A. (1985) Regulation of capsular polysaccharide synthesis in Escherichia coli K-12: characterization of three regulatory genes. J. Bacteriol. 162, 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jutras B. L., Bowman A., Brissette C. A., Adams C. A., Verma A., Chenail A. M., and Stevenson B. (2012) EbfC (YbaB) is a new type of bacterial nucleoid-associated protein and a global regulator of gene expression in the lyme disease spirochete. J. Bacteriol. 194, 3395–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dillon S. C., and Dorman C. J. (2010) Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 185–195 [DOI] [PubMed] [Google Scholar]
- 45. Cooley A. E., Riley S. P., Kral K., Miller M. C., DeMoll E., Fried M. G., and Stevenson B. (2009) DNA-binding by Haemophilus influenzae and Escherichia coli YbaB, members of a widely-distributed bacterial protein family. BMC Microbiol. 9, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.