

Abstract
Scarcely understood defects lead to asthenozoospermia, which results in poor fertility outcomes. Incomplete knowledge of these defects hinders the development of new therapies and reliance on interventional therapies, such as in vitro fertilization, increases. Sperm cells, being transcriptionally and translationally silent, necessitate the proteomic approach to study the sperm function. We have performed a differential proteomics analysis of human sperm and seminal plasma and identified and quantified 667 proteins in sperm and 429 proteins in seminal plasma data set, which were used for further analysis. Statistical and mathematical analysis combined with pathway analysis and self-organizing maps clustering and correlation was performed on the data set.
It was found that sperm proteomic signature combined with statistical analysis as opposed to the seminal plasma proteomic signature can differentiate the normozoospermic versus the asthenozoospermic sperm samples. This is despite the results that some of the seminal plasma proteins have big fold changes among classes but they fall short of statistical significance. S-Plot of the sperm proteomic data set generated some high confidence targets, which might be implicated in sperm motility pathways. These proteins also had the area under the curve value of 0.9 or 1 in ROC curve analysis.
Various pathways were either enriched in these proteomic data sets by pathway analysis or they were searched by their constituent proteins. Some of these pathways were axoneme activation and focal adhesion assembly, glycolysis, gluconeogenesis, cellular response to stress and nucleosome assembly among others. The mass spectrometric data is available via ProteomeXchange with identifier PXD004098.
Sperm motility is a cornerstone of male fertility and defects in motility are associated with poor fertility outcomes. Most cases of asthenozoospermia are labeled idiopathic as the underlying defect is not fully known, which necessitates management by intervention. Poor motility of the sperm usually requires the interventional therapeutic measures such as gamete intrafallopian transfer or in vitro fertilization. These interventions are not completely efficient in addition to being expensive and unavailable in many countries. The defects leading to asthenozoospermia are not understood, which needs to change, to allow for innovative treatment options directed at the root cause of this problem. Mature sperm are believed to be transcriptionally and translationally silent (1), which demands proteomic studies (as opposed to genomic or transcriptomic studies) to understand the underlying pathways giving rise to sperm motility.
Differential proteomics can help a great deal to better understand the human sperm motility, as it is very easy and non-invasive procedure to collect the human seminal fluid. This presents an opportunity to study asthenozoospermia on proteome scale. Quantitative differential proteomics associated with statistical analyses will provide us with proteins, significantly different among the two classes of sperm (normozoospermic (NZS)1 and asthenozoospermic (AZS)), which will have a role in the sperm motility pathways. Such studies will elucidate the pathways underlying the sperm motility, which can lead to identification of novel treatment avenues. Human seminal plasma and sperm have an ongoing exchange of protein factors between them. This exchange regulates the temporal aspects of various processes in successful fertilization such as acrosome reaction, formation of oviductal sperm reservoir and gamete interaction (2). Various seminal plasma proteins have also been implicated in sperm motility (3). There have been some studies on human sperm and seminal plasma for identification of biomarkers of AZS (4–6). However, low level of overlap between the results of these studies, as well as lack of statistical power, restrict the formation of a consensus on the topic. This demands more studies for identifying proteins having roles in the human sperm motility. We have performed a differential proteomic study on NZS and AZS seminal plasma and sperm samples. Quantified protein expression (667 proteins quantified in sperm cell sample and 429 in seminal plasma samples) was analyzed by ANOVA followed by principle component analysis. Orthogonal Projections to Latent Structures Discriminant Analysis (OPLS-DA) based modeling was performed and subsequent S-Plot identified the proteins being significantly different among the samples. Receiver operating curve (ROC) analysis was performed on these significantly different proteins and area under the curve was calculated. Various other analyses such as multiple pathway analyses combined with self-organizing maps analysis identified the clusters of covariant proteins and associated pathways.
2. MATERIALS AND METHODS
Sample Collection and Preprocessing
Human semen samples collected from Department of Laboratory Medicine, All India Institute of Medical Sciences, New Delhi, India were processed as described below. Institutional Ethics Committee approved the study with the reference number IEC/NP-147/01.05.2014, RP-33/2014. The World Health Organization (WHO) 2010 recommendations for semen analysis were followed. Samples were categorized as NZS based on sperm count >15 million/ml, sperm motility >40% and normal spermatozoa morphology. Samples having less than 40% sperm motility were characterized as AZS. The age of the semen donors was between 20–40 years. The clinical parameters of the semen donors are given in supplemental Table S1. Samples with the visual presence of leukocytes were not included in the study. Samples were centrifuged at 2000 × g for 20 min at 4 °C to separate spermatozoa from seminal plasma. The resulting pellet was washed with PBS three times and centrifugation was repeated each time to completely remove seminal plasma. Seminal plasma was further centrifuged at 10,000 × g for 20 min to remove cell debris and other impurities if any. The clear supernatants obtained were lyophilized and stored at −80 °C till further use.
Further Processing and Trypsin Digestion
Lyophilized sperm and seminal plasma were rehydrated on a thermomixer for 2 h (25 °C) in 0.1% Rapigest SF (Waters, Manchester, UK) for seminal plasma, whereas the concentration of Rapigest for sperm samples were 0.5%. Samples were sonicated (at 50% output) by a probe sonicator (UP200H, Dr. Hielscher GmBH, Germany) for 5 cycles of 3 s each. The protein concentration was determined with Pierce BCA assay kit (Thermo Fisher Scientific, Waltham, MA). The liquid containing the 30 μg total protein was aliquoted and boiled at 100 °C in a water bath for 10 min. After cooling down the samples, dithiothreitol (DTT) was added to the final concentration of 5 mm to the samples and they were incubated at 65 °C in a thermomixer for 30 min. Samples were again cooled down and iodoacetamide was added to the final concentration of 15 mm and samples incubated for 30 min at 25 °C with shaking. Fifteen mm DTT (final concentration) was again added to the samples to quench the remaining Iodoacetamide and prevent overalkylation. One μg of the Trypsin Gold (Promega, Southampton, UK) was added to each sample and the mixture was incubated at 37 °C overnight. Next day, the samples were cleaned by C18 spin columns (Pierce, Thermo Fisher Scientific) according to manufacturer's protocol. Elution was dried in the speed vacuum (Savant, Thermo Fisher Scientific) and reconstituted in 86 μl of 0.1% formic acid containing 50fmol of Hi3 peptide mixture (Waters) per 4 μl as a spike-in standard for quantitation of proteins.
UPLC-MS
Four μl samples, equivalent to ∼1.4 μg total protein, was injected to nano Acquity UPLC (Ultra Performance Liquid Chromatography) - system (Waters). TRIZAIC nanoTile 85 μm x 100 mm HSS-T3u wTRAP was used as separating device prior to mass spectrometer. Samples were loaded, trapped and washed for 2 mins with 8.0 μl/min with 1% B. The analytical gradient used is as follows: 0–1 min 1% B, at 2 min 5% B, at 65 min 30% B, at 78 min 50% B, at 80 min 85% B, at 83 min 85% B, at 84 min 1% B and at 90 min 1% B with 450 nL/min. Buffers were made to UPLC-grade chemicals (Sigma-Aldrich); Buffer A: 0.1% formic acid in water and Buffer B: 0.1% formic acid in acetonitrile.
The data was acquired in DIA (data independent acquisition) fashion using HDMSE-mode with Synapt G2-S HDMS (Waters Corporation). HDMSE mode included ion mobility spectroscopy (IMS). The collected data range was 100–2000 m/z, scan time 1 s, IMS wave velocity 650 m/s, collision energy was ramped in trap between 20 to 60 V. Calibration was done with Glu1-Fibrinopeptide B MS2 fragments and as a lock mass, Glu1-Fibrinopeptide B precursor ion was used during the runs.
The samples were run as triplicates and further analysis was done with, Progenesis QI for Proteomics - software (Nonlinear Dynamics, Newcastle, UK).
Data Analysis
The raw files were imported to Progenesis QI for proteomics software (Version V2, Nonlinear Dynamics) using lock mass correction with 785.8426 m/z, corresponding to doubly charged Glu1-Fibrinopeptide B. Default parameters for peak picking and alignment algorithm were used. The software facilitated the peptide identification with Protein Lynx Global Server (PLGS version 3.0) and label-free quantification (7).
The peptide identification was done against Uniprot human FASTA sequences (UniprotKB Release 2015_09, 20205 sequence entries) with (CLPB_ECOLI (P63285)), ClpB protein sequence inserted for label-free quantification. Modifications used were as follows: fixed at cysteine (carbamidomethyl) and variable in methionine (oxidation). Trypsin was used as digesting agent and one missed cleavage was allowed. Fragment and peptide error tolerances were set to auto and FDR to less than 4%. Auto-tolerances are calculated by PLGS automatically depending on the resolution of the run. For example, the first run had resolution of 17170.87 and the precursor tolerance was 5.8 ppm and fragment tolerance was 14.6 ppm. The false discovery rate in an Ion Accounting search is less than the specified rate (default = 4%) all the way through the search, right up to the last protein identified. When the false discovery rate exceeds the specified FDR value the search stops. The FDR is determined using a randomized version of the specified sequence data bank. One or more ion fragments per peptide, three or more fragments per protein and one or more peptides per protein were required for ion matching. These are default parameters in the software.
The identified proteins are grouped as one according to parsimony principle and also peptides unique to the protein are reported. Parsimony principle states that protein hits are reported as the minimum set that accounts for all observable peptides. Progenesis QI for proteomics does not take a strict parsimonious approach because of over-stringency as has been pointed out before (8). However, for resolution of conflicts, if two proteins contain some common peptides, protein with fewer peptides is subsumed into the protein with higher number of peptides that are a superset of the subsumed protein's peptides. All relevant proteins are listed as a group under the lead protein with greatest coverage or the highest score when the coverages of two or more proteins are equal. Quantitation is performed using the lead identity peptide data. More details about this approach can be accessed on the software website (www.nonlinear.com).
The proteins were considered different if they have a fold change 2 or more and an ANOVA p value 0.05 or less. The ANOVA calculation assumes that the conditions are independent and applies the statistical test that assumes the means of the conditions are equal.
The label-free protein quantitation was done with Hi-N method (7). In every injection, the sample contained also 50 fmol of six CLPB_ECOLI (P63285, ClpB protein) peptides (Hi3 E. coli Standard, Waters). Hi3 peptides are used for normalizing the peptide abundancies and relative quantitation was based on all the non-conflicting peptides found. The peptide ranking is done across all the runs. The abundancies of the peptides are averaged to provide a signal to the protein. Workings of the Progenesis softwares have been described in details on the software website (www.nonlinear.com) and also in published literature (9).
Peptide Statistics by Progenesis QI Proteomics
Explanation about some of the common terms used in peptide statistics is given below.
Q value: tells us the expected proportion of false positives if that peptide ion's p value is chosen as the significance threshold.
Power: can be defined as the probability of finding a real difference if it exists. 80% or 0.8 is considered an acceptable value for power. The Power Analysis is performed independently for each peptide ion, using the expression variance, sample size and difference between the means.
Experimental Design and Statistical Rationale
We studied 5 NZS and 8 AZS spermatozoa samples as well as 7 NZS and 10 AZS seminal plasma samples. Categorization parameters are given in Sample Collection and Preprocessing and clinical measurement of the samples are given in supplemental Table S1. Samples were compared among the classes, NZS and AZS. Differences between controls and cases were evaluated with ANOVA on a protein-to-protein basis. Principle component analysis was done with Progenesis QI for proteomics. EZinfo 3.0.3.0 (Release date Dec 02, 2014, Umetrics, Sweden) is a separate statistical package that can be used with Progenesis QI for proteomics. The data was imported into the EZinfo and supervised OPLS-DA modeling was performed, which gave us the variance versus correlation plot (S-Plot). Default parameters were used. ROC curve analysis was also performed on some of the significantly different proteins predicted by S-Plot in case of sperm proteomic data set. Analyze-It program, which works with Microsoft Excel, was used with all the default parameters. This is an exploratory/discovery based study to propose protein targets implicated in defects of AZS.
Pathway Analysis
The pathway analysis was done by three different methods. Literature was searched for several proteins from data set leading to sperm motility pathways and it is presented as a figure in Results. Integrated Molecular Pathway Level Analysis (IMPaLA) was used for pathway over representation analysis by their web-based service. The method and rationale behind the approach has been published previously (10). Ingenuity pathway analysis (Ingenuity Systems, Redwood City, CA) was used for performing core analysis on the sperm cells proteomic data set with default parameters of the software. The results (canonical pathways) are presented in Results as a figure.
Self-Organizing Maps (SOM)
The objective of the SOM algorithm is to find prototype vectors that represent the input data set and at the same time realize a continuous mapping from input space to a lattice (11). This lattice consists of a defined number of “neurons.” All the SOM calculations and analysis were done in MATLAB based on the following basic principle. The basic principle behind the SOM algorithm is that the weight vectors of neurons that are first initialized randomly, come to represent a number of original measurement vectors during an iterative data input process. The iterative process is carried out by a sequential regression process. For each observation x(t), where t = 1, 2, … is the step index, we first identify the index c of some reference model, which represents the best match in terms of Euclidean distance by the condition,
| (Eq. 1) |
Here, the index i ranges over all reference models on the map. The construction ‖x − y‖ represents the Euclidean distance between feature vectors x and y. Next, all reference models on the map are updated with the following regression rule where model index c is the reference model index as computed above.
| (Eq. 2) |
Here hci is the neighborhood function that is defined as follows:
| (Eq. 3) |
Where |c − i| represents the distance between the best matching reference model c and some other reference model i on the map, β is the neighborhood distance and α is the learning rate. This regression is usually repeated over the available observations.
Data Repository
The raw files were converted with MSConvert (ProteoWizard) to mzML-files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (12) partner repository with the data set identifier PXD004098.
3. RESULTS
Metadata
Sixteen samples were included in the study according to the criterion described in Methods. The samples had varying sperm counts and motility parameters as seen in the supplemental Table S1. Every sample was run three times and the samples, where three technical replicates were not similar (where the alignment of triplicates was retrospectively visibly poor in Progenesis QI for proteomics after they did not group closely in principal component analysis), were excluded from the further analysis. Three such not similar triplicates were excluded from further analysis and they are indicated in the supplemental Table S1. Sperm from sample A4 were not analyzed because there was too little protein recovered from it.
Proteomic Analysis
Human sperm cells and seminal fluid samples were analyzed in HDMSE mode followed by database search and the corresponding results are reported below.
Sperm Proteomic Analysis
For quantification only proteins with 2 or more unique peptides were considered and only these 667 proteins were taken for further analysis. Fold change ranged from 79.4 to 1 when NZS had the highest mean and from 19.6 to 1 when the AZS had the highest mean. All protein abundances were compared by ANOVA among all the samples and the p value ranged from 5.99 × 10−13 to 0.999. The confidence score of protein identifications ranged from 1157.067 to 4.842. Ten of the most downregulated and up-regulated proteins in AZS in human sperm cell samples and human seminal plasma samples are presented in Table I (complete data in supplemental Table S2, protein coverages in supplemental Table S10). Some of the proteins having p values higher than 0.05 are also present in bold letters in Table I and II as they are not considered significantly different among the samples despite having the big fold change. This is mainly because ANOVA was considered as the major criterion for establishing real differences between the groups. This is a good strategy as we can see in supplemental Table S3 that one NZS sample for neutrophil defensin 1 protein has unusually high protein abundance (compared with AZS), which results in high mean and subsequently high fold change. Other samples in the NZS, however have low expression of this protein. ANOVA helps identify proteins having consistent expression across the samples with real fold changes.
Table I. Top 10 up- and downregulated proteins in asthenozoospermia samples compared to NZS in human sperm cell samples. Peptide count is the total peptides found for corresponding proteins and unique peptides is the number of unique peptides out of the total peptides found. Confidence score, ANOVA p value, maximum fold change, disease class of the highest and lowest mean condition and full name of the protein are also reported in separate columns.
| Accession | Peptide count | Unique peptides | Confidence score | ANOVA (p) | Max fold change | Highest mean condition | Lowest mean condition | Protein Name |
|---|---|---|---|---|---|---|---|---|
| Top 10 Downregulated proteins in Asthenozoospermia in human sperm cell samples | ||||||||
| P13727 | 5 | 4 | 29.3207 | 5.89E-06 | 79.40088 | Normal | Astheno | Bone marrow proteoglycan |
| P59665;P59666 | 7 | 6 | 51.7033 | 0.125966 | 21.76551 | Normal | Astheno | Neutrophil defensin 1 |
| Q9BYX7 | 10 | 2 | 62.5462 | 6.93E-13 | 17.17476 | Normal | Astheno | Putative beta-actin-like protein 3 |
| P62736;C9JFL5;F6QUT6;F6UVQ4;P63267;P68032 | 29 | 3 | 207.7618 | 5.79E-07 | 15.40699 | Normal | Astheno | Actin, aortic smooth muscle |
| Q92766 | 7 | 3 | 39.7539 | 1.78E-06 | 8.022462 | Normal | Astheno | Ras-responsive element-binding protein 1 |
| E9PN67;C9J066;Q8N4C6 | 5 | 4 | 28.2629 | 7.18E-11 | 6.170855 | Normal | Astheno | Ninein |
| H0YCJ7;E9PJH4;E9PK82;E9PL09;E9PPU1;E9PQ96;F2Z2S8;H0YEU2;H0YF32;P23396 | 3 | 2 | 15.195 | 0.025676 | 5.929772 | Normal | Astheno | 40S ribosomal protein S3 (Fragment) |
| Q12906;K7ELV3 | 7 | 2 | 42.8884 | 7.07E-07 | 5.785136 | Normal | Astheno | Interleukin enhancer-binding factor 3 |
| Q8NCQ7;A0A0A0MT24 | 3 | 2 | 16.7889 | 1.45E-06 | 5.648057 | Normal | Astheno | Protein PROCA1 |
| A6NNN6;E7ETA6;Q15154 | 7 | 2 | 35.0439 | 0.006358 | 5.557926 | Normal | Astheno | Pericentriolar material 1 protein |
| Top 10 Upregulated proteins in Asthenozoospermia in Human sperm cell samples | ||||||||
| Q9NY65;C9J2C0;V9GZ17 | 37 | 3 | 181.908 | 6.35E-06 | 19.69428 | Astheno | Normal | Tubulin alpha-8 chain |
| A7MCY6 | 3 | 2 | 14.8265 | 8.59E-05 | 17.9325 | Astheno | Normal | TANK-binding kinase 1-binding protein 1 |
| P20151 | 11 | 5 | 67.5844 | 1.46E-07 | 10.12548 | Astheno | Normal | Kallikrein-2 |
| Q9H0I9 | 4 | 3 | 30.5099 | 0.001624 | 9.963466 | Astheno | Normal | Transketolase-like protein 2 |
| Q05639 | 10 | 2 | 63.3446 | 8.28E-05 | 8.014055 | Astheno | Normal | Elongation factor 1-alpha 2 |
| G3V1V0;B7Z6Z4;F8VPF3;F8VZU9;F8W180;F8W1R7; G3V1Y7;G8JLA2;J3KND3;P60660 | 4 | 2 | 32.5303 | 0.011136 | 7.863896 | Astheno | Normal | Myosin light polypeptide 6 |
| Q15722 | 2 | 2 | 9.3132 | 0.935433 | 7.747275 | Astheno | Normal | Leukotriene B4 receptor 1 |
| P28340;M0R2B7 | 4 | 3 | 21.6549 | 2.12E-07 | 7.521456 | Astheno | Normal | DNA polymerase delta catalytic subunit |
| P04792;C9J3N8;F8WE04 | 8 | 3 | 70.4424 | 5.95E-08 | 7.519383 | Astheno | Normal | Heat shock protein beta-1 |
| P32119;A6NIW5 | 13 | 4 | 83.2372 | 0.000142 | 6.928101 | Astheno | Normal | Peroxiredoxin-2 |
Abundances of NZS and AZS quantified proteins (667 proteins) in sperm cell proteomic data set were used for performing principal component analysis as described in Methods. Upper panel shows the PCA biplot when all the proteins were considered for PCA. There is a tendency for separation but the samples do not completely segregate in the plot. When only housekeeping proteins (FC 1.0 to 1.29) were used for PCA (middle panel, Fig. 1) NZS versus AZS again does not segregate into two completely separate classes, which is expected. However, when the proteins passing the cutoff of ANOVA p value below 0.05 and fold change more than 2, were considered for PCA (lower panel, Fig. 1) there was a complete separation of the samples into two components. The NZS samples (blue) were clustered together tightly whereas a bigger cluster was observed for AZS samples, which was separate from NZS cluster.
Fig. 1.

Sperm cells normal versus asthenozoospermic samples. Purple dots are for the asthenozoospermic samples and blue dots are for the normozoospermic samples. Upper panel is the PCA when all the proteins quantitated were considered for the PCA, middle panel is when only housekeeping proteins (FC 1.0 to 1.29) were considered for PCA. Lower panel is when only the proteins having ANOVA p value less than 0.05 and fold change more than 2 in either condition were considered for PCA.
When the samples were divided into three classes based on the motility of the sperm parameter (0%, 10–30%, 50–60% motility) and then used for PCA, it is shown in Fig. 2. The reason for classifying the samples into three motility classes was to see if there is a continuous variation in the data set from high sperm motility (50–60%) to moderate (10–30% sperm motility) to no sperm motility (0% sperm motility). PCA biplot can visualize this information easily in a graphical manner. The total proteins PCA is shown in upper panel of Fig. 2, PCA of housekeeping proteins is shown in the middle panel and the lower panel represents the PCA when only housekeeping proteins and proteins having ANOVA p value below 0.05 and fold change above 2 were used respectively (Fig. 2). The NZS samples again cluster together tightly, separated a little bit from 10–30% motility class and completely from 0% motility classes. There is a progression seen in the clustering of the samples from 0% to 10–30% to 50–60% motility.
Fig. 2.

Sperm cells motility classes PCA: Purple dots are samples having 10–30% sperm motility, blue dots are samples having 50–60% sperm motility and orange dots are samples having 0% sperm motility. Upper panel is the PCA when all the proteins quantitated were considered for the PCA, middle panel is when only housekeeping proteins (FC 1.0 to 1.29) were considered for PCA. Lower panel is when only the proteins having ANOVA p value less than 0.05 and fold change more than 2 in either condition were considered for PCA.
Seminal Plasma Proteomic Analysis
In human seminal plasma samples, 429 proteins were quantified with two or more peptides (supplemental Table S3). Seven hundred twenty-six peptides were identified by differences in IMS-drift time. The fold change ranged from 3.16 to 1 when the highest mean was set to NZS samples and from 9.68 to 1 when the highest mean was set to AZS samples. Top 10 upregulated and downregulated proteins in AZS samples are reported in Table II with corresponding ANOVA p values and confidence score.
Table II. Top 10 upregulated and downregulated protein in AZS in human seminal plasma. Peptide count is the total peptides found for corresponding proteins and unique peptides is the number of unique peptides out of the total peptides found. Confidence score, ANOVA p value, maximum fold change, disease class of the highest and lowest mean condition and full name of the protein are also reported in separate columns.
| Accession | Peptide count | Unique peptides | Confidence score | ANOVA (p) | Max fold change | Highest mean condition | Lowest mean condition | Description |
|---|---|---|---|---|---|---|---|---|
| Top 10 Downregulated proteins in Asthenozoospermia in human seminal plasma samples | ||||||||
| A0A0A0MRT2 | 3 | 2 | 13.3256 | 0.018601 | 3.161278 | normal | astheno | WW domain-containing adapter protein with coiled-coil |
| K7ESQ0;K7EMH9 | 3 | 2 | 15.94 | 0.004213 | 3.092424 | normal | astheno | Ran-binding protein 3 (Fragment) |
| Q9BQ16;A0A0A0MTJ2;B4DG04;E7EMP8; E7ENM6;H0YA19 | 8 | 6 | 40.1268 | 0.000845 | 2.755599 | normal | astheno | Testican-3 |
| Q8N0Y7 | 6 | 2 | 41.5525 | 0.000126 | 2.472531 | normal | astheno | Probable phosphoglycerate mutase 4 |
| Q8TF46;E9PS35 | 3 | 2 | 15.3761 | 0.0478 | 2.322317 | normal | astheno | DIS3-like exonuclease 1 |
| Q9NY33;E9PKK8;E9PNX5;E9PPK9;E9PQ14; G3V180;G3V1D3 | 3 | 2 | 14.9251 | 0.001695 | 2.228232 | normal | astheno | Dipeptidyl peptidase 3 |
| Q13163 | 3 | 2 | 18.7701 | 0.000966 | 2.113316 | normal | astheno | Dual specificity mitogen-activated protein kinase kinase 5 |
| P55072 | 5 | 4 | 31.8791 | 0.000223 | 2.080158 | normal | astheno | Transitional endoplasmic reticulum ATPase |
| Q8IWB6;J3QLK3 | 6 | 3 | 29.0166 | 2.47E-08 | 2.007586 | normal | astheno | Inactive serine/threonine-protein kinase TEX14 |
| Q9NWH9;H0YLE6;H0YLW7;H0YMR6;H0YMW8; H0YNF3;H7BXE3 | 30 | 13 | 154.3292 | 0.000323 | 1.986234 | normal | astheno | SAFB-like transcription modulator |
| Top 10 Upregulated proteins in Asthenozoospermia in Human seminal plasma samples | ||||||||
| Q9BUD6 | 2 | 2 | 12.0373 | 2.08E-06 | 9.681792 | astheno | normal | Spondin-2 |
| Q075Z2 | 2 | 2 | 11.3504 | 0.380548 | 7.606171 | astheno | normal | Binder of sperm protein homolog 1 |
| I3L1D5;O15296 | 3 | 3 | 11.2812 | 0.01337 | 6.428859 | astheno | normal | Arachidonate 15-lipoxygenase B |
| P30044 | 4 | 3 | 21.4712 | 8.52E-06 | 5.718718 | astheno | normal | Peroxiredoxin-5, mitochondrial |
| O75503;A0A024R644 | 3 | 2 | 14.9273 | 0.253421 | 5.42119 | astheno | normal | Ceroid-lipofuscinosis neuronal protein 5 |
| A6NES4;A0A087WT58;C9IYW5 | 3 | 2 | 15.7691 | 0.019931 | 4.815401 | astheno | normal | Maestro heat-like repeat-containing protein family member 2A |
| P62140;C9J9S3;C9JP48;E7ETD8;F5H037;F8WE71 | 3 | 2 | 21.701 | 0.002846 | 4.655673 | astheno | normal | Serine/threonine-protein phosphatase PP1-beta catalytic subunit |
| Q8WWZ1 | 4 | 4 | 24.2537 | 0.069253 | 3.762251 | astheno | normal | Interleukin-1 family member 10 |
| P04217;M0R009 | 6 | 4 | 37.2903 | 0.001377 | 3.474544 | astheno | normal | Alpha-1B-glycoprotein |
| A4FU69;C9J1E6 | 3 | 2 | 24.0961 | 3.60E-06 | 3.350488 | astheno | normal | EF-hand calcium-binding domain-containing protein 5 |
In the seminal plasma when the same PCA analysis was done as described for sperm cells in Fig. 1, total proteins PCA did not cluster differentially (upper panel, Fig. 3). When only housekeeping proteins were used for PCA, as expected, the two classes did not cluster differentially (middle panel, Fig. 3). And unlike sperm cell samples, even the most changing proteins PCA (PCA done with proteins having ANOVA p value less than 0.05 and FC > 2) did not cluster significantly differentially (lower panel, Fig. 3). This shows that sample groups in seminal plasma data set are not significantly different as selecting only the significant proteins should generally show a good separation. The samples divided into three motility classes (sample showing 0%, 10–30% and 50–60% sperm motility) and then used for PCA (supplemental Fig. S1) did not show any significantly better results compared with only two classes (Fig. 3).
Fig. 3.

Seminal plasma PCA Normozoospermia versus Asthenozoospermia: Purple dots are normozoospermic samples and blue dots are asthenozoospermic samples. Upper panel is the PCA when all the proteins quantitated were considered for the PCA, middle panel is when only housekeeping proteins (FC 1.0 to 1.29) were considered for PCA. Lower panel is when only the proteins having ANOVA p value less than 0.05 and fold change more than 2 in either condition were considered for PCA.
Orthogonal Statistical Validation of Proteomic Data
To ascertain whether differentiation in the PCA space was real or erroneous, we performed Orthogonal Projections to Latent Structures Discriminant Analysis (OPLS-DA). If the separation between the two classes is real in the PCA space, then OPLS-DA should be able to pull out some proteins that are really variable between the classes. Reverse would be true if the separation was not real. This would serve as the validation of the PCA separation. Further, to access the validity of the OPLS-DA model, we employed area under the curve (AUC) values in receiver operating curve (ROC) analysis for proteins declared significantly different by OPLS-DA. This double validation strategy would ensure that only the true significantly different proteins between the two classes are identified.
Orthogonal Projections to Latent Structures Discriminant Analysis (OPLS-DA)
OPLS-DA is a modeling technique that compares the markers coming from two different groups of samples. From this modeling, an S-Plot can be generated that has two axis, x axis is the measure of amount of change in a particular analyte and y axis is a measure of significance of the analyte in the two group comparison. When the number of variables is too big, such as in a shotgun proteomic experiment, this analysis can be used to filter out the proteins that are most significantly different among the two groups. OPLS-DA can find out the predictive and uncorrelated variance in two classes of samples (13).
Sperm Cell Proteomic Data Set
An OPLS-DA modeling was performed on sperm cell proteomics data set. The resulting S-plot is shown in Fig. 4, which gave, as output, the most different proteins among the two classes. Proteins passing the threshold of 0.80 for p(corr) values were considered as significantly different among the two classes. These proteins and their associated characteristics are summarized in Table III. All these proteins are downregulated in AZS sperm cells.
Fig. 4.

S-plot of the semen cells data set: S-Plot obtained from OPLS-DA regression analysis. Discriminating component of the OPLS-DA model is shown in the S-plot, which shows the relationship between the correlation p(corr) and the covariance (p). Data are log10 transformed and mean centered.
Table III. Proteins significantly different in the S-Plot are shown in the table. Uniprot accession or a group of accessions is shown as primary Id. Peptides are the total number of peptides found for the said protein and unique peptides are number of unique peptides out of the total peptides. Confidence score, ANOVA p value, highest and lowest mean condition, full name of the protein, covariance (p[1]) and correlation (p(corr)[1]) are shown in the table.
| Primary ID | Peptides | Unique peptides | Confidence Score (protein) | ANOVA p | Max FC | Highest Mean | Lowest Mean | Protein Name | p[1] | p(corr)[1] |
|---|---|---|---|---|---|---|---|---|---|---|
| O15031;H0Y7X5 | 7 | 2 | 39.38 | 5.99E-13 | 2.51 | Normal | Astheno | Plexin-B2 | 0.02 | 0.89 |
| Q9BYX7 | 10 | 2 | 62.55 | 6.93E-13 | 17.17 | Normal | Astheno | Putative beta-actin-like protein 3 | 0.05 | 0.86 |
| E9PN67;C9J066;Q8N4C6 | 5 | 4 | 28.26 | 7.18E-11 | 6.17 | Normal | Astheno | Ninein | 0.04 | 0.85 |
| Q92576;E7ER40 | 13 | 7 | 66.94 | 1.60E-08 | 2.49 | Normal | Astheno | PHD finger protein 3 | 0.05 | 0.84 |
| P63167;F8VRV5;F8VXI7;F8VXL2 | 11 | 5 | 67.77 | 4.03E-08 | 4.79 | Normal | Astheno | Dynein light chain 1, cytoplasmic | 0.13 | 0.82 |
| Q8NCQ7;A0A0A0MT24 | 3 | 2 | 16.79 | 1.45E-06 | 5.65 | Normal | Astheno | Protein PROCA1 | 0.03 | 0.81 |
| Q9NQT6 | 20 | 13 | 113.61 | 7.14E-09 | 1.66 | Normal | Astheno | Fascin-3 | 0.11 | 0.80 |
| J3QSU1;F5H5K1;J3KTP0;J3QLI7;Q96QE4 | 9 | 6 | 54.92 | 6.26E-09 | 3.80 | Normal | Astheno | Leucine-rich repeat-containing protein 37B | 0.10 | 0.80 |
Seminal Plasma Proteomic Data Set
In seminal plasma proteomic analysis none of the proteins passed the 0.80 cutoff for significance. No significant proteins were found for seminal plasma data set, therefore from here onwards, the seminal plasma data set was not considered for further analyses described in the study.
Receiver Operating Characteristics (ROC) Curve Analysis
Proteins found most significant in OPLS-DA S-Plot for sperm cell proteomic data set were analyzed by ROC curve and the area under the curve (AUC) was calculated. These calculations with default options were done with Analyze-it program, which works with Microsoft Excel. ROC curve analysis was employed to further validate the diagnostic value of the OPLS-DA model and to check if proteins suggested by S-Plot, as being most significantly different between the NZS and AZS in sperm cell proteomic data set, hold true in ROC curve analysis as well (i.e. having high AUC values). Most of these proteins had the perfect AUC of 1 with two of them having 0.97 and 0.93 (Dynein light chain 1 and Fascin-3 respectively). One protein for which the fold change in sperm cell proteomic data set was very close to 1 was also used in the analysis as validation control for calculations by this method. The AUC for this protein was 0.52, which was expected as this protein should not be able to classify the two classes of samples. All these calculations are shown in Table IV.
Table IV. ROC curve was drawn, for the proteins found to be significantly different in S-Plot of sperm proteomic data set and one housekeeping protein, by Analyse-it program, which works with Microsoft Excel. Uniprot accession, full protein name, Area under the curve (AUC), 95% confidence interval (95% CI) and standard error (S.E.) are given in the table.
| Uniprot accession | Protein name | AUC | 95% CI | S.E. |
|---|---|---|---|---|
| O15031;H0Y7X5 | Plexin-B2 | 1 | 1–1 | 0 |
| Q9BYX7 | Putative beta-actin-like protein 3 | 1 | 1–1 | 0 |
| E9PN67;C9J066;Q8N4C6 | Ninein | 1 | 1–1 | 0 |
| Q92576;E7ER40 | PHD finger protein 3 | 1 | 1–1 | 0 |
| P63167;F8VRV5;F8VXI7;F8VXL2 | Dynein light chain 1, cytoplasmic | 0.975 | 0.906–1.044 | 0.035 |
| Q8NCQ7;A0A0A0MT24 | Protein PROCA1 | 1 | 1–1 | 0 |
| Q9NQT6 | Fascin-3 | 0.925 | 0.766–1.084 | 0.081 |
| J3QSU1;F5H5K1;J3KTP0;J3QLI7;Q96QE4 | Leucine-rich repeat-containing protein 37B | 1 | 1–1 | 0 |
| A0A087WW73 | Phosphoinositide phospholipase | 0.525 | 0.175–0.875 | 0.178 |
Pathway Analysis
We did pathway analysis using several tools including manually finding the information in literature, because the databases these tools use are different and the algorithms to find the pathways are different. Our objective here was to find the relevant pathways from a comprehensive list. The 8 proteins from Table III, having the highest significance in terms of differential classification of NZS and AZS samples in OPLS-DA and ROC curve analysis in sperm cell samples were used for extensive literature search to delineate the pathways related to sperm motility. A schematic diagram of the results of this literature search, which was able to connect many of these sperm proteins to pathways related to sperm motility and/or migration, are shown in Fig. 5. In addition to literature search, two more tools were used for pathway analysis of proteins in sperm and seminal plasma namely IMPaLA (10) and IPA.
Fig. 5.
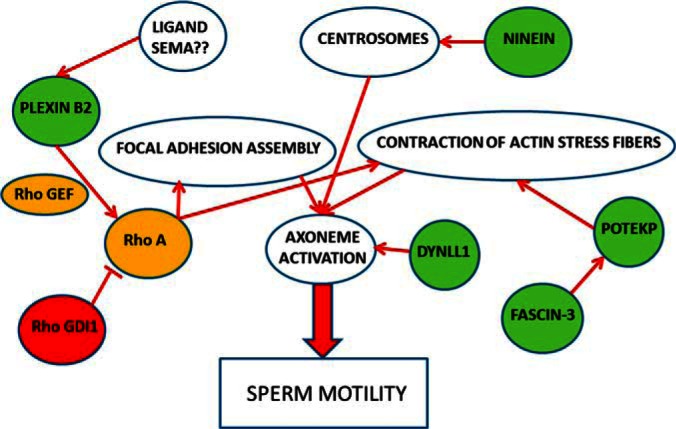
Five proteins having the significance of statistical analysis were connected to pathways related to sperm motility. These pathways were found by extensive literature mining and the corresponding references are given in the Discussion where role of all these proteins are described in more detail. Green circles are the proteins that are downregulated in asthenozoospermic sperm cell samples and red are up-regulated. The yellow circles signify the proteins that are involved in these pathways but were not found in our data set.
Integrated Molecular Pathway Level Analysis (IMPaLA)
IMPaLA tool was used for pathway over representation analysis in data set of sperm proteins sorted according to highest mean in AZS (right panel, Fig. 6) and then proteins sorted according to highest mean in NZS (left panel Fig. 6). The results are shown in Fig. 6. The full pathway over representation results of sperm cell proteins are shown in supplemental Table S4. In highest mean NZS samples, Glycolysis, Glucose metabolism and Gluconeogenesis were the major pathways found. However, in highest mean AZS samples cellular response to stress, nucleosome assembly, histone demethylation and packaging of telomere ends were the major pathways.
Fig. 6.

IMPaLA pathway over-representation analysis of sperm cell proteomic data set: pathway over representation analysis was performed separately on proteins list having highest mean in normozoospermia and asthenozoospermia. The top 10 pathways enriched in both the conditions are shown here with p values of the pathways enriched in blue and Q value in red.
Ingenuity Pathway Analysis (IPA)
Ingenuity pathway analysis was performed on sperm cell and seminal plasma proteomics data and the part of the results are shown in Fig. 7. Top canonical pathways found in sperm proteomic data set with -logp value above threshold can be seen in the figure with the most significant being glycolysis, gluconeogenesis, unfolded protein response and others such as protein ubiquitnation pathway, cellular effects of sildenafil, aldosterone signaling and eNOS signaling.
Fig. 7.

Canonical pathways: Total proteins in sperm cell proteomic data set were analyzed for Ingenuity Pathway Analysis “Core analysis” and the top canonical pathway enriched are shown here. Horizontal orange line running through the bars is the threshold for p value for these pathways's enrichment. Color coding for positive and negative z-score and for pathways with no activity pattern available are shown in the figure.
Validation of Pathways by Clustering Proteins by Self Organizing Maps (SOM) Followed by Pathway Analysis
Sperm cell proteomic data set was divided into three categories with samples having 50–60% motility in one category and 10–30 and 0% motility in another two separate categories. Only the means of the samples falling in these categories were taken further for analysis. The main reason for dividing the data set into three categories instead of two used in other analyses was to see if there was a continuous progression of disease phenotype from 50–60% motility to 10–30% to 0% or vice-versa (or in other words, if the proteomic data set can reveal or reflect the severity of the disease in question). These categories were also formed in PCA for the same reason. After categorization, a numerical data set was formed with three variables, i.e. “0% motility,” “10–30% motility,” and “50–60% motility.” Scaling of these variables is the first step before analyzing the data completely, because the SOM algorithm uses Euclidian metric to measure distances between vectors. If one variable has values in the range of (0, …, 1000) and another in the range of (0, …, 1) the former will almost completely dominate the map organization because of its greater impact on the distances measured. Therefore, the data set is normalized using histogram technique.
The next step includes the initialization and training the neural network. In the algorithm, we initialize the map with a size of (26,5) and the training is done in two phases: (a) rough training with large (initial) neighborhood radius and large (initial) learning rate; (b) fine-tuning with small radius and learning rate. The batch training algorithm is iterative, but instead of using a single data vector at a time, the whole data set is presented to the map before any adjustments are made. In each training step, the data set is partitioned according to the Voronoi regions of the map weight vectors, i.e. each data vector belongs to the data set of the map unit to which it is closest. The training results are illustrated in supplemental Fig. S2. From the component planes, it can be seen that the 0% motility and 10–30% motility data are related to each other. However, 50–60% motility does not show strong relation with either of the motility classes (supplemental Fig. S2). The correlation analysis can be better visualized in Fig. 8, where the data points are in the upper three panel and map prototype values on the lower three panels.
Fig. 8.

Correlation analyses of the sperm cell proteomic data set divided in three motility categories. Panel A, B, and C are datapoints in untrained map and D, E, and F are the trained map (50–60% motility (x axis) versus 0% motility (y axis) is panel A and D, 50–60% motility (x axis) versus 10–30% motility (y axis) is panel B and E and 10–30% motility (x axis) versus 0% motility (y axis) is panel C and F. Data points are the protein abundances with each point corresponding to one protein Id.
Fig. 8 shows the correlation analysis between different variables. Degree of the straightness of the line between variables demonstrates correlation (more straight line means stronger correlation). Note that the variable values have been denormalized. Taking a closer look at Fig. 8, it shows the aforesaid correlation outcomes from supplemental Fig. S2. Based on these analyses, the clustering of the data was done. supplemental Fig. S3 shows the Davies-Boulding index that indicates there are five clusters. The clusters on the map are also illustrated. Using this clusters information, respective data belonging to a particular cluster was extracted. The extracted clusters are given in supplemental Tables S5–S9, in which each Table is a separate cluster.
To convert these clusters into biological information, pathway over representation analysis (IMPaLA) of all the clusters was done. The resulting pathways (only those which passed the threshold of 0.05 p value) were compared with their pathway counterparts from original NZS and AZS data set. Five clusters and three parts of the original data set (highest mean AZS and highest mean NZS conditions as well as housekeeping proteins, FC 1–1.3) were compared with each other and the results are shown in Fig. 9. When all the clusters are compared with each other, it can be seen that major number of the pathways enriched in each cluster are unique. Cluster 1 protein pathways mainly overlapped with the housekeeping proteins. Cluster 2, 3 and 5 on the other hand, had majority of the pathways common with AZS samples and cluster 4 had overlapping pathways with NZS samples.
Fig. 9.

Five clusters found by SOM in sperm cell proteomic data set are shown here. Pathway over-representation analysis was done on clusters and highest mean NZS and highest mean AZS and housekeeping proteins using IMPaLA tool. These pathways were compared with each other and the comparisons are shown in the form of Venn diagram.
4. DISCUSSION
Twenty-four percent of infertile men present with isolated AZS (14) caused by various factors including varicocele, infection or genetic causes (15–17). Many cases are however idiopathic in nature and no specific causes can be ascribed to the condition (18). Further studies are needed to understand the sperm motility better for treatment/management of such cases. Sperm cells are transcriptionally and translationally silent (1), which calls for proteomic studies to find proteins implicated in aberrant and/or altered motility of sperm cells associated with idiopathic AZS. Advantages of sperm proteomic analysis and points to be considered have been recently reviewed (19).
We have performed shotgun proteomic analysis of the sperm cells and seminal plasma proteins in NZS and AZS samples. We have included 667 proteins for quantification in sperm cell samples and 429 proteins in seminal plasma samples. Quantification was performed label-free by the software Progenesis-QI for proteomics. The quantification of these proteins was followed by rigorous statistical/mathematical data analysis including principal component analysis, OPLS-DA (S-Plot), ROC Curve analysis and self-organizing maps analysis. The Principal Component Analysis (PCA) in Progenesis QI for proteomics determines the principal axis of abundance variation for individual proteins, which can easily identify the outliers. This is also a good method to study technical replicates as they should be close on a PCA biplot. As described previously in the metadata section of the results, we have removed such non-performing technical replicates from the further analyses. Axes in PCA biplot represent the directionality of most variation through the data. PCA biplot can distinguish if two or more classes of samples have little or more variation compared with each other. It can be used to visualize the differences among the classes in a simple manner.
The proteomic signature of sperm cell samples was able to separate the two classes in PCA as shown in Figs. 1 and 2. However, the separation was not very significant or complete in seminal plasma proteomic data (Fig. 3 and supplemental Fig. S1). Going back to the proteomic data sets, this was probably because of the higher ANOVA p values (compared with sperm cell proteomic data set) observed for majority of the proteins in human seminal plasma proteins. Proteins with lower ANOVA p values will be expected to lie further apart on a PCA whereas those with higher ANOVA p values, meaning low variance, will be expected to lie close to each other. ANOVA, therefore is an easy potential predictor of PCA and OPLS-DA performance and these techniques have been combined in some instances previously (20).
When the variance among two classes is low, majority of the variables fall into uncorrelated variance class and makes it difficult to find the predictive variance that can classify the two samples (13). However, very low p values were observed for more proteins in the human sperm cell proteomic data set and OPLS-DA modeling and the corresponding S-plot was able to pull out multiple proteins that are candidates for implication in sperm motility pathways (Table III). None of the proteins from seminal plasma passed the p(Corr) threshold of 0.80 in the S-plot originating from OPLS-DA modeling. The AUC for seminal plasma top differing proteins (according to fold change) gave the highest value of 0.87 for spondin-2 (compared with AUC of 1 for most of the significantly different proteins in sperm cell) and much less for other top proteins (data not shown). It is clear from all these analyses that human sperm cell proteomic signature as opposed to human seminal plasma proteomic signature can differentiate the NZS and AZS groups easily.
The 8 significant proteins in S-plot of sperm cell proteomic data set were all downregulated in AZS samples. We did an extensive literature search for finding the common pathways converging to sperm motility (Fig. 5). Five of these proteins were found to be related to the sperm motility pathways having multiple nodes. Ninein is a protein that helps assemble the centrosomes (21, 22), which regulate the axoneme activation (23) which, in turn, is responsible for sperm motility (24). Fascin-3 is an actin bundling protein and together they regulate actin stress fiber contraction (25), which is important for axoneme activation converging to sperm motility. Plexin-B2 is a cell surface receptor (downregulated in AZS sperm cells in our data), which, upon ligand based activation, activates the RhoA protein (26) leading to focal adhesion assembly and contraction of actin stress fibers (27), once again converging to axoneme activation and sperm motility pathway. RhoGDI, which blocks this action of RhoA (28), was found to be up-regulated in AZS sperm cells in our study (supplemental Table S2). Many of these proteins are being implicated in the sperm motility pathways for the first time and these interesting targets need further validation in future studies. It has previously been found that OPLS-DA is a good tool for new hypothesis formation in terms of classifying the classes of samples (13) and many a times, it results in outputs that cannot be extracted by other types of analyses in terms of biological importance.
The most significantly different proteins in S-plot for sperm cells proteomic data set and some of the top proteins in seminal plasma data set having the highest fold change differences were analyzed by ROC curve analysis (Table IV, and data not shown for seminal plasma proteins) to validate the predictive power of these proteins to disease classes. In all the comparative proteomics studies (discovery type or targeted) whether searching for biomarkers or proteins significantly different among the classes, disease versus the control samples, ROC curve can play important role. It works as a binary classification technique and evaluates the performances of individual proteins to classify the samples. Traditional use of AUC from ROC curve has been in the biomarker discovery or validation studies however it basically suggests the significantly different proteins among the sample classes (reflected in higher AUC). Therefore, it can also be used for probing out the mechanistic insight from discovery proteomic studies. It can help produce protein targets, which can be, either tested as biomarkers or, used for mechanistic studies to establish their roles in the disease. However, the typical sample size for this kind of study that benefits from ROC curve analysis is 30–50 when the AUC values are closer to 1 (29). Our study has 17 seminal plasma samples and 13 corresponding sperm cell samples and we realize it is a limited study. However, it does give an important impetus for future studies to validate our findings in larger sample sets and also in clarifying roles of these proteins in mechanistic studies to understand the process underlying the motility of the human sperm.
In the SOM analysis of the sperm cells proteomic data set we were able to cluster the data of three sperm motility categories into five clusters. Correlation analysis showed that 50–60% motility did not correlate with the 10–30% or 0% motility classes (Fig. 8). However, 10–30 and 0% motility classes correlated strongly (Fig. 8). This is indicative of the phenomenon where downgradation from high to low motility is continuous and abundance of key proteins or the whole proteomic signature of the classes can differentiate the classes and reflect this downgradation. In-line with this fact the optimal threshold cutoff found by ROC curve analysis for the most significantly different proteins in OPLS-DA S-Plot can prove to be a strong indicator of this progression. These proteins, when going below or above certain abundance in sperm cells may lead to lower motility. This is a significant result however we realize that this needs to be validated in larger set of samples. The five clusters found in the SOM analysis of the sperm proteomic data set were found to separately contain largely pathways related to either NZS or AZS or housekeeping proteins (Fig. 9). This suggests that functional analysis of the clusters in terms of pathway over-representation analysis is a good strategy to find the biological relevance of the clustering techniques.
Going back to the integration of all these analyses techniques, proteins having lower AUC in the ROC curve analysis (Dyenin light chain 1 and Fascin3) are also the proteins that have lower p(corr) values in S-Plot (however, still high enough) than most other proteins in the Table (Table III). These two proteins also have higher ANOVA p values than most other proteins in table (however, still low enough, See Table III and Table IV). These techniques of data analysis seem to have some degree of crosstalk with each other and relevance in one can be validated by relevance in the other. Appropriate statistical and/or mathematical analyses have the power to reveal proteins having the significant differences in a binary classification that are not otherwise visible in a data set based only on FC. OPLS-DA is very powerful technique for binary data sets however validation analyses needs to be carried out (such as ROC Curve analysis) to judge the validity of the model.
In summary and conclusion, we have found that sperm motility pathway defects are reflected in sperm proteomic signature using appropriate statistical methods. Seminal fluid proteomic data set is not reflective a great deal about these defects, particularly in terms of mechanistic insights converging to appropriate pathways. Further studies to elucidate the sperm motility pathways and defects thereof could be preferably carried out on sperm samples, even though some proteins from seminal plasma might still have roles to play. ANOVA p values are a good indicator of the proteins different among the two classes, which is also validated by other statistical techniques such as OPLS-DA and ROC curve analysis. Fold change, alone, should not be a criterion to find the significantly different proteins among the two classes of samples. We propose a moderate number of proteins targets (Sperm cells proteomic data set, See Table III and IV) whose abundance is significantly predictive of the defects of the sperm motility pathways. These high confidence targets need to be further validated in future studies with larger sample sets and also in regard to their role in the sperm motility in individual protein-based mechanistic studies.
Supplementary Material
Footnotes
Author contributions: M.S. designed research; M.S., S.J., A.T., and A.S. performed research; M.S., S.J., T.J., A.T., S.S., and S.Y. contributed new reagents or analytic tools; M.S., S.J., T.J., S.S., S.Y., and R.R. analyzed data; M.S., S.J., and R.R. wrote the paper.
* Ashima Sinha thanks Department of Science & Technology, Government of India, New Delhi for research grant under Women Scientist Scheme-A (No. SR/WOS-A/LS-72/2014).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- NZS
- normozoospermia(ic)
- AUC
- area under the curve
- AZS
- asthenozoospermia(ic)
- BCA
- bicinchoninic acid assay
- CI
- confidence interval
- DIA
- data independent acquisition
- DTT
- dithiothreitol
- FC
- fold change
- FDR
- false discovery rate
- HDMS
- high definition mass spectrometer
- IMPaLA
- integrated molecular pathway level analysis
- IMS
- ion mobility spectroscopy
- IPA
- ingenuity pathway analysis
- OPLS-DA
- orthogonal projections to latent structures discriminant analysis
- PCA
- principle component analysis
- ROC
- receiver operating curve
- SE
- standard error
- SF
- surfactant
- SOM
- self organizing maps
- UPLC
- ultra performance liquid chromatography
- WHO
- World Health Organization.
REFERENCES
- 1. Amaral A., Castillo J., Ramalho-Santos J., and Oliva R. (2014) The combined human sperm proteome: cellular pathways and implications for basic and clinical science. Human Reproduction Update 20, 40–62 [DOI] [PubMed] [Google Scholar]
- 2. Carballada R., and Esponda P. (1998) Binding of seminal vesicle proteins to the plasma membrane of rat spermatozoa in vivo and in vitro. Int. J. Androl. 21, 19–28 [DOI] [PubMed] [Google Scholar]
- 3. Ding Z., Qu F., Guo W., Ying X., Wu M., and Zhang Y. (2007) Identification of sperm forward motility-related proteins in human seminal plasma. Mol. Reprod. Dev. 74, 1124–1131 [DOI] [PubMed] [Google Scholar]
- 4. Amaral A., Paiva C., Attardo Parrinello C., Estanyol J. M., Ballescà J. L., Ramalho-Santos J., and Oliva R. (2014) Identification of Proteins Involved in Human Sperm Motility Using High-Throughput Differential Proteomics. J. Proteome Res. 13, 5670–5684 [DOI] [PubMed] [Google Scholar]
- 5. Shen S., Wang J., Liang J., and He D. (2013) Comparative proteomic study between human normal motility sperm and idiopathic asthenozoospermia. World J. Urol. 31, 1395–1401 [DOI] [PubMed] [Google Scholar]
- 6. Zhao C., Huo R., Wang F. Q., Lin M., Zhou Z. M., and Sha J. H. (2007) Identification of several proteins involved in regulation of sperm motility by proteomic analysis. Fertility Sterility 87, 436–438 [DOI] [PubMed] [Google Scholar]
- 7. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., and Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE : A virtue of parallel ms acquisition. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 8. Serang O., Moruz L., Hoopmann M. R., and Käll L. (2012) Recognizing uncertainty increases robustness and reproducibility of mass spectrometry-based protein inferences. J. Proteome Res. 11, 5586–5591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Luca A., Henry M., Meleady P., and O'Connor R. (2015) Label-free LC-MS analysis of HER2+ breast cancer cell line response to HER2 inhibitor treatment. DARU J. Pharmaceut. Sci. 23, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamburov A., Cavill R., Ebbels T. M., Herwig R., and Keun H. C. (2011) Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 27, 2917–2918 [DOI] [PubMed] [Google Scholar]
- 11. Kohonen T. (1995) Self-organizing Maps, Springer [Google Scholar]
- 12. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., and Hermjakob H. (2015) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44(D1), D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stenlund H., Gorzsás A., Persson P., Sundberg B., and Trygg J. (2008) Orthogonal projections to latent structures discriminant analysis modeling on in situ FT-IR spectral imaging of liver tissue for identifying sources of variability. Analytical Chemistry 80, 6898–6906 [DOI] [PubMed] [Google Scholar]
- 14. Luconi M., Forti G., and Baldi E. (2006) Pathophysiology of sperm motility. Frontiers in bioscience : a journal and virtual library 11, 1433–1447 [DOI] [PubMed] [Google Scholar]
- 15. Gdoura R., Kchaou W., Chaari C., Znazen A., Keskes L., Rebai T., and Hammami A. (2007) Ureaplasma urealyticum, Ureaplasma parvum, Mycoplasma hominis and Mycoplasma genitalium infections and semen quality of infertile men. BMC Infect. Diseases 7, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pasqualotto F. F., Sundaram A., Sharma R. K., Borges E. Jr, Pasqualotto E. B., and Agarwal A. (2008) Semen quality and oxidative stress scores in fertile and infertile patients with varicocele. Fertil Steril 89, 602–607 [DOI] [PubMed] [Google Scholar]
- 17. Jaiswal D., Sah R., Agrawal N. K., Dwivedi U. S., Trivedi S., and Singh K. (2012) Combined Effect of GSTT1 and GSTM1 Polymorphisms on Human Male Infertility in North Indian Population. Reproductive Sciences 19, 312–316 [DOI] [PubMed] [Google Scholar]
- 18. Ortega C., Verheyen G., Raick D., Camus M., Devroey P., and Tournaye H. (2011) Absolute asthenozoospermia and ICSI: what are the options? Human Reproduction Update 17, 684–692 [DOI] [PubMed] [Google Scholar]
- 19. Agarwal A., Bertolla R. P., and Samanta L. (2016) Sperm proteomics: potential impact on male infertility treatment. Expert Rev. Proteomics 13, 285–296 [DOI] [PubMed] [Google Scholar]
- 20. de Haan J. R., Wehrens R., Bauerschmidt S., Piek E., van Schaik R. C., and Buydens L. M. (2007) Interpretation of ANOVA models for microarray data using PCA. Bioinformatics 23, 184–190 [DOI] [PubMed] [Google Scholar]
- 21. Dammermann A., and Merdes A. (2002) Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 159, 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delgehyr N., Sillibourne J., and Bornens M. (2005) Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J. Cell Sci. 118, 1565–1575 [DOI] [PubMed] [Google Scholar]
- 23. Avidor-Reiss T., and Gopalakrishnan J. (2013) Cell cycle regulation of the centrosome and cilium. Drug Discovery Today. Disease Mechanisms 10, e119–e124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Porter M. E., and Sale W. S. (2000) The 9 + 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J. Cell Biol. 151, F37–F42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elkhatib N., Neu M. B., Zensen C., Schmoller K. M., Louvard D., Bausch A. R., Betz T., and Vignjevic D. M. (2014) Fascin plays a role in stress fiber organization and focal adhesion disassembly. Curr. Biol. 24, 1492–1499 [DOI] [PubMed] [Google Scholar]
- 26. Azzarelli R., Pacary E., Garg R., Garcez P., van den Berg D., Riou P., Ridley A. J., Friedel R. H., Parsons M., and Guillemot F. (2014) An antagonistic interaction between PlexinB2 and Rnd3 controls RhoA activity and cortical neuron migration. Nat. Commun. 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chrzanowska-Wodnicka M., and Burridge K. (1996) Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hiraoka K., Kaibuchi K., Ando S., Musha T., Takaishi K., Mizuno T., Asada M., Ménard L., Tomhave E., and Didsbury J. (1992) Both stimulatory and inhibitory GDPGTP exchange proteins, smg GDS and rho GDI, are active on multiple small GTP-binding proteins. Biochem. Biophys. Res. Commun. 182, 921–930 [DOI] [PubMed] [Google Scholar]
- 29. Obuchowski N. A. (2000) sample size tables for receiver operating characteristic studies. Am. J. Roentgenol. 175, 603–608 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.