

Abstract
Carnosic acid (CA) and carnosol (CS) are two structurally related diterpenes present in rosemary herb (Rosmarinus officinalis). Although several studies have demonstrated that both diterpenes can scavenge free radicals and interfere in cellular processes such as cell proliferation, they may not necessarily exert the same effects at the molecular level. In this work, a shotgun proteomics study based on stable isotope dimethyl labeling (DML) and nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) has been performed to identify the relative changes in proteins and to gain some light on the specific molecular targets and mechanisms of action of CA and CS in HT-29 colon cancer cells. Protein profiles revealed that CA and CS induce different Nrf2-mediated response. Furthermore, examination of our data revealed that each diterpene affects protein homeostasis by different mechanisms. CA treatment induces the expression of proteins involved in the unfolded protein response in a concentration dependent manner reflecting ER stress, whereas CS directly inhibits chymotrypsin-like activity of the 20S proteasome. In conclusion, the unbiased proteomics-wide method applied in the present study has demonstrated to be a powerful tool to reveal differences on the mechanisms of action of two related bioactive compounds in the same biological model.
Carnosic acid (CA)1 and carnosol (CS) are two structurally related orthodiphenolic compounds with abietane carbon skeleton containing hydroxyl groups at positions C-11 and C-12 that belong to the abietanes family of naturally occurring diterpenes in the popular Lamiaceae herbs, rosemary (Rosmarinus officinalis), and sage (Salvia officinalis) (1). CS has a lactone moiety across the B ring, whereas CA has a free carboxylic acid group (supplemental Fig. S1). Both diterpenes are capable of directly scavenging free radicals (2) and are also regarded as “proelectrophilic” compounds that become active electrophiles after oxidation to their quinone forms. It has been recognized that CA and CS quinones react with a critical thiol in Keap1, causing it to release Nrf2 transcription factor that may enter the nucleus for subsequent activation of ARE (antioxidant-response element)-mediated transcription of an array of proteins that protect against oxidative stress (3, 4). In fact, this effect has been considered as the predominant cause for the observed protective activity of both diterpenes in studies on their role in central nervous system (5). Besides their well-known property for indirectly increasing endogenous cellular antioxidant defenses via activation of the Keap1/Nrf2/ARE cascade, these compounds have a potential to modulate other multiple mechanisms causing a broad range of effects in cellular functions and biological outcomes depending on the cell model under study and the experimental conditions (6). For instance, CA and CS may interfere with a range of different cellular processes related to cell proliferation, invasiveness, tumorigenesis, and survival of cancer cells. Although CA and CS seem to share comparable antiproliferative potency they may not necessarily exert the same exact effects at the molecular level (7–11). Indeed, the wide spectrum of molecular targets of CA and CS in cancer cells has been recently reviewed by Petiwala and Johnson (12). For instance, CS has been shown to target Bcl-2 (13), CREB-binding protein/p300 (14), NF-kB and c-Jun (15), β-catenin (16), p21 (9), ERK1/2 (17), Jak2/Src-STAT3 (18), AMPK-mTOR (19), androgen receptor and estrogen receptor α (20, 21), among others. In the case of CA, its anticancer activity has also been linked to different molecular targets. For example, CA blocked the epithelial to mesenchymal transformation by inhibiting Akt phosphorylation and the secretion of several proteins involved in the invasiveness of melanoma (18) and colon adenocarcinoma cells (22). In another work, CA induced apoptosis through reactive oxygen species mediated p38 activation in neuroblastoma cells (4). In human prostate carcinoma cells, CA increased PP2A activity, leading to Akt and NF-kB signaling inhibition and induction of apoptosis (23). In addition, CA has been shown to sensitize human renal carcinoma Caki cells to TRAIL-induced apoptosis through down-regulation of c-FLIP and Bcl-2 at the post-translational levels and induction of DR5, Bim and PUMA transcription, events that were attributed to the up-regulation of CHOP and ATF4 typically observed in cells under ER stress (24). Recent works have reported the involvement of autophagy cell death in the antiproliferative activity of CS and CA in different cancer cell lines (17, 25). In this regard, CA-induced autophagic cell death has been closely linked to negative regulation of the Akt/mTOR pathway in human hepatoma cells (25). Furthermore, blockage of Akt signaling by PTEN expression seems to be a part of the causative mechanism for the antiproliferative effect of CA in leukemia cells (26). CS has been also implicated in the inhibition of PI3K/Akt and mTOR signaling pathway mediated by AMPK activation in G2 phase-arrested prostate cancer cells (19). Taken together, all the reported pleiotropic cellular and molecular effects conferred to these rosemary diterpenes support the notion that the underlying mechanisms of action of these compounds are complex and diverse. Recently, foodomics has demonstrated to be a useful strategy to cover the identification of a wide range of molecular changes induced by rosemary compounds in in vitro cell models. In this line of work, comprehensive transcriptomic and metabolomic analyses helped on identifying global changes induced by rosemary polyphenols on colon cancer and leukemia cells (27–29). As an example, previous results obtained in our laboratory have shown that a CA-enriched rosemary extract transcriptionally trigger a strong Nrf2-mediated antioxidant response in addition to the unfolded protein response (UPR) to alleviate ER stress (29). Furthermore, the recent application of comprehensive proteomics based on nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) combined with stable isotope dimethyl labeling (DML) has generated new insights regarding the role of autophagy and proteostasis in the cellular response to rosemary polyphenols, demonstrating the suitability of this proteomics strategy for the investigation of the mechanisms of action of dietary compounds in cancer cells (30). In the present work, we have applied DML and nano-LC-MS/MS to investigate global protein changes in HT-29 colon cancer cells in response to individual rosemary diterpenes, CA and CS. The objectives of this study were to: (i) identify changes in relative abundance of proteins altered by CA and CS exposures over the time; and (ii) detect differences between the protein profiles obtained in CA- and CS-treated cells in order to shed light on the specific molecular targets and mechanisms of action of each diterpene in colon cancer cells.
EXPERIMENTAL PROCEDURES
Chemicals
ACN, methanol (MeOH), formic acid (FA), ammonia solution, NaCl and urea were obtained from Merck (Darmstadt, Germany). Ethanol was provided by VWR Chemicals (Fontenay-sous-Bois, France). Acetone, EDTA, protease inhibitor mixture, PBS, n-octyl-β-d-glucopyranoside (BOG), triethyl ammonium bicarbonate (TEAB), sodium metavanadate (NaVO4), NaF, β-Glycerophosphate, sodium pyrophosphate, (37%, v/v), iodoacetamide (IAA), DTT, CA, CS, sucrose, MgCl2, KCl, adenosine 5′-triphosphate disodium salt hydrate (ATP), digitonine and MG-132 were purchased from Sigma Aldrich (St. Louis, MO). Trypsin/Lys-C Mix (Mass Spec Grade V5072) was purchased from Promega (Madison) and deuterated formaldehyde CD2O (20% (v/v)) was obtained from ISOTEC (Miamisburg, OH). Sodium cyanoborohydride (NaBH3CN) was purchased from Fluka (Buchs, Switzerland). Ultrapure water was prepared by Milli-Q water purification system (Millipore, Bedford, MA). N-Succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC) chymotrypsin-like substrate and purified human erythrocytes 20S proteasome were purchased from Enzo Life Sciences (Plymouth Meeting, PA).
Cell Culture
Colon adenocarcinoma HT-29 cells obtained from ATCC (American Type Culture Collection, LGC Promochem, UK) were grown in McCoy's 5A supplemented with 10% (v/v) heat-inactivated FBS, 50 U/ml penicillin G, and 50 U/ml streptomycin at 37 °C in humidified atmosphere and 5% CO2. When cells reached ∼50% confluence, they were trypsinized, neutralized with culture medium, seeded at 10, 000 cells/cm2 in 78 cm2 cell culture dishes and allowed to adhere overnight at 37 °C for 24 h.
Flow Cytometry Analysis
To study the cell cycle distribution, HT-29 cells were treated with cytostatic concentrations of CA or CS in complete culture medium for 24 h. After the treatment, cells were trypsinized, washed with PBS, and fixed with 70% (v/v) cold ethanol at −20 °C for at least 24 h. Then, fixed cells were resuspended in 0.5 ml of PI/RNase staining buffer (BD Pharmingen, San Diego, CA), incubated for 15 min in the dark, and analyzed on a Gallios flow cytometer equipped with a blue (488 nm) laser (Beckman Coulter, Miami, FL). Events were gated for peak width and area to exclude subcellular debris and aggregates. A total of 10,000 events were recorded for each sample and a frequency histogram of peak area was generated and analyzed using Cylchred (V.1.0.0.1) software (University of Wales College of Medicine, Cardiff, U.K.). Results are provided as the mean ± S.E. of three independent experiments.
Western Blot Analysis
For Western blot analysis, 40 μg of protein lysates from HT-29 cells treated for 24 h with different concentrations of CA (GI50, TGI or LC50) or vehicle (0.2% (v/v) DMSO) were separated in 12% Bis-Tris SDS-polyacrylamide gels by electrophoresis, and transferred to nitrocellulose membranes using Trans-Blot® S.D. Semi-Dry Transfer Cell (Bio-Rad Laboratories, Hercules, CA). The membranes were blocked for 1 h at room temperature with 5% (v/v) nonfat dry milk in TBST (20 mm Tris, 150 mm NaCl and 0.1% (v/v) Tween 20). Incubation with specific primary antibodies against HSPA5 protein (dilution 1:500) from Cell Signaling (Danvers, MA) (Cat# 3177) and β-actin protein (dilution 1:10000) from Sigma (Cat# A2066) was performed in blocking buffer overnight at 4 °C. Horseradish peroxidase-conjugated anti-IgG from Sigma (Cat# A9169) was used as secondary antibody. The specific proteins were detected using an enhanced chemiluminescence kit (GE Healthcare, U.K.) according to the manufacturer's instructions. Results are shown as the expression ratio of HSPA5 normalized to β-actin between the treated and control samples, and a two-sample t test was applied considering significant differences when p value < 0.05.
Determination of 26S and 20S Proteasome Activity
To determine the 26S and 20S proteasome chymotrypsin-like activity after diterpene treatment, HT-29 cells were incubated with cytostatic concentrations of CA, CS, or vehicle (0.2% (v/v) DMSO) for different times. As a positive control, cells were incubated with 1 μm of MG-132, a specific inhibitor of the proteasome activity. After the treatment, the 20S and 26S proteasome activities were measured as previously described (31). Cells were trypsinized, washed with PBS and divided equally into two aliquots. To evaluate the 20S proteasome activity, one aliquot was resuspended in 300 μl of lysis buffer (50 mm Tris titrated by HCl to pH 7.5, 250 mm sucrose, 5 mm MgCl2, 1 mm DTT, 0.5 mm EDTA, and 0.025% digitonin). To evaluate the 26S activity, the second aliquot was resuspended in lysis buffer containing 2 mm ATP. ATP prevents dissociation of the 26S proteasome into its components and ensures its maximal activity. Cells were incubated on ice for 5 min, followed by centrifugation at 20,000 × g for 15 min at 4 °C. The supernatants were collected and the protein concentration in the cell lysates was determined using Bio-Rad DCTM (Bio-Rad Laboratories). To measure the 20S proteasome chymotrypsin-like activity, 4 μg of protein extract were incubated in 200 μl of assay buffer (50 mm Tris titrated by HCl to pH 7.5, 40 mm KCl, 5 mm MgCl2, 1 mm DTT), for 45 min at 37 °C with 100 μm fluorogenic peptide substrate Suc-LLVY-AMC. The ATP-dependent 26S proteasome chymotrypsin-like activity was estimated using the same procedure as for the 20S, but 2 mm ATP was added to the reaction mixtures. After incubation, hydrolyzed 7-amino-4-methylcoumarin (AMC) was measured in a microplate reader (Synergy HT, BioTek Instruments, Winooski, Vermont), using an excitation filter of 360 nm and an emission filter of 460 nm. Results are provided as the mean ± S.E. of the proteasome activity relative to the control of three independent experiments, and ANOVA with Fisher LSD post hoc test was applied considering significant differences when p < 0.05.
To determine the inhibitory chymotrypsin-like activity of diterpenes in purified 20S proteasome, 200 ng of purified human erythrocytes 20S proteasome were incubated with 100 μm Suc-LLVY-AMC in 200 μl of assay buffer (50 mm Tris titrated by HCl to pH 7.5), for 45 min at 37 °C with or without different concentrations of CA, CS, or MG-132. Hydrolyzed AMC was quantified as described above, and IC50 (50% inhibitory concentration) was calculated from three independent experiments using SigmaPlot (version 12.5) software (Systat Software Inc., Erkrath, Germany).
Quantitative Reverse Transcription PCR (RT-qPCR)
To determine the expression ratios of PSMC1 gene in response to CS treatment, HT-29 cells were incubated with a cytostatic concentration of CS or vehicle (0.2% (v/v) DMSO) for different times (2, 6 or 24 h). After the treatment, RNA was isolated from cells using TRIzol Plus RNA Kit (Invitrogen, Spain) according to manufacturer's protocol. Starting amounts of 0.5 μg of total RNA were reverse transcribed using Transcriptor First Strand cDNA Synthesis kit with oligo(dT) primers (Roche Diagnostics, Barcelona, Spain). Quantitative PCR was performed using LightCycler® 480 Real-Time PCR and LightCycler® 480 SYBR Green I (Roche Diagnostics). The primer sequences (5′-3′) used for PSMC1 transcript detection were PSMC1-F: TTCCGAGTTGCTGAAGAACA, and PSMC1-R: ATCCATCCAACTGGTTCAGC (32); and for GAPDH transcript detection were GAPDH-F: ATCCATCCAACTGGTTCAGC and GAPDH-R: ATCCATCCAACTGGTTCAGC (29). Results are shown as the expression ratio of PSMC1 normalized to GAPDH between the treated and control cells, and a two-sample t test was applied considering significant differences when p value < 0.05.
Experimental Design and Sample Preparation for Proteomics Analysis
For proteomic experiments, HT-29 cells were incubated with different concentrations (GI50, 50% growth inhibition; TGI, total growth inhibition, LC50, 50% lethal concentration) of two polyphenols (CA, CS) or vehicle (0.2% (v/v) DMSO), for 2, 6, or 24 h. Three biological replicates were used in the experiments, obtaining a total of 63 samples. After incubation, cells were trypsinized and washed with 1 ml of cold PBS, and 1 × 106 cells were lysed with 300 μl of lysis buffer (6 m urea, 1% BOG, 0.15 m NaCl, 1.3 mm EDTA, 1 mm NaVO4, 5 mm NaF, 2.5 mm sodium-pyrophospate, and 5 mm β-Glycerophosphate in PBS) supplemented with 10 μl of protease inhibitor mixture. The samples were incubated for 60 min at 4 °C during mild agitation, sonicated for 30 min at 0 °C in water bath (Elma, Singen, Germany) and centrifuged at 10,000 × g at 4 °C for 15 min (Sigma, Osterode am Harz, Germany). Protein concentration was measured using Bio-Rad DCTM assay and 10 μg of proteins were incubated at 4 °C for 90 min in 500 μl of ice-cold tributylphosphate/acetone/methanol mixture (1:12:1, v/v/v). The precipitate was centrifuged for 15 min (3,000 × g at 4 °C), washed with 1 ml cold acetone, and finally air-dried. The resulting pellets were dissolved in 20 μl of 1% (w/v) BOG with 20% (v/v) ACN in 0.1 m TEAB, and the proteins were reduced with 10 μl of 45 mm DTT at 56 °C for 15 min and alkylated with 10 μl of 100 mm IAA for 15 min in the dark. For protein digestion, samples were incubated at 37 °C overnight in darkness with 0.5 μg of LysC/trypsin solution (5% w/w, of total protein content). Samples were dried in SpeedVac to remove ACN, resuspended in 70 μl of 0.1 m TEAB and water saturated ethyl-acetate was used to extract BOG (33). The tryptic peptide mixtures were reconstituted in 70 μl of 0.1 m TEAB and dimethyl labeling was performed as previously described (34). Briefly, 4 μl of regular formaldehyde CH2O (4%, v/v) was added to control samples and 4 μl of deuterated formaldehyde CD2O (4%, v/v) was added to treated samples, marking them as light and medium respectively. After vortexing, 4 μl of freshly prepared 0.6 m NaBH3CN solution was added to each sample and incubated for 60 min at room temperature with mild agitation. The reaction was finished by adding 16 μl of 1% (v/v) ammonia solution and 8 μl of 5% (v/v) FA was added to consume the excess of the labeling reagents. Finally, light and medium samples were mixed together and desalted on Isolute C18 solid phase extraction columns (1 ml, 50 mg capacity, Biotage, Uppsala, Sweden). After desalting, peptides were dried in a SpeedVac and redissolved in 0.1% (v/v) FA to a concentration of 1 μg/μl prior to nano-LC-MS/MS.
Nano-LC-MS/MS Analysis and Protein Database Searches
Five-microliter aliquots containing ∼ 1 μg of tryptic peptides were injected into a nano-LC-MS/MS system consisting of EASY-nLC II (Thermo Fisher Scientific, Bremen, Germany) coupled via nanoelectrospray ionization ion source to Orbitrap Velos Pro™ mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The peptide separations were performed on in-house packed uncoated fused silica emitters (PicoTipTM emitter, length 150 mm, 75 μm i.d., 375 μm o.d., tip opening 5 ± 1 μm, New Objective, Woburn, MA). The emitters were packed with a methanol slurry of reversed-phase, fully end-capped Reprosil-Pur C18-AQ 3 μm resin (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) using a PC77 pressure injection cell (Next Advance, Averill Park, NY). The separations were performed at a flow rate of 250 nL/min with mobile phases A (water with 0.1% (v/v) formic acid) and B (acetonitrile with 0.1% (v/v) formic acid). A 97-min gradient from 4% B to 30% B followed by 8 min from 30% B to 48% B, 6 min from 48% B to 75% B and a washing step with 75% B for 3 min was used. The mass spectrometer was operated in positive ion mode with unattended data-dependent acquisition mode, in which the mass spectrometer automatically switches between acquiring a high-resolution survey mass spectrum in the Orbitrap (resolving power 60,000 fwhm) and consecutive low-resolution, collision-induced dissociation fragmentation of up to ten of the most abundant ions in the ion trap using normalized collision energy of 35.0 eV. Ions that were once selected for acquisition were dynamically excluded for 30 s for further fragmentation. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (35) via the PRIDE partner repository with the data set identifier PXD004253. Annotated spectra are available for inspection (search key c7xhnyxgyf) via the MS-Viewer tool (36) of the Protein Prospector (http://prospector.ucsf.edu/prospector/mshome.htm).
All MS raw files were collectively processed with MaxQuant (version 1.5.2.8) (http://www.coxdocs.org/doku.php?id=maxquant:start) (37) applying the Andromeda search engine with the following adaptions (38). The false discovery rate (FDR) was set to 1% for both proteins and peptides and we specified a minimum length of seven amino acids. MaxQuant scored peptides for identification based on a search with a maximum mass deviation of precursor and fragment masses of up to 20 ppm and 0.5 Da. The Andromeda search engine was used for the MS/MS spectra search against a concatenated forward and reversed version of the Uniprot human database (downloaded on February 11, 2015, containing 89,909 entries and 245 frequently detected contaminants) for quantitative study. Enzyme specificity was set as C-terminal to Arg and Lys, also allowing cleavage at proline bonds and a maximum of two missed cleavages. Carbamidomethylation of cysteine residues was set as fixed modification whereas oxidation of methionine, phosphorylation of serine/threonine/tyrosine and protein N-acetylation were allowed as variable modifications. For dimethyl labeling, DimethylLys0 and DimethylNter0 were set as light labels, and DimethylLys4 and DimethylNter4 were set as medium labels. A minimum peptide ratio count of two and at least one “razor peptide” was required for quantification. After protein quantification, each data set was normalized to the median of the ratios to correct for mixing of medium and light labeled cells at 1:1 ratios, and to enable a better comparison between the different conditions.
Statistical and Bioinformatics Analysis
Prior to any statistical analysis using Perseus software (http://www.coxdocs.org/doku.php?id=perseus:start), identifications flagged as reverse, potential contaminants, or proteins identified only by site modification were excluded for further analysis, and the relative protein abundance was transformed to the log2 scale. To identify the differentially expressed proteins in treated cells with respect to the control group, a 1.5-fold cutoff in relative protein abundance and a p value < 0.05 (one sample t test) were applied in those proteins identified in at least two biological replicates. The lists of differentially expressed proteins were uploaded in the bioinformatics tool Ingenuity Pathway Analysis (IPA; Qiagen, Redwood City, CA) to perform causal upstream regulator (UR) and functional enrichment analysis. In UR analysis, the activation state of each regulator (such as transcription factors) is predicted based on global direction of changes in the different experimental condition for previously published targets of this regulator. Significance of the activation or deactivation of molecules predicted by UR analysis was tested by the Fisher Exact test p value, considering only the predictions with significant p value < 0.05, and regulation z-score < −2 or > 2, for deactivation and activation, respectively. Functional enrichment analysis was used to identify over-represented molecular and cellular functions in the protein data sets. Significance of the molecular and cellular functions was tested by the Fisher's exact test p value.
RESULTS AND DISCUSSION
CA and CS Treatments, Protein Identification, and Quantification
The antiproliferative activity of CA and CS was previously determined and reported in (10). The response parameters GI50 and TGI, indicators for cytostaticity, and LC50, indicative for cytotoxicity, were used as reference to prepare the working concentrations in the proteomics experiments. Based on reported data, GI50, TGI and LC50 were 33.3 ± 2.8, 47.7 ± 2.9, and 69.1 ± 3.9 μm for CA, and 41.6 ± 1.7, 52.7 ± 3.0, and 70.6 ± 4.0 μm for CS, respectively.
The study of cell cycle distribution can provide useful information to determine the mechanism by which both diterpenes induce growth inhibition. The effect of rosemary diterpenes in cell cycle progression has shown to be cell type- and concentration-dependent. For instance, it has been reported that CA and CS induce G2/M phase arrest in Caco-2 cells (9), but CA and CS block cell cycle before and after prometaphase, respectively. A recent study in our laboratory suggested that the effects of a CA-enriched rosemary extract on cell cycle distribution are highly dependent on the extract concentration (11). In the present work, we used the maximum cytostatic concentration (TGI) calculated for CA and CS to investigate the possible changes on HT-29 cell cycle distribution exerted by these compounds using flow cytometry analysis. As shown in Fig. 1, incubation of exponentially growing HT-29 cells with TGI concentrations of each diterpene for 24 h resulted in a substantial inhibition of cell cycle progression at different cell cycle phases. Namely, CA induced an obvious G1 arrest on HT-29 cells, represented by the accumulation of cells in the G1 phase (67.6% ± 1.2) with a concomitant decrease in the percentage of cells in the G2/M phase (10.5% ± 1.5) with respect to untreated control cells (57.0% ± 1.5 and 17.3% ± 1.6, respectively). On the other side, CS caused G2/M arrest as observed by the significant increase in the cell population in that phase (42.8% ± 3.9), with a simultaneous decrease in the percentage of cells in the G1 (46.5% ± 3.6) and S phases (10.8% ± 0.3). These results give evidences that the molecular and cellular mechanisms underlying the antiproliferative activity of CA and CS are potentially distinct in HT-29 cell line.
Fig. 1.
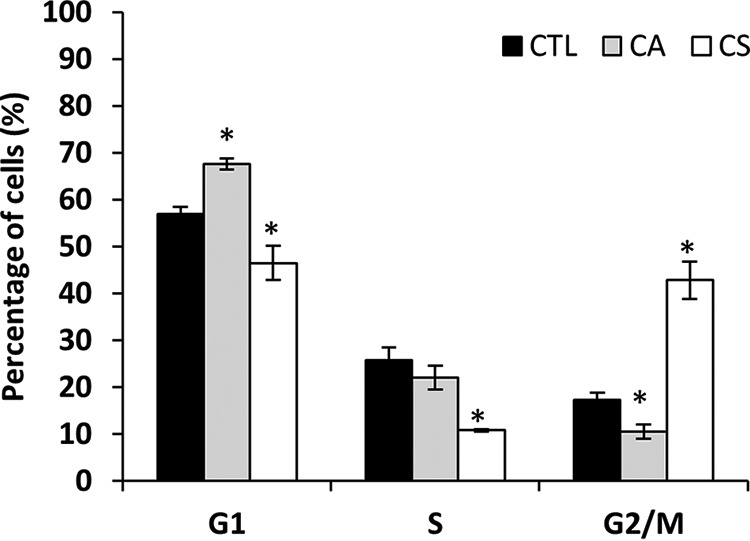
Cell cycle distribution determined by flow cytometry of HT-29 cells incubated with or without TGI concentration of CA or CS for 24 h (* indicates significant differences between the treated and control samples as determined by t test, p < 0. 05).
To get insights into the antiproliferative activity of CA and CS, the global protein changes in HT-29 cells in response to each diterpene were investigated using quantitative proteomics platform based on DML combined with nano-LC-MS/MS. We aimed at identifying temporal changes in protein levels in HT-29 cell lysates in response to different cytostatic and cytotoxic concentrations of diterpenes. To achieve this, cells were incubated with three different concentrations of the CA and CS corresponding to the three different response parameters, GI50, TGI, and LC50, as well as with the vehicle (0.2% (v/v) DMSO, as controls) for different times (2, 6 and 24 h). In these proteomic experiments, quantitative comparison based on stable isotope dimethyl labeling was performed on the eighteen sets of experiments, using “light” for control cells and “medium” labels and CA- or CS-treated cells (Fig. 2). Using three biological replicates per group, equal protein amounts of light-labeled and medium-labeled samples were mixed to generate 54 samples for further analysis. All mixtures were then analyzed using nano-LC-MS/MS analysis. MS raw data files were then simultaneously processed with the MaxQuant software for FDR-controlled peptide and protein identification and dimethyl labeling-based quantification (the identification and quantification of the peptides and proteins are found in supplemental Tables S1 and S2, respectively). A total of 1952 distinct proteins were identified (protein level FDR < 1%) with an average of 697 quantified proteins after performing all the analyses (see supplemental Table S3). The number of proteins identified in common across the 18 experimental conditions was 248. Relative abundance ratios were calculated for all proteins to screen the significantly altered proteins in each experimental condition, considering all the combinations of three variables (diterpene type, concentration and exposure time; see supplemental Tables S4 and S5). In addition, and although the aim of the present work is not the study of the phosphoproteome, 226 phosphorylated peptides (after excluding reverse and potential contaminants, phosphosite assignment is as reported by Andromeda) were detected without performing any phosphopeptide enrichment (supplemental Table S6). In this study, the restrictive criterion to consider a protein as significantly changed upon diterpene exposure included a 1.5-fold change cutoff in relative protein abundance, equivalent to log2 fold change of ± 0.585; and a p value < 0.05 (one sample t test). According to this, the total number of proteins displaying significant changes upon CA and CS treatments was 76 and 57, respectively (Table I and Table II). Among them, a cluster of 26 proteins were common to both treatments.
Fig. 2.

Scheme of the DML method and nano-LC-MS/MS analysis followed for the proteomics study. HT-29 cells were incubated with different concentrations (GI50, TGI, LC50) of two polyphenols (CA, CS) or vehicle for 2, 6. or 24 h. After protein extraction and enzymatic digestion, peptides from control and treated samples were labeled as “light” and “medium” respectively, and mixed 1:1 prior to nano-LC-MS/MS analysis.
Table I. Log2 ratio of the significantly altered proteins after the incubation of HT-29 cells with GI50, TGI and LC50 concentrations of CA and CS for 2 and 6 hours.
| 2 h |
6 h |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TGI |
LC50 |
GI50 |
TGI |
LC50 |
||||||
| Gene name | log2 ratio | Gene name | log2 ratio | Gene name | log2 ratio | Gene name | log2 ratio | Gene name | log2 ratio | |
| CA | EFHD2 | −0.63 | SLC25A3 | 0.59 | MISP | −0.61 | SDHA | 0.80 | HMOX1 | 2.26 |
| RPS24 | 0.68 | SQSTM1 | 0.68 | |||||||
| RPS26 | 0.63 | |||||||||
| CS | HMOX1 | 3.46 | HMOX1 | 3.54 | HMOX1 | 3.04 | ||||
| HSPA1A | 1.22 | DNAJB1 | 1.70 | HSPA1A | 1.73 | |||||
| SQSTM1 | 0.67 | HSPA1A | 1.53 | HSPH1 | 0.92 | |||||
| PSMC4 | 0.63 | HSPH1 | 0.88 | SQSTM1 | 0.91 | |||||
| POF1B | −0.88 | SLC2A1 | 0.63 | |||||||
| SLC3A2 | 0.60 | |||||||||
Table II. Significantly altered proteins after the incubation of HT-29 cells with GI50, TGI, and LC50 concentrations of CA and CS for 24 hours.
| Name | CA |
CS |
Name | CA |
CS |
Name | CA |
CS |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | GI50 | TGI | LC50 | |||
| Antioxidant defence | Unfolded Protein Response (tRNA charging and AAs metabol.) | Chromatine | ||||||||||||||||||
| GCLCa | 0.92 | 1.23 | 1.29 | – | – | – | AARSc | n.s. | 0.62 | 0.96 | – | n.s. | n.s. | HMGN2 | – | n.s. | n.s. | −1.16 | – | n.s. |
| GCLMa | – | 1.37 | 1.53 | n.s. | 1.11 | 1.54 | WARSc | 0.88 | n.s. | 1.15 | – | – | n.s. | HIST1H2AJ | n.s. | n.s. | n.s. | – | −1.42 | n.s. |
| TXNRD1a | 1.08 | 1.41 | 1.41 | 0.85 | 1.13 | 1.28 | GARSc | 0.63 | 0.99 | 1.21 | n.s. | n.s. | n.s. | CBX3 | n.s. | n.s. | n.s. | n.s. | n.s. | −0.61 |
| TXN1 | 0.61 | n.s. | n.s. | n.s. | n.s. | n.s. | SARSc | n.s. | 0.94 | 1.17 | – | – | – | Other functions | ||||||
| SRXN1a | – | – | 2.63 | – | – | – | YARSc | n.s. | – | 0.60 | – | – | n.s. | TMEM109 | n.s. | n.s. | −0.71 | n.s. | n.s. | n.s. |
| MT2Aa | n.s. | n.s. | 1.48 | n.s. | n.s. | 1.04 | SLC3A2c | 0.82 | 1.49 | 1.78 | n.s. | 0.72 | 1.02 | GLG1 | n.s. | – | −0.90 | n.s. | n.s. | n.s. |
| NADPH generation | SLC1A5c | n.s. | n.s. | n.s. | n.s. | 0.59 | n.s. | TMPO | n.s. | n.s. | −0.65 | n.s. | n.s. | n.s. | ||||||
| SLC2A1 | n.s. | 0.94 | n.s. | n.s. | n.s. | n.s. | ASNSc | 1.63 | – | 2.47 | – | – | 1.62 | KPNA2 | n.s. | −0.83 | −1.19 | n.s. | n.s. | n.s. |
| ME1a | 0.85 | 0.91 | 0.94 | n.s. | – | 0.91 | PSAT1c | 0.59 | 0.98 | 1.09 | – | n.s. | 0.80 | STMN1 | n.s. | −0.65 | −0.78 | n.s. | n.s. | n.s. |
| IDH1a | n.s. | n.s. | 0.75 | n.s. | n.s. | n.s. | E2F-targets | GMDS | −0.60 | n.s. | n.s. | – | – | – | ||||||
| PGDa | 0.69 | 0.77 | 0.79 | n.s. | n.s. | n.s. | RRM1d | n.s. | −0.88 | – | n.s. | n.s. | n.s. | HN1L | n.s. | – | n.s. | n.s. | n.s. | −0.69 |
| G6PDa | n.s. | 0.68 | n.s. | n.s. | n.s. | n.s. | COPS8d | n.s. | −0.71 | n.s. | n.s. | n.s. | n.s. | RAB1A | n.s. | n.s. | n.s. | – | – | −0.97 |
| Detoxification metabolism | TOP2Ad | n.s. | −1.33 | −2.46 | n.s. | n.s. | n.s. | SLC12A2 | – | n.s. | – | – | – | −1.87 | ||||||
| AKR1B10a | 1.52 | 1.82 | 1.61 | 1.37 | 1.58 | n.s. | RBBP4d | n.s. | n.s. | −0.85 | n.s. | n.s. | n.s. | ALDH18A1 | n.s. | n.s. | −0.59 | n.s. | – | −0.66 |
| AKR1C1 | 1.83 | 2.24 | 2.94 | 1.79 | n.s. | 1.30 | CSDE1d | n.s. | n.s. | −0.66 | n.s. | n.s. | n.s. | S100P | n.s. | n.s. | 1.40 | n.s. | n.s. | n.s. |
| AKR1C3 | 1.26 | 1.37 | 1.31 | n.s. | n.s. | 0.71 | Pyrimidine and purine nucleotides biosynthesis | GFPT1 | n.s. | n.s. | 1.06 | n.s. | n.s. | n.s. | ||||||
| Ubiquitin-proteasomal degradation | CAD | n.s. | n.s. | −0.64 | n.s. | −0.67 | n.s. | TOP1c | n.s. | n.s. | 0.60 | n.s. | n.s. | n.s. | ||||||
| PSMC1a | n.s. | n.s. | n.s. | n.s. | 0.73 | 0.76 | GMPS | n.s. | n.s. | −0.61 | n.s. | n.s. | n.s. | LRRC59 | n.s. | n.s. | 0.59 | n.s. | n.s. | n.s. |
| PSMC2a | n.s. | n.s. | n.s. | n.s. | 0.67 | 0.72 | HPRT1 | n.s. | n.s. | −0.61 | – | – | n.s. | ANXA5 | n.s. | n.s. | 0.62 | n.s. | n.s. | n.s. |
| PSMC3a | n.s. | n.s. | n.s. | n.s. | 0.72 | n.s. | DNA replication/cell proliferation | GOT1 | n.s. | – | 0.74 | n.s. | n.s. | n.s. | ||||||
| PSMC5a | n.s. | – | n.s. | n.s. | – | 0.87 | NASP | n.s. | n.s. | −0.80 | n.s. | n.s. | n.s. | RCN1 | 0.66 | – | n.s. | – | – | n.s. |
| PSMA4a | n.s. | n.s. | n.s. | n.s. | n.s. | 0.69 | NAP1L1 | n.s. | n.s. | −0.60 | n.s. | n.s. | n.s. | CAPN2 | 0.62 | – | n.s. | – | – | – |
| PSMA7a | n.s. | n.s. | n.s. | n.s. | 0.63 | n.s. | MKI67 | n.s. | −1.32 | n.s. | – | n.s. | −1.07 | CLIC4 | – | – | 0.72 | – | – | – |
| PSMD1a | n.s. | n.s. | n.s. | – | – | 0.62 | SERPINB5 | 0.60 | – | n.s. | – | – | – | UGDHa | 0.97 | 1.06 | 1.06 | n.s. | n.s. | n.s. |
| PSMD2a | n.s. | – | – | – | – | 0.75 | Cellular adhesion | SERPINH1 | – | n.s. | – | – | – | 0.87 | ||||||
| VCP | n.s. | n.s. | n.s. | n.s. | n.s. | 0.64 | GCNT3 | – | – | 1.79 | – | – | – | ACSL5 | n.s. | – | – | – | – | 0.83 |
| Chaperones | CD44 | n.s. | n.s. | n.s. | n.s. | n.s. | −0.92 | CD59 | n.s. | n.s. | n.s. | n.s. | n.s. | 0.80 | ||||||
| HSP90AA1 | n.s. | n.s. | n.s. | n.s. | 0.78 | 0.81 | MUC5AC | n.s. | n.s. | n.s. | n.s. | n.s. | −0.69 | CALB2 | n.s. | n.s. | n.s. | 0.70 | n.s. | n.s. |
| HSPH1 | n.s. | n.s. | n.s. | 0.87 | 0.96 | 1.33 | CNN2 | n.s. | −1.04 | −1.40 | n.s. | n.s. | −1.05 | S100A14 | n.s. | n.s. | n.s. | n.s. | 0.82 | n.s. |
| HSPA1A | n.s. | n.s. | 0.68 | 1.18 | 1.48 | 2.29 | POF1B | n.s. | −0.96 | −1.65 | n.s. | −0.96 | – | C1QBP | n.s. | n.s. | n.s. | – | 0.61 | n.s. |
| SQSTM1 | 1.42 | 1.78 | 2.52 | 1.59 | 1.74 | 1.77 | Cytoskeleton | NAMPT | 0.65 | 1.10 | 1.41 | n.s. | 0.93 | 0.96 | ||||||
| Unfolded Protein Response (ERAD) | SLC9A3R1 | n.s. | n.s. | −0.69 | n.s. | n.s. | −0.59 | SFN | 0.61 | 0.64 | 1.39 | n.s. | 1.07 | 1.23 | ||||||
| SEC61Bb | n.s. | n.s. | 0.83 | – | n.s. | n.s. | MSN | n.s. | n.s. | n.s. | n.s. | 0.64 | n.s. | ANXA1 | 0.81 | 1.07 | 1.52 | n.s. | n.s. | 0.79 |
| HYOU1b | n.s. | 0.59 | 1.21 | n.s. | n.s. | n.s. | VIL1 | n.s. | n.s. | n.s. | n.s. | −0.73 | n.s. | PSAP | 0.73 | n.s. | 1.19 | n.s. | n.s. | 1.27 |
| SSR1b | n.s. | n.s. | 0.65 | – | – | n.s. | TUBA1C | n.s. | n.s. | n.s. | n.s. | – | 0.59 | RAB10 | n.s. | n.s. | 0.64 | – | n.s. | 0.84 |
| SSR4b | n.s. | n.s. | 0.64 | – | – | n.s. | ||||||||||||||
| HSPA5b,c | n.s. | 0.65 | 1.21 | n.s. | n.s. | 0.59 | ||||||||||||||
| HSP90B1b,c | n.s. | n.s. | 0.65 | n.s. | n.s. | n.s. | ||||||||||||||
| HSPA9b | n.s. | n.s. | 0.59 | n.s. | n.s. | n.s. | ||||||||||||||
Proteins whose expression is regulated by: aNrf2;
b XBP1;
c ATF4;
d E2F1. n.s., not significant.
Then, the concordance of a protein data set obtained in the present study and microarray data obtained under identical cell culture conditions in a previous work (30) was evaluated to gain further insights into these data. Namely, owing to the availability of the gene expression microarray data set (30), the selected conditions for correlation analysis were the untreated and treated cells with the GI50 concentration of CA for 24 h. Transcriptomic data overlapped 92.1% of proteomic data, representing 744 proteins for which mRNA and protein changes were compared, providing a Pearson's correlation coefficient of 0.540 (supplemental Fig. S2). Of these common molecules, 95.6% were not altered with statistical significance, whereas 1.2% and 1.7% were significantly altered at the protein or mRNA level, respectively. It is widely accepted that protein abundance depends not only on transcription rates of the gene but also on additional regulatory mechanisms, such as mRNA stability, protein degradation, and translational regulation (39). Furthermore, differences in sensitivity, dynamic range or ambiguity in identification, among other sources of variability associated with the measurement of proteins and transcripts, may also contribute to the potential discordance between mRNA and protein abundances. In the present study, when we considered only the 23 statistically significant proteins (shown in Table II), the direction of the changes in 20 proteins (AKR1B10, AKR1C1, AKR1C3, ANXA1, ASNS, CAPN2, GARS, GCLC, ME1, NAMPT, PGD, PSAP, PSAT1, SERPINB5, SFN, SLC3A2, SQSTM1, TXNRD1, UGDH, and WARS) correlated well with the detected changes in their mRNAs levels. Only three of the significant hits (TXN, GMDS, and RCN1) were not found significantly altered at the transcript level.
Early Changes in HT-29 Proteome Upon Diterpene Exposure
In order to study the dynamic of protein expression in HT-29 cells during the course of CA and CS treatments, the ratio of the significantly changed proteins obtained at different incubation times were examined. Three exposure duration times of 2, 6, and 24 h were chosen in order to analyze very early, early, and late proteomic perturbations, respectively, occurring in treated cells with different concentrations of individual diterpenes (Table I and Table II). Most of the changes were detected after 24 h incubations with CA and CS. Indeed, none of the significantly altered proteins (p value < 0.05) after 2 h treatment with GI50 concentration of CA passed the log fold change threshold established in the present study, and only EFHD2 and SLC25A3 were significantly altered by using the TGI and LC50 concentrations of CA, respectively. Interestingly, CS did not induce detectable significant changes in the relative abundance of any protein after 2 h; however, after 6 h of treatment, all the tested CS concentrations up-regulated the protein markers for the Nrf2-mediated antioxidant response (HMOX1 and SQSTM1), as well as three cytosolic chaperones, DNAJB1, HSPH1, and HSPA1A, being the latter accumulated in a concentration-dependent fashion (Table I). In contrast, only the highest concentration of CA altered the levels of the oxidative stress markers, HMOX1 and SQSTM1 in HT-29 cells. Altogether, these results suggest that HT-29 cells trigger the antioxidant response upon exposure to both diterpenes, CA and CS; however, the observed different temporal and concentration effect of CS versus CA on markers for Nrf2-signaling might be accounted for a faster activation in cells treated with CS. In addition, the early up-regulation of different cytosolic chaperones in CS-treated cells may be indicative of proteotoxic stress at very early exposure times.
CA and CS Induce Different Pattern of Nrf2-dependent Proteins in HT-29 Cells
In order to identify the molecular and cellular functions that might be altered in response to each diterpene, functional enrichment analysis was performed on the protein data sets using IPA. These results (supplemental Fig. S3) indicated that the most significant (Fisher's exact test p value <0.05) over-represented molecular and cellular functions in data sets obtained in CA treatments were associated with cell death and survival, whereas a significant number of proteins related to cellular compromise was identified in the data set obtained from CS-treated cells. Next, causal upstream regulator analyses of the protein profiles obtained after 24 h exposures to CA and CS were performed using IPA bioinformatics tool to obtain a broader picture of the potential transcriptional regulators that could be operating in the response of HT-29 cells to diterpenes. In agreement with the well-known ability of CA and CS to activate Nrf2 signaling (3, 4), causal analysis predicted significant activation of Nrf2 under most of the tested conditions with both diterpenes (Table III). To further determine whether Nrf2 activation by CA and CS follows the same transcriptional program, the relative abundance of the proteins that support Nrf2 activation by both diterpenes was examined (Table II). In general, CA exerted changes that were slightly higher in SQSTM1 (also known as p62), as well as in molecules related with the antioxidant response and detoxication metabolism. In addition, a greater number of proteins related with NADPH generation were altered by CA treatments when compared with those affected by CS. On the other side, only CS-treated cells showed altered levels of eight different proteins belonging to 19S and 20S proteasome complexes. The results obtained suggest that although both diterpenes appear to affect antioxidant endogenous defenses, CA and CS induce different Nrf2-mediated response, revealing potential relevant differences in their mode of action. Nrf2 signaling pathway has become the subject of an intense research in last years (40). Recent evidences highlight the complexity of Nrf2 signaling pathway, such as for example the crosstalk with other pathways such as UPR or autophagy (41, 42). Thus, depending on the cellular context and type of activation, Nrf2 signaling can be prolonged by different mechanisms other than the classical Nrf2-Keap1, such as for example Keap1 sequestration by p62, that may have profound biological consequences (43).
Table III. Prediction of transcription factors by UR analysis (IPA software) in HT-29 cells treated with GI50, TGI and LC50 concentrations of CA and CS for 24 hours.
| GI50 |
TGI |
LC50 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TFa | z-score | p value | Proteins | TFa | z-score | p value | Proteins | TFa | z-score | p value | Proteins | |
| CA | NFE2L2 | 2.889 | 7.72·10−11 | UGDH, TXNRD1, SQSTM1, SLC2A1, PSAT1, PGD, ME1 GCLM, GCLC, G6PD, AKR1B10 | NFE2L2 | 3.193 | 1.33·10−12 | UGDH, TXNRD1, SQSTM1, SLC2A1, PSAT1, PGD, ME1, GCLM, GCLC, G6PD, AKR1B10 | NFE2L2 | 3.028 | 1.80·10−11 | UGDH, TXNRD1, SRXN1, SQSTM1, PSAT1, PGD, ME1, HSPA9,HSP90B1, GCLM, GCLC, AKR1B10 |
| E2F1 | −2.000 | 5.83·10−8 | UGDH, TOP2A, RRM1, COPS8, PSAT1, SLC3A2, STMN1, TXNRD1 | ATF4 | 2.611 | 3.27·10−11 | WARS, SLC3A2, SARS, PSAT1, HSPA5, HSP90B1, ASNS, AARS, GARS | |||||
| XBP1 | 2.417 | 7.52·10−7 | SSR4, SSR1, SEC61B, HYOU1, HSPA5, HSP90B1 | |||||||||
| CS | NFE2L2 | 2.756 | 1.80·10−9 | TXNRD1, SQSTM1, PSMC3, PSMC1, PSMA7, HSP90AA1, GCLM, AKRQB10 | NFE2L2 | 2.799 | 4.84·10−11 | TXNRD1, SQSTM1, PSMD1, PSMC1, PSMA4, PSAT1, ME1, HSP90AA1, GCLM | ||||
a TF, Transcription Factor.
CA Activates UPR Proteins and Downregulates Proteins Transcriptionally Controlled by E2F1
A detailed examination of the proteomic profiles obtained after 24 h treatments and IPA results (Tables II and III) showed other interesting differences on the cellular response to both diterpenes. Specifically, CA treatment caused up-regulation of proteins transcriptionally controlled by ATF4 and XBP1, along with ER chaperones. Proteins supporting activation of XBP1 and ATF4 included relevant molecules involved in ER-associated degradation (ERAD) system, ER chaperones with key role in UPR, amino-acyl tRNA synthetases related with tRNA charging function, and molecules involved in essential amino acid uptake and metabolism (Table II). Among the altered proteins, the glucose-regulated protein of 78 kDa (also known as HSPA5/Bip/GRP78) was found up-regulated in a concentration dependent manner after 24-h exposure with CA. It has been well established that up-regulation of HSPA5 is a marker of ER stress and a central regulator of the activation of UPR transducers (44). The elevation in HSPA5 relative abundance in cells treated with the different CA concentrations compared with control cells was confirmed by Western blot using commercially available antibodies against HSPA5 protein and β-actin (supplemental Fig. S4). Consistent with the quantification by mass spectrometry, the relative levels of HSPA5 protein were significantly increased in CA-treated cells with TGI and LC50 concentrations at 24 h of treatments. It is well recognized that ATF4 can be activated/induced independently of the UPR. For instance, ATF4 activation is an important event within the integrated stress response (ISR) that can be triggered by amino acid starvation, heavy metals and heme deficiency, and viral infection, as well as ER stress (45). However, the ISR does not seem to involve XBP1 activation. In our study, the coincident activation of ATF4 and XBP1, which are respective regulators of two independent signaling branches of UPR, in CA-treated cells is coherent with UPR activation.
Until recently, activation of the UPR signaling was unequivocally indicative of ER-luminal sensing of unfolded or misfolded protein accumulation. However, some recent works have described an alternative activation of the UPR that involves other types of signals, such as some mitogenic hormones, that do not originate in the ER lumen (46, 47). This alternative UPR activation appears to act in an “anticipatory” mode to cope with a future requirement for increased protein folding capacity because of protein secretion or cell proliferation, instead of the more classical “reactive” mode of UPR aimed at alleviating pre-existing ER stress (48). In the anticipatory mode of UPR pathway, hormones induce a moderate and transient increase in intracellular calcium that results in weak activation of the UPR (does not strongly activate PERK-ATF4 signaling). In our study, such UPR signaling pattern does not seem compatible with the observed elevated levels of the eleven ATF4-regulated proteins in CA-treated cells. In support of the activation of the reactive mode of UPR by CA treatment, several published studies suggest that CA induces ER stress in various cell types (21, 24, 29, 49). Also, it has been shown that the UPR contributes signals to cell death pathways in CA-treated cells as consequence of severe or unresolved ER stress that determine cell fate (21, 24), a mechanism that does not occur in the anticipatory UPR (48). It has been suggested that this effect was caused by a redox imbalance because of the elevation of intracellular reactive oxygen species (ROS) levels, a condition that is generally believed to alter ER homeostasis (24). In this regard, high concentrations (>30 μm) of CA have been shown to induce ROS generation in HT-29 and other cell types (4, 11). Numerous environmental, physiological and pathological insults, as well as nutrient fluctuations, disrupt the ER protein-folding environment to cause ER stress (50). Recent studies carried out in our laboratories have demonstrated that a CA-enriched rosemary extract also induces intracellular ROS and UPR activation in colon cancer cells (29, 30). Our present results suggest that CA is one of the bioactive compounds in the extract contributing to the activation of such response to stress. However, further detailed investigation of this aspect will be required to elucidate how this diterpene can contribute to ER stress.
Furthermore, IPA causal analysis predicted deactivation of E2F1 transcription factor in cells treated with TGI concentration of CA (Table III). E2F1 belongs to the E2F family of transcription factors that, in combination with retinoblastoma (Rb) family tumor suppressor proteins, controls DNA replication and cell cycle progression. The Rb/E2F pathway plays a pivotal role in regulating the initiation of DNA replication, and disruption of the pathway is common in virtually all human cancers (51). Although E2F is involved in a variety of cellular activities, the best understood function of E2F is to regulate transcription of genes involved in the transition from G1 to S phase, regulators of S phase entry and components of the DNA replication machinery. Our observations indicate that transcriptional targets of E2F1 essential for DNA synthesis (RRM1, TOP2A) and cell cycle progression (COPS8, RBBP4) were down-regulated in CA-treated cells (Table II). In support of a decreased E2F1 transcriptional activity, our data shows that the tumor suppressor SERPINB5 (also known as maspin) was up-regulated in CA-treated cells. Published data suggest that maspin controls cell cycle; specifically, it has been shown that maspin down-regulation by E2F1 activation accelerates cell cycle progression in gastric cancer (52). CA treatment additionally induced down-regulation of various proteins directly involved in the generation of pyrimidine (CAD) and purine (GMPS, HPRT1) nucleotides needed for DNA synthesis, as well as proteins involved in DNA replication (NAP1L1, NASP) and cell proliferation maintenance (MKI67), suggesting that CA treatment attenuates all these cellular processes. In agreement with previous transcriptomic studies carried out on CA-enriched extracts (29), our data show that the response mediated by down-regulation of E2F1 transcriptional activity constitutes a reasonable link to the G1 phase arrest observed in CA-treated cells, and provides a potential explanation for the inhibitory effect of this diterpene on HT-29 cell proliferation. Also, connected with some of these observations, the expression levels of TOP2A, MKI67 and E2F1 have recently shown to provide valuable prediction in the prognosis of cancer (53). Our data also showed that CS down-regulated various proteins, such as CBX3, HIST1H2AJ, and HMGN2, with recognized key roles in epigenetic control of chromatin structure and gene expression. Particularly, CBX3 has recently shown to promote colon cancer growth by directly repressing expression of p21, an inhibitor of cellular proliferation in response to DNA damage (54).
Rosemary Diterpenes Altered Cell Adhesion and Cytoskeletal Proteins
Other findings of our present study revealed the alteration of proteins with a key role in cell adhesion and colorectal cancer (Table II). For instance, LC50 CA treatment caused substantial increase in the relative abundance of GCNT3, a protein that has been shown to suppress cell adhesion, motility, and invasion of colon cancer cells (55). GCNT3 is frequently expressed at low levels in the majority of colorectal carcinomas whereas its overexpression dramatically inhibits colon cancer cell growth in vitro and in vivo. Such effects have been associated with its ability to induce carbohydrate changes on the cells surface affecting cell-extracellular matrix interactions. In recent years, GCNT3 expression has gained relevance and it has been recently proposed as a promising biomarker for colon cancer to monitor the response to chemotherapy (56). The GCNT3 up-regulation in CA-treated cells observed in the present study is coincident with other published data associating the GCNT3-inducing effect of CA and CA-enriched rosemary extracts with their antitumor effect on colon and pancreatic cancer cell lines (56). In contrast, the relative abundance of CD44 and MUC5AC proteins significantly decreased in CS-treated cells with LC50. CD44 is a cell surface marker for cancer stem cells in various tumors and a major adhesion molecule for the extracellular matrix. It has been implicated in tumor cell invasion and metastasis (57) and its ablation triggers growth arrest in proliferative tumor cells (58). De novo synthesis of MUC5AC, a secreted gel-forming mucin normally found in the stomach, has been reported in colon carcinomas (59). This mucin is involved in the formation of a biological inhibitory complex toward E-cadherin, a key component for the maintenance of cell-cell adhesion. In HT-29 cells, the loss of MUC5AC expression has been linked with a gain of function of E-cadherin, resulting in the loss of their invasiveness (60). Among the altered cytoskeletal proteins, it is interesting to note the down-regulation of SLC9A3R1 by CA and CS treatments. This protein is a known scaffold for epidermal growth factor receptor (EGF-R), and has also been suggested as marker of colorectal cancer progression on the basis of its expression and subcellular localization (61). Specifically, this marker appears to play a central role in maintaining the integrity and capacity of EGF-R proliferative signaling pathways in HT-29 cells (62). SLC9A3R1 downregulation has been reported to shut down the entire pathway and the cells revert to a less proliferative phenotype. Taken together, our results show that CA and CS treatments modulate proteomic changes that affect cytoskeleton, cell-surface and secreted molecules. The observed changes suggest that rosemary diterpenes affect cell adhesion to extracellular matrix, and therefore, reduce the invasive potential of HT-29 cells. Similarly, Barni et al. have demonstrated that CA inhibited the cell adhesion and migration functions in Caco-2 cells; although in that case the effects were associated to the decreased activity of secreted proteases and down-regulation of COX-2 expression (22).
CS Inhibits Proteasome Activity in HT-29 Cells
As mentioned above, our data obtained at 24 h indicate that CS modulates the levels of various protein subunits which are critical for 26S proteasome functions. The 26S proteasome is a multienzymatic protease complex with a central role in the ubiquitin-proteasome system, which is responsible for turnover and removal of intracellular abnormal proteins (63) and has a pivotal role in the regulation of many cellular processes including signal transduction, transcription, stress responses, cell differentiation, and metabolic adaptation (64–66). The 26S proteasome consists of one 20S core proteasome with multicatalytic activity and two 19S regulatory caps (67–69). The 19S regulatory caps contain the lid, which is responsible for recognition and docking of polyubiquitinated proteins into the 20S complex, and the base, which has ATPase activity needed for the unfolding and linearization of proteins (70). The 20S core can also exist in a free form that is often referred to as the 20S proteasome, and in contrast with the 26S proteasome, the 20S proteasome functions independently of ATP and is unable of degrading polyubiquitinylated proteins (71).
Unlike normal cells, several cancer cells show aberrant increased proteasomal activity that promotes the degradation of tumor suppressor proteins and cell cycle proteins favoring cancer cell survival, high proliferation rates, and development of drug resistance (72–82). In our present work, to further examine whether the changes in the relative abundance of proteasome subunits induced by CS accounted for changes in the proteasome activity, the chymotrypsin-like activity of the proteasome was assessed in cells after diterpenes exposure for different times (2, 6 and 24 h). MG-132, a well-known proteasome inhibitor was used as positive control. After incubations, 26S and 20S proteasome activities were assayed in cell extracts with the Suc-LLVY-AMC labeled peptide under ATP-stimulated and ATP-independent conditions, respectively. Data revealed that CS treatment decreased both proteasome activities at the earliest time assayed (2 h) in HT-29 cells (Fig. 3A and 3B). Proteasome activity further decreased down to 50.5% upon exposure to CS for 6 h, but it increased in the window from 6 to 24 h to levels close below those detected in untreated control cells. In contrast, CA treatment did not induce significant changes in either 26S or 20S proteasome activity. We therefore hypothesize that CS directly inhibits the 20S proteasome catalytic activity. To test this hypothesis, rosemary diterpenes were tested for their capacity to inhibit chymotrypsin-like activity in 20S purified proteasome. As shown in Fig. 3C, the profile of proteasome inhibition shows that CA failed to inhibit proteasome activity when tested to concentrations up to 50 μm whereas CS inhibited proteasomal function with an IC50 value of 16.5 μm (Fig. 3D). Compared with the stronger inhibitory activity of MG-132 (IC50∼70 nm, Fig. 3E), CS was ∼200-fold less potent, suggesting that CS is a weak inhibitor of 20S proteasome activity. The inhibitory activity of CS observed in HT-29 cells and the successive activity recovery correlated with proteasome subunit up-regulation detected at 24 h. To investigate whether the changes in the relative abundance of proteasome subunits were determined by gene expression, the transcriptional profile of PSMC1 gene, that encodes a 19S proteasome subunit, was analyzed over the time course of the experiment using RT-qPCR (see supplemental Fig. S5). Gene expression data indicated a strong induction (more than 2-fold) of PSMC1 gene at 6 h after initial exposure to CS that was sustained until 24 h. As mentioned above, proteomic data revealed a significant increase (0.73 as log2 ratio, equivalent to 1.7-fold) in the relative abundance of PSMC1 protein at 24 h, suggesting that the observed change in the proteasome subunit is related with changes in gene expression. These results are in full agreement with other recent works evidencing that cells increase the expression of proteasome subunits in response to partial proteasome inhibition as a compensatory mechanism to raise proteasome content (83, 84). Although reversible proteasome inhibition by CS cannot be discarded in the present study, the detected elevation of proteasome abundance provides reasonable evidence to explain, at least in part, the observed recovery of proteasome activity after 24 h of exposure to CS. In this regard, different reports suggest that Nrf1 and Nrf2 transcription factors control the induction of proteasome subunits genes in several cell types (85, 86). However, it appears that only Nrf1 up-regulates proteasome genes upon proteasome inhibition (32). It has been recently observed that the compensatory response mediated by proteasome inhibition and Nrf1 also involves the up-regulation of VCP (also known as p97), a protein with a key role in the Nrf1 translocation for subsequent processing and activation (84, 87). Consistent with Nrf1 activation, our proteomic data indicated increased levels of VCP/p97 suggesting that up-regulation of proteasome subunits in CS-treated cells could be potentially mediated by Nrf1 transcriptional activity. Proteasome inhibition frequently results in G2/M phase cell-cycle arrest and, ultimately, in cell death (72, 88–90), which is in good agreement with the observed effects induced by CS in our present study. Such deleterious effects induced by proteasome inhibition, are frequently more significant in neoplastic cells than in normal cells (91). In last years, the proteasome has emerged as an attractive therapeutic target for the treatment of cancer (92). Some proteasome inhibitors have shown to be particularly effective in the sensitizing drug-resistant tumors (78, 93). However, their clinical use is limited by their typical elevated toxicity. It has been suggested that proteasome inhibitors from natural food sources with low toxicity can be potential anticancer agents (93). According to our results, CS could be a promising proteasome inhibitor of comparable potency to other dietary polyphenols, such as genistein, kaempferol, quercetin, and myricetin, with reported inhibitory effects on the 20S proteasome proteolytic activity (94, 95). With regard to their chemical structures, CA and CS are very similar in that they are ortho-diphenolic diterpenes with abietane carbon skeleton containing hydroxyl groups at positions C-11 and C-12. The only existing structural difference is that CS has a lactone moiety across the B ring, whereas CA has a free carboxylic acid group. Thus, it may be reasonable that proteasome inhibitory activity exerted by CS may be linked to the lactone moiety. In this regard, a nonpeptide inhibitor bearing a β-lactone moiety was reported to irreversibly react with the proteasome's active site threonines and inhibit its chymotrypsin-like activity (96). Following a similar mechanism, the ester bond in (-)epigallocatechin-3-gallate and other green tea polyphenol derivatives was found to have high susceptibility toward a nucleophilic attack by the proteasome leading to the acylation of the reactive site threonine and subsequent inhibition of the catalytic activity (97). This allows us to speculate that the ester bond found in the lactone moiety of CS may be involved in its inhibitory activity. However, further research will help to elucidate the structure-activity relationship of CS with proteasome inhibition activity.
Fig. 3.

Inhibition of the proteasome activity in HT-29 cells after incubation with TGI concentration of CA or CS for 2, 6 and 24 h. A, 26S proteasome activity; B, 20S proteasome activity. Concentration dependent inhibition of the chymotrypsin-like activity of the purified 20S proteasome. C, Carnosic acid; D, Carnosol; E, MG-132.
Our proteomics data also indicated a strong pattern of HSPA1A and HSP90AA1 accumulation in CS-treated cells compared with control (Table I and II), suggesting the activation of cytoprotective heat shock response to alleviate loss of protein homeostasis in the cytosolic compartment. Interestingly, HSPA1A gene expression is controlled via rapid activation of heat shock factor-1 (HSF1) and is normally induced by proteasome inhibitors (98). Recent data suggest that HSPA1A overexpression protects cancer cells from proteasome inhibitors toxicity by promoting lysosomal integrity (99). To target this survival response, chaperone inhibitors alone or in combination with proteasome inhibitors have been widely investigated as a therapeutic strategy in some cancers; however, because of the induction of other chaperones as compensatory mechanisms, this approach has not progressed to clinical trials. This is in line with recent strategies that highlight the advantage of targeting the master transcription factor HSF1 to enhance the impact of proteasome inhibition (100).
CONCLUSIONS
Collectively, our results suggest that although both CA and CS activate the Nrf2 pathway, they induce distinct Nrf2-mediated transcriptional programs that may potentially constitute different mechanisms of action. The proteomic study carried out in the present work suggests that CA and CS cause cellular stress by negatively altering cell proteostasis. However, a detailed examination of our data reveals that each diterpene affects protein homeostasis by different mechanisms. Thus, cellular response to CA involved reactive UPR activation, a signaling network commonly triggered by ER stress. On the other hand, HT-29 cells activated the cytoprotective heat shock response, constituted by increased levels of cytosolic chaperones that potentially alleviate the loss protein homeostasis upon CS-mediated proteasome inhibition. In our present work, we have successfully demonstrated that CS directly inhibits chymotrypsin-like activity of the 20S proteasome. Other concurrent protein changes affecting DNA synthesis and replication, cell cycle progression, cell adhesion and cytoskeleton functions, among others, corroborate the “pleiotropic” character of the effects exerted by both diterpenes at the molecular level. In summary, our unbiased proteome-wide strategy has proven to be a powerful tool to reveal differences on the mechanisms of action of two related bioactive compounds in the same biological model.
Supplementary Material
Acknowledgments
A.V. thanks the Ministerio de Economía y Competitividad for his FPI pre-doctoral fellowship (BES-2012-057014).
Footnotes
Author contributions: A.V., V.G., and A.C. designed research; A.V. and K.A. performed research; K.A., C.S., and J.B. contributed new reagents or analytic tools; A.V. and V.G. analyzed data; V.G. and A.C. wrote the paper.
* This work was supported by the project AGL2014-53609-P (Ministerio de Economía y Competitividad, Spain) and S2013/ABI-2728 (Comunidad de Madrid). The Swedish Research Council (Vetenskapsrådet), 2011-4423 and 2015-4870 to J.B. is acknowledged for financial support.
 This article contains supplemental material.
This article contains supplemental material.
The authors declare that there is no conflict of interest.
Supplemental Data Related To This Article: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD004253.
1 The abbreviations used are:
- CA
- carnosic acid
- AMC
- 7-amino-4-methylcoumarin
- BOG
- n-octyl-β-d-glucopyranoside
- CS
- carnosol
- DML
- dimethyl labeling
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- FDR
- false discovery rate
- GI50
- 50% growth inhibition
- IC50
- 50% inhibitory concentration
- IPA
- Ingenuity Pathway Analysis
- ISR
- integrated stress response
- LC50
- 50% lethal concentration
- nano-LC-MS/MS
- nano-liquid chromatography-tandem mass spectrometry
- Rb
- retinoblastoma
- ROS
- reactive oxygen species
- RT-qPCR
- quantitative reverse transcription PCR
- Suc-LLVY-AMC
- N-Succinyl-Leu-Leu-Val-Tyr-AMC
- TGI
- total growth inhibition
- UPR
- unfolded protein response
- UR
- upstream regulator.
REFERENCES
- 1. González M. A. (2015) Aromatic abietane diterpenoids: their biological activity and synthesis. Nat. Prod. Rep. 32, 684–704 [DOI] [PubMed] [Google Scholar]
- 2. Aruoma O. I., Halliwell B., Aeschbach R., and Löligers J. (1992) Antioxidant and pro-oxidant properties of active rosemary constituents: carnosol and carnosic acid. Xenobiotica 22, 257–268 [DOI] [PubMed] [Google Scholar]
- 3. Satoh T., Kosaka K., Itoh K., Kobayashi A., Yamamoto M., Shimojo Y., Kitajima C., Cui J., Kamins J., Okamoto S., Izumi M., Shirasawa T., and Lipton S. A. (2008) Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cyteines on Keap1. J. Neurochem. 104, 1116–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsai C. W., Lin C. Y., Lin H. H., and Chen J. H. (2011) Carnosic acid, a rosemary phenolic compound, induces apoptosis through reactive oxygen species-mediated p38 activation in human neuroblastoma IMR-32 cells. Neurochem. Res. 36, 2442–2451 [DOI] [PubMed] [Google Scholar]
- 5. Satoh T., McKercher S. R., and Lipton S. A. (2013) Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 65, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. González-Vallinas M., Reglero G., and Ramírez de Molina A. (2015) Rosemary (Rosmarinus officinalis L.) extract as a potential complementary agent in anticancer therapy. Nutr. Cancer 67, 1221–1229 [DOI] [PubMed] [Google Scholar]
- 7. Takahashi T., Tabuchi T., Tamaki Y., Kosaka K., Takikawa Y., and Satoh T. (2009) Carnosic acid and carnosol inhibit adipocyte differentiation in mouse 3T3-L1 cells through induction of phase2 enzymes and activation of glutathione metabolism. Biochem. Biophys. Res. Commun. 382, 549–554 [DOI] [PubMed] [Google Scholar]
- 8. Tamaki Y., Tabuchi T., Takahashi T., Kosaka K., and Satoh T. (2010) Activated glutathione metabolism participates in protective effects of carnosic acid against oxidative stress in neuronal HT22 cells. Planta Med. 76, 683–688 [DOI] [PubMed] [Google Scholar]
- 9. Visanji J. M., Thompson D. G., and Padfield P. J. (2006) Induction of G2/M phase cell cycle arrest by carnosol and carnosic acid is associated with alteration of cyclin A and cyclin B1 levels. Cancer Lett. 237, 130–136 [DOI] [PubMed] [Google Scholar]
- 10. Valdés A., García-Cañas V., Simó C., Ibáñez C., Micol V., Ferragut J. A., and Cifuentes A. (2014) Comprehensive Foodomics study on the mechanisms operating at various molecular levels in cancer cells in response to individual rosemary polyphenols. Anal. Chem. 86, 9807–9815 [DOI] [PubMed] [Google Scholar]
- 11. Valdés A., García-Cañas V., Koçak E., Simó C., and Cifuentes A. (2016) Foodomics study on the effects of extracellular production of hydrogen peroxide by rosemary polyphenols on the anti-proliferative activity of rosemary polyphenols against HT-29 cells. Electrophoresis 37, 1–10 [DOI] [PubMed] [Google Scholar]
- 12. Petiwala S. M., and Johnson J. J. (2015) Diterpenes from rosemary (Rosmarinus officinalis): Defining their potential for anti-cancer activity. Cancer Lett. 367, 93–102 [DOI] [PubMed] [Google Scholar]
- 13. Dörrie J., Sapala K., and Zunino S. J. (2001) Carnosol-induced apoptosis and downregulation of Bcl-2 in B-lineage leukemia cells. Cancer Lett. 170, 33–39 [DOI] [PubMed] [Google Scholar]
- 14. Subbaramaiah K., Cole P. A., and Dannenberg A. J. (2002) Retinoids and carnosol suppress cyclooxygenase-2 transcription by CREB-binding protein/p300-dependent and -independent mechanisms. Cancer Res. 62, 2522–2530 [PubMed] [Google Scholar]
- 15. Huang S. C., Ho C. T., Lin-Shiau S. Y., and Lin J. K. (2005) Carnosol inhibits the invasion of B16/F10 mouse melanoma cells by suppressing metalloproteinase-9 through down-regulating nuclear factor-kappa B and c-Jun. Biochem. Pharmacol. 69, 221–232 [DOI] [PubMed] [Google Scholar]
- 16. Moran A. E., Carothers A. M., Weyant M. J., Redston M., and Bertagnolli M. M. (2005) Carnosol inhibits beta-catenin tyrosine phosphorylation and prevents adenoma formation in the C57BL/6J/Min/+ (Min/+) mouse. Cancer Res. 65, 1097–1104 [PubMed] [Google Scholar]
- 17. Al Dhaheri Y., Attoub S., Ramadan G., Arafat K., Bajbouj K., Karuvantevida N., AbuQamar S., Eid A., and Iratni R. (2014) Carnosol induces ROS-mediated beclin1-independent autophagy and apoptosis in triple negative breast cancer. PLoS ONE 9:e109630, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park K. W., Kundu J., Chae I. G., Kim D. H., Yu M. H., Kundu J. K., and Chun K. S. (2014) Carnosol induces apoptosis through generation of ROS and inactivation of STAT3 signaling in human colon cancer HCT116 cells. Int. J. Oncol. 44, 1309–1315 [DOI] [PubMed] [Google Scholar]
- 19. Johnson J. J., Syed D. N., Heren C. R., Suh Y., Adhami V. M., and Mukhtar H. (2008) Carnosol, a dietary diterpene, displays growth inhibitory effects in human prostate cancer PC3 cells leading to G2-phase cell cycle arrest and targets the 5′-AMP-activated protein kinase (AMPK) pathway. Pharm. Res. 25, 2125–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson J. J., Syed D. N., Suh Y., Heren C. R., Saleem M., Siddiqui I. A., and Mukhtar H. D. (2010) Disruption of androgen and estrogen receptor activity in prostate cancer by a novel dietary diterpene carnosol: implications for chemoprevention. Cancer Prev. Res. 3, 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petiwala S. M., Berhe S., Li G., Puthenveetil A. G., Rahman O., Nonn L., and Johnson J. J. (2014) Rosemary (Rosmarinus officinalis) extract modulates CHOP/GADD153 to promote androgen receptor degradation and decreases xenograft tumor growth. PLoS ONE 9, e89772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barni M. V., Carlini M. J., Cafferata E. G., Puricelli L., and Moreno S. (2012) Carnosic acid inhibits the proliferation and migration capacity of human colorectal cancer cells. Oncol. Rep. 27, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kar S., Palit S., Ball W. B., and Das P. K. (2012) Carnosic acid modulates Akt/IKK/NF-κB signaling by PP2A and induces intrinsic and extrinsic pathway mediated apoptosis in human prostate carcinoma PC-3 cells. Apoptosis 17, 735–747 [DOI] [PubMed] [Google Scholar]
- 24. Min K. J., Jung K. J., and Kwon T. K. (2014) Carnosic acid induces apoptosis through reactive oxygen species-mediated endoplasmic reticulum stress induction in human renal carcinoma Caki cells. J. Cancer Prev. 19, 170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao Q., Liu H., Yao Y., Geng L., Zhang X., Jiang L., Shi B., and Yang F. (2015) Carnosic acid induces autophagic cell death through inhibition of the Akt/mTOR pathway in human hepatoma cells. J. Appl. Toxicol. 35, 485–492 [DOI] [PubMed] [Google Scholar]
- 26. Wang R., Cong W. H., Guo G., Li X. X., Chen X. L., Yu X. N., and Li H. (2012) Synergism between carnosic acid and arsenic trioxide on induction of acute myeloid leukemia cell apoptosis is associated with modulation of PTEN/Akt signaling pathway. Chin. J. Integr. Med. 18, 934–941 [DOI] [PubMed] [Google Scholar]
- 27. Valdés A., Simó C., Ibáñez C., Rocamora-Reverte L., Ferragut J. A., García-Cañas V., and Cifuentes A. (2012) Effect of dietary polyphenols on K562 leukemia cells: A Foodomics approach. Electrophoresis 33, 2314–2327 [DOI] [PubMed] [Google Scholar]
- 28. Ibáñez C., Valdés A., García-Cañas V., Simó C., Celebier M., Rocamora-Reverte L., Gómez-Martínez A., Herrero M., Castro-Puyana M., Segura-Carretero A., Ibáñez E., Ferragut J. A., and Cifuentes A. (2012) Global Foodomics strategy to investigate the health benefits of dietary constituents. J. Chromatogr. A 1248, 139–153 [DOI] [PubMed] [Google Scholar]
- 29. Valdés A., Sullini G., Ibáñez E., Cifuentes A., and García-Cañas V. (2015) Rosemary polyphenols induce unfolded protein response and changes in cholesterol metabolism in colon cancer cells. J. Funct. Foods 15, 429–439 [Google Scholar]
- 30. Valdés A., Artemenko K. A., Bergquist J., García-Cañas V., and Cifuentes A. (2016) Comprehensive proteomic study of the antiproliferative activity of a polyphenol-enriched rosemary extract on colon cancer cells using nanoliquid chromatography-orbitrap MS/MS. J. Proteome Res. DOI: 10.1021/acs.jproteome.6b00154 [DOI] [PubMed] [Google Scholar]
- 31. Kisselev A. F., and Goldberg A. L. (2005) Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 398, 364–378 [DOI] [PubMed] [Google Scholar]
- 32. Radhakrishnan S. K., Lee C. S., Young P., Beskow A., Chan J. Y., and Deshaies R. J. (2010) Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell. 38, 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yeung Y. G., and Stanley E. R. (2010) Rapid detergent removal from peptide samples with ethyl acetate for mass spectrometry analysis. Curr. Protoc. Protein Sci. 16, 16.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boersema P. J., Raijmakers R., Lemeer S., Mohammed S., and Heck A. J. (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 [DOI] [PubMed] [Google Scholar]
- 35. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker P. R., and Chalkley R. J. (2014) MS-viewer: a web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 38. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 39. Waters K. M., Pounds J. G., and Thrall B. D. (2006) Data merging for integrated microarray and proteomic analysis. Brief. Funct. Genomic Proteomic. 5, 261–272 [DOI] [PubMed] [Google Scholar]
- 40. Suzuki T., and Yamamoto M. (2015) Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 88, 93–100 [DOI] [PubMed] [Google Scholar]
- 41. Eletto D., Chevet E., Argon Y., and Appenzeller-Herzog C. (2014) Redox controls UPR to control redox. J. Cell Sci. 127, 3649–3658 [DOI] [PubMed] [Google Scholar]
- 42. Levonen A. L., Hill B. G., Kansanen E., Zhang J., and Darley-Usmar V. M. (2014) Redox regulation of antioxidants, autophagy, and the response to stress: implications for electrophile therapeutics. Free Radic. Biol. Med. 71, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dodson M., Redmann M., Rajasekaran N. S., Darley-Usmar V., and Zhang J. (2015) KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 469, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee A. S. (2005) The ER chaperone and signling regulator GRP78/Bip as a monitor of endosplasmic reticulum stress. Methods 35, 373–381 [DOI] [PubMed] [Google Scholar]
- 45. Wek R. C., Jiang H. Y., and Anthony T. G. (2006) Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11 [DOI] [PubMed] [Google Scholar]
- 46. Yu L., Andruska N., Zheng X., and Shapiro D. J. (2016) Anticipatory activation of the unfolded protein response by epidermal growth factor is required for immediate early gene expression and cell proliferation. Mol. Cell Endocr. 422, 31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Andruska N., Zheng X., Yang X., Helferich W. G., and Shapiro D. J. (2015) Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene 34, 3760–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shapiro D. J., Livezey M., Yu L., Zheng X., and Andruska N. (2016) Anticipatory UPR Activation: A Protective Pathway and Target in Cancer. Trends Endocrinol. Metab. 27, 731–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yan M., Li G., Petiwala S. M., Householter E., and Johnson J. J. (2015) Standardized rosemary (Rosmarinus officinalis) extract induces Nrf2/sestrin-2 pathway in colon cancer cells. J. Func. Foods 13, 137–147 [Google Scholar]
- 50. Wang M., and Kaufman R. J. (2014) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 [DOI] [PubMed] [Google Scholar]
- 51. Nevins J. R. (2001) The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10, 699–703 [DOI] [PubMed] [Google Scholar]
- 52. Kim M., Ju H., Lim B., and Kang C. (2012) Maspin genetically and functionally associates with gastric cancer by regulating cell cycle progression. Carcinogenesis 33, 2344–2350 [DOI] [PubMed] [Google Scholar]
- 53. Malhotra S., Lapointe J., Salari K., Higgins J. P., Ferrari M., Montgomeryet K., van de Rijn M., Brooks J. D., and Pollack J. R. (2011) A tri-marker proliferation index predicts biochemical recurrence after surgery for prostate cancer. PLoS ONE 6, e20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu M., Huang F., Zhang D., Ju J., Wu X. B., Wang Y., Wang Y., Wu Y., Nie M., Li Z., Ma C., Chen X., Zhou J. Y., Tan R., Yang B. L., Zen K., Zhang C. Y., Chen Y. G., and Zhao Q. (2015) Heterochromatin protein HP1γ promotes colorectal cancer progression and is regulated by miR-30a. Cancer Res. 75, 4593–4604 [DOI] [PubMed] [Google Scholar]
- 55. Huang M. C., Chen H. Y., Huang H. C., Huang J., Liang J. T., Shen T. L., Lin N. Y., Ho C. C., Cho I. M., and Hsu S. M. (2006) C2GnT-M is downregulated in colorectal cancer and its re-expression causes growth inhibition of colon cancer cells. Oncogene 25, 3267–3276 [DOI] [PubMed] [Google Scholar]
- 56. González-Vallinas M., Molina S., Vicente G., Zarza V., Martín-Hernández R., García-Risco M. R., Fornari T., Reglero G., and Ramírez de Molina A. (2014) Expression of microRNA-15b and the glycosyltransferase GCNT3 correlates with antitumor efficacy of Rosemary diterpenes in colon and pancreatic cancer. PLoS ONE 9, e98556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu H., Tian Y., Yuan X., Wu H., Liu Q., Pestell R. G., and Wu K. (2015) The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets Ther. 8, 3783–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ishimoto T., Nagano O., Yae T., Tamada M., Motohara T., Oshima H., Oshima M., Ikeda T., Asaba R., Yagi H., Masuko T., Shimizu T., Ishikawa T., Kai K., Takahashi E., Imamura Y., Baba Y., Ohmura M., Suematsu M., Baba H., and Saya H. (2011) CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 19, 387–400 [DOI] [PubMed] [Google Scholar]
- 59. Bara J., Chastre E., Mahiou J., Singh R. L., Forgue-Lafitte M. E., Hollande E., and Godeau F. (1998) Gastric M1 mucin, an early oncofetal marker of colon carcinogenesis, is encoded by the MUC5AC gene. Int. J. Cancer 75, 767–773 [DOI] [PubMed] [Google Scholar]
- 60. Truant S., Bruyneel E., Gouyer V., De Wever O., Pruvot F. R., Mareel M., and Huet G. (2003) Requirement of both mucins and proteoglycans in cell-cell dissociation and invasiveness of colon carcinoma HT-29 cells. Int. J. Cancer 104, 683–694 [DOI] [PubMed] [Google Scholar]
- 61. Hayashi Y., Molina J. R., Hamilton S. R., and Georgescu M. M. (2010) NHERF1/EBP50 is a new marker in colorectal cancer. Neoplasia 12, 1013–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kruger W. A., Monteith G. R., and Poronnik P. (2013) NHERF-1 regulation of EGF and neurotensin signalling in HT-29 epithelial cells. Biochem. Biophys. Res. Commun. 432, 568–573 [DOI] [PubMed] [Google Scholar]
- 63. Glickman M. H., and Ciechanover A. (2002) The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 82, 373–428 [DOI] [PubMed] [Google Scholar]
- 64. Schmidt M., and Finley D. (2014) Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 1843, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sengupta S., and Henry R. W. (2015) Regulation of the retinoblastoma-E2F pathway by the ubiquitin-proteasome system. Biochim. Biophys. Acta 1849, 1289–1297 [DOI] [PubMed] [Google Scholar]
- 66. Demasi M., Netto L. E., Silva G. M., Hand A., de Oliveira C. L., Bicev R. N., Gozzo F., Barros M. H., Leme J. M., and Ohara E. (2013) Redox regulation of the proteasome via S-glutathionylation. Redox Biol. 2, 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peters J. M. (1994) Proteasomes: protein degradation machines of the cell. Trends Biochem. Sci. 19, 377–382 [DOI] [PubMed] [Google Scholar]
- 68. Peters J. M., Franke W. W., and Kleinschmidt J. A. (1994) Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J. Biol. Chem. 269, 7709–7718 [PubMed] [Google Scholar]
- 69. McDougall G. J., Ross H. A., Ikeji M., and Stewart D. (2008) Berry extracts exert different antiproliferative effects against cervical and colon cancer cells grown in vitro. J. Agric. Food Chem. 56, 3016–3023 [DOI] [PubMed] [Google Scholar]
- 70. Shen M., Schmitt S., Buac D., and Dou Q. P. (2013) Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin. Ther. Targets 17, 1091–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Coux O., Tanaka K., and Goldberg A. L. (1996) Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65, 801–847 [DOI] [PubMed] [Google Scholar]
- 72. Adams J. (2002) Proteasome inhibition: a novel approach to cancer therapy. Trends Mol. Med. 8, S49–S54 [DOI] [PubMed] [Google Scholar]
- 73. An B., Goldfarb R. H., Siman R., and Dou Q. P. (1998) Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 5, 1062–1075 [DOI] [PubMed] [Google Scholar]
- 74. Lopes U. G., Erhardt P., Yao R., and Cooper G. M. (1997) p53-dependent induction of apoptosis by proteasome inhibitors. J. Biol. Chem. 272, 12893–12896 [DOI] [PubMed] [Google Scholar]
- 75. Nam S., Smith D. M., and Dou Q. P. (2001) Tannic acid potently inhibits tumor cell proteasome activity, increases p27 and Bax expression, and induces G1 arrest and apoptosis. Cancer Epidemiol. Biomarkers Prev. 10, 1083–1088 [PubMed] [Google Scholar]
- 76. Wada M., Kosaka M., Saito S., Sano T., Tanaka K., and Ichihara A. (1993) Serum concentration and localization in tumor cells of proteasomes in patients with hematologic malignancy and their pathophysiologic significance. J. Lab. Clin. Med. 12, 215–231 [PubMed] [Google Scholar]
- 77. Kumatori A., Tanaka K., Inamura N., Sone S., Ogura T., Matsumoto T., Tachikawa T., Shin S., and Ichihara A. (1990) Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl. Acad. Sci. U.S.A. 87, 7071–7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kisselev A. F., and Goldberg A. L. (2001) Proteasome inhibitors: from research tools to drug candidates, Chem. Biol. 8, 739–758 [DOI] [PubMed] [Google Scholar]
- 79. Afjehi-Sadat L., Gruber-Olipitz M., Felizardo M., Slavc I., and Lubec G. (2004) Expression of proteasomal proteins in ten different tumor cell lines. Amino Acids 27, 129–140 [DOI] [PubMed] [Google Scholar]
- 80. Ishibashi Y., Hanyu N., Suzuki Y., Yanai S., Tashiro K., Usuba T., Iwabuchi S., Takahashi T., Takada K., Ohkawa K., Urashima M., and Yanaga K. (2004) Quantitative analysis of free ubiquitin and multi-ubiquitin chain in colorectal cancer. Cancer Lett. 211, 111–117 [DOI] [PubMed] [Google Scholar]
- 81. Marfella R., D'Amico M., Esposito K., Baldi A., Di Filippo C., Siniscalchi M., Sasso F. C., Portoghese M., Cirillo F., Cacciapuoti F., Carbonara O., Crescenzi B., Baldi F., Ceriello A., Nicoletti G. F., D'Andrea F., Verza M., Coppola L., Rossi F., and Giugliano D. (2006) The ubiquitin-proteasome system and inflammatory activity in diabetic atherosclerotic plaques: effects of rosiglitazone treatment. Diabetes 55, 622–632 [DOI] [PubMed] [Google Scholar]
- 82. Deng J., Carlson N., Takeyama K., Dal Cin P., Shipp M., and Letai A. (2007) BH3 Profiling Identifies Three Distinct Classes of Apoptotic Blocks to Predict Response to ABT-737 and Conventional Chemotherapeutic Agents. Cancer Cell 12, 171–185 [DOI] [PubMed] [Google Scholar]
- 83. Bugno M., Daniel M., Chepelev N. L., and Willmore W. G. (2015) Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim. Biophys. Acta 1849, 1260–1276 [DOI] [PubMed] [Google Scholar]
- 84. Sha Z., and Goldberg A. L. (2014) Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr. Biol. 24, 1573–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kwak M. K., Cho J. M., Huang B., Shin S., and Kensler T. W. (2007) Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 43, 809–817 [DOI] [PubMed] [Google Scholar]
- 86. Steffen J., Seeger M., Koch A., and Krüger E. (2010) Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell. 40, 147–158 [DOI] [PubMed] [Google Scholar]
- 87. Radhakrishnan S. K., den Besten W., and Deshaies R. J. (2014) p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 3, e01856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dou Q. P., and Li B. (1999) Proteasome inhibitors as potential novel anticancer agents. Drug Resist. Updat. 2, 215–223 [DOI] [PubMed] [Google Scholar]
- 89. Gottesman S., Wickner S., and Maurizi M. R. (1997) Protein quality control: triage by chaperones and proteases. Genes Dev. 11, 815–823 [DOI] [PubMed] [Google Scholar]
- 90. Bence N. F., Sampat R. M., and Kopito R. R. (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555 [DOI] [PubMed] [Google Scholar]
- 91. Kazi A., Daniel K. G., Smith D. M., Kumar N. B., and Dou Q. P. (2003) Inhibition of the proteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochem. Pharmacol. 66, 965–976 [DOI] [PubMed] [Google Scholar]
- 92. Landis-Piwowar K. R., Milacic V., Chen D., Yang H., Zhao Y., Chan T. H., Yan B., and Dou Q. P. (2006) The proteasome as a potential target for novel anticancer drugs and chemosensitizers. Drug Resist. Updat. 9, 263–273 [DOI] [PubMed] [Google Scholar]
- 93. Shen M., Chan T. H., and Dou Q. P. (2012) Targeting tumor ubiquitin-proteasome pathway with polyphenols for chemosensitization. Anticancer Agents Med. Chem. 12, 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen D., Daniel K. G., Chen M. S., Kuhn D. J., Landis-Piwowar K. R., and Dou Q. P. (2005) Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem. Pharmacol. 69, 1421–1432 [DOI] [PubMed] [Google Scholar]
- 95. Shim S. H. (2011) 20S proteasome inhibitory activity of flavonoids isolated from Spatholobus suberectus. Phytother. Res. 25, 615–618 [DOI] [PubMed] [Google Scholar]
- 96. Dick L. R., Cruikshank A. A., Destree A. T., Grenier L., McCormack T. A., Melandri F. D., Nunes S. L., Palombella V. J., Parent L. A., Plamondon L., and Stein R. L. (1997) Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J. Biol. Chem. 272, 182–188 [DOI] [PubMed] [Google Scholar]
- 97. Nam S., Smith D. M., and Dou Q. P. (2001) Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J. Biol. Chem. 276, 13322–13330 [DOI] [PubMed] [Google Scholar]
- 98. Akerfelt M., Trouillet D., Mezger V., and Sistonen L. (2007) Heat shock factors at a crossroad between stress and development. Ann. N.Y. Acad. Sci. 1113, 15–27 [DOI] [PubMed] [Google Scholar]
- 99. Qi W., White M. C., Choi W., Guo C., Dinney C., McConkey D. J., and Siefker-Radtke A. (2013) Inhibition of inducible heat shock protein-70 (hsp72) enhances bortezomib-induced cell death in human bladder cancer cells. PLoS ONE 8, e69509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shah S. P., Lonial S., and Boise L. H. (2015) When Cancer Fights Back: Multiple Myeloma, Proteasome Inhibition, and the Heat-Shock Response. Mol. Cancer Res. 13, 1163–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.