

Abstract
Many DNA binding proteins utilize one-dimensional (1D) diffusion along DNA to accelerate their DNA target recognition. Although 1D diffusion of proteins along DNA has been studied for decades, a quantitative understanding is only beginning to emerge and few chemical tools are available to apply 1D diffusion as a design principle. Recently, we discovered that peptides can bind and slide along DNA – even transportingcargo along DNA. Such molecules are known as molecular sleds. Here, to advance our understanding of structure-function relationships governing sequence nonspecific DNA interaction of natural molecular sleds and to explore the potential for controlling sliding activity, we test the DNA binding and sliding activities of chemically modified peptides and analogs, and show that synthetic small molecules can slide on DNA. We found new ways to control molecular sled activity, novel small molecule synthetic sleds, and molecular sled activity in N-methylpyrrole/N-methylimidazole polyamides that helps explain how these moleculeslocate rare target sites.
Keywords: Single Molecule Imaging, Molecular Sleds, Small Molecule, Polyamide, Drug Delivery
Table of Contents
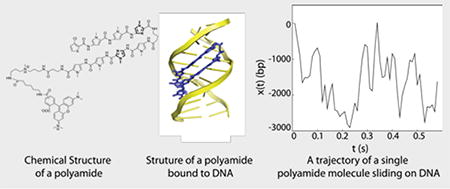
Synthetic small moleculeswith a wide range of chemical functionalitycan slide on DNA.
Nature modulates biochemical reactivity and logic states by controlling dimensionality. A widely recognized example is 3D to 2D Reduction-of-Dimensionality (RD) by targeting factors to cellular membranes and acceleratingtheir interaction. 3D to 1D RD occurs by targeting proteins to 1D on DNA where one-dimensional diffusion along DNA (including sliding and hopping processes) accelerates DNA target recognition.[1] Although 1D transport of proteins along DNA has been of interest for decades, few chemical tools are available to engineer RD to 1D for applications in biotechnology, for example to enable better control of transcription, enhanced nucleic acid enzymes, and improved small molecule activity in cells.
Recent work on adenovirus proteins led to the first identification of protein-protein interactions driven by RD to 1D[2] and the first molecular sled, the peptide pVIc (GVQSLKRRRCF)[3]. Molecular sleds are small DNA-binding molecules such as peptides that slide along sequence-nonspecific DNA and can translocate cargo along DNA. A wide range of basicpolypeptide sequences, including nuclear localization signals (NLS), exhibit these activities[4].
Molecules that slide on DNA are of interest as tools for accelerating and controlling biochemical processes in 1D because of their small sizes and fast diffusion.[5] Sliding-based approaches differ from previously reported DNA-scaffolding strategies utilizing sequence-specific DNA binding proteins[6] or nucleic acid hybridization[7] to co-localize reacting species because interactions between molecular sleds and DNA are sequence-nonspecific, the DNA-targeting moieties are very small, and the reactants are highly mobile on the DNA template. The discovery of small peptides with robust sliding activity raises the possibility that other classes of small molecules may slide.
The mechanism by which N-methylpyrrole/N-methylimidazole polyamides, programmable sequence-specific DNA-binding small molecules, localize to target sites in vivo remains unexplored – in particular, it is unclear whether polyamides undergo sliding activity on DNAlike protein transcription factors(TFs). Despite predictive models describing the affinity and specificity for target site binding in vitro[8], it has been difficult to predict their in vivo efficacy[9]. Recently, a new model predicting in vivo binding based on site clustering in ~400 bp windows (including lower-affinity sites) showed superior performance.[10] Such deviation from independent-site thermodynamic models suggests that other factors, such as mechanisms that couple binding interactions linearly along the DNA, non-equilibrium effects, epigenomiceffects, and/or other active cellular processes, affect the activity of polyamides in vivo.
Here, to establish a broader understanding of molecular sled transport along DNA, we assay a diverse set of modified peptides and DNA binding small molecules for 1D diffusion and sliding using a previously established single-molecule assay[11] (supplementary methods, Fig. S2&S3, Movie S1). In this assay, biotin-λ-DNA (approximately 48,500 base pairs long) is immobilized onto a flow channel surface by one end and a laminar flow is applied to flow stretch the DNA. The stretched DNA molecules serve as spatially extended templates for the analysis of binding and transport activity of molecular sledslabeled with fluorescent dye molecules. The trajectories of single molecules are tracked by time-lapse total internal fluorescence imaging. The raw images are analyzed by using a custom single particle tracking software[4] to identify trajectories of single molecule diffusing along DNA. Theone-dimensional diffusion constant (D1) value characterizing motion along the DNA strand is then estimated by a covariance-based method[12].
Peptidesliding activity is sensitive to C-terminal modification
We previously observed that the presence of a free N-terminal amine increases the sliding activity and DNA affinity of peptidyl sleds (Table S1)[4]. Here we show an analogous effect on converting a C-terminal carboxylate to an amide, which doubles the D1 value of TMR-LKLRLRLR (Fig. 1B) and increases the binding strength (see Kd values in Table S2), consistent with the increased net charge on the modified peptide. We observed the same trends in D1 and Kd values for TMR-LRLRLRLR and TMR-LRLRLRLK upon C-terminal amidation (Fig. 1B, Table S2), and further that this modificationrenders D1 more sensitive to amino acid sequence (Fig. S6A) than previously observed (supplementary text).
Figure 1.
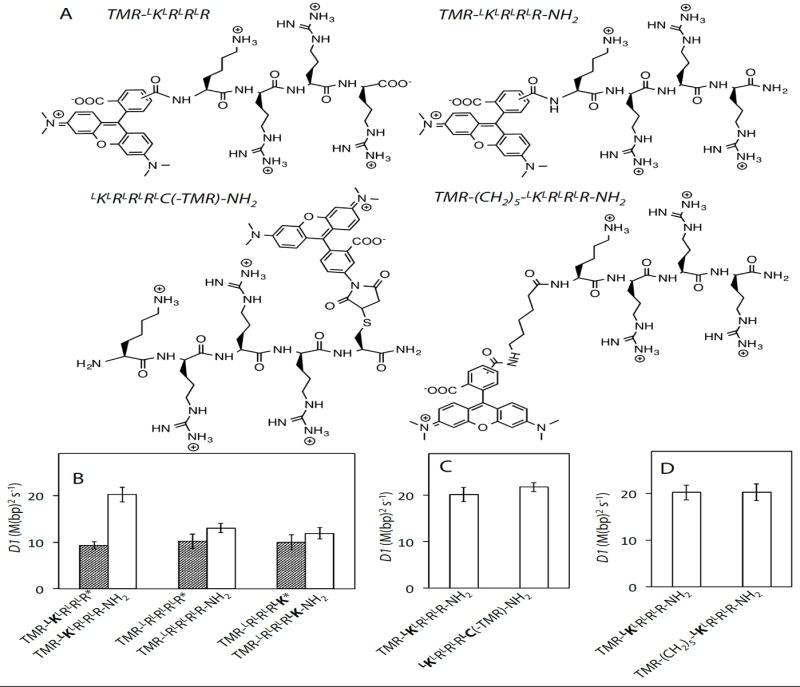
A) Selected chemical structures. B-D) One-dimensional diffusion constants (D1) of peptidyl sleds in 2 mMNaCl buffer(all buffers are pH 7.4). TMR was conjugated to the N-terminal amines of all peptides except for LKLRLRLRLC(-TMR)-NH2. Error bars represent 95% confidence intervalsin all figures. *indicates data that were previously published[4] and re-analyzed here. See Supplementary Table S2 for D1 values.
Knowing that either converting a C-terminal carboxylate to an amide or retaining a free N-terminal amine can enhance the sliding activity of peptidyl sleds, we were curious whether these effects could be combined. Fig. 1C and Table S2 show that the D1 and Kd values of LKLRLRLRLC(-TMR)-NH2, which has a free N-terminal amine and a C-terminal amide (TMR is conjugated to the Cysside chain) are not significantly different than the corresponding values for TMR-LKLRLRLR-NH2 which has only a C-terminal amide modification, indicating that the N-terminal and C-terminal effects are non-additive.
That C-terminal amidation has similar effects on activity asremoving an N-terminal R-group is a curious finding. The acid-base properties of amines and amides are distinct, but each can act as a hydrogen bond donor. These effects are also intriguing because both alterations intensify the interaction of the peptide with DNA, which might be expected to increase “friction” for sliding and thus reduce D1 (in contrast to observations). When we previously increased peptide-DNA affinity by increasing the number of basic side chains on a peptide, we saw a decrease in D1[4]. Thus, there seems to be a unique effect of the peptide termini on sliding kinetics, one that we speculate may be related to the hydrogen bonding potential of the terminal groups.
Side chain length in sliding peptides has strong effects on DNA affinity but not sliding activity
We assayed the activity of tetra-arginine-like peptides with side chain lengths both shorter (LRS: L-2-amino-3-guanidinopropionic acid) and longer (LRL: L-homo-arginine) than arginine. Previous structural modeling based on studies of pVIc in complex with the adenoviral proteinase (AVP; the enzyme it activates catalytically and endows with sliding activity) described the four basic residues of pVIc (KRRR) spanning the major groove with backbone in a β-sheet-like configuration.[3] We subsequently presented an alternative model with lysine and arginine side chains inserted into the minor groove, which is much narrower than the major groove.[4] Fig. 2B shows that the Kd values of tetra-arginine-like peptides rapidly decrease as the side chain length increases. The fact that peptides with shorter side chains slide just about as fast as peptides with longer side chains that bind more tightly (Fig. 2C) indicates that fast sliding does not require side chains to reach far enough into the DNA minor groove for optimal binding.
Figure 2.
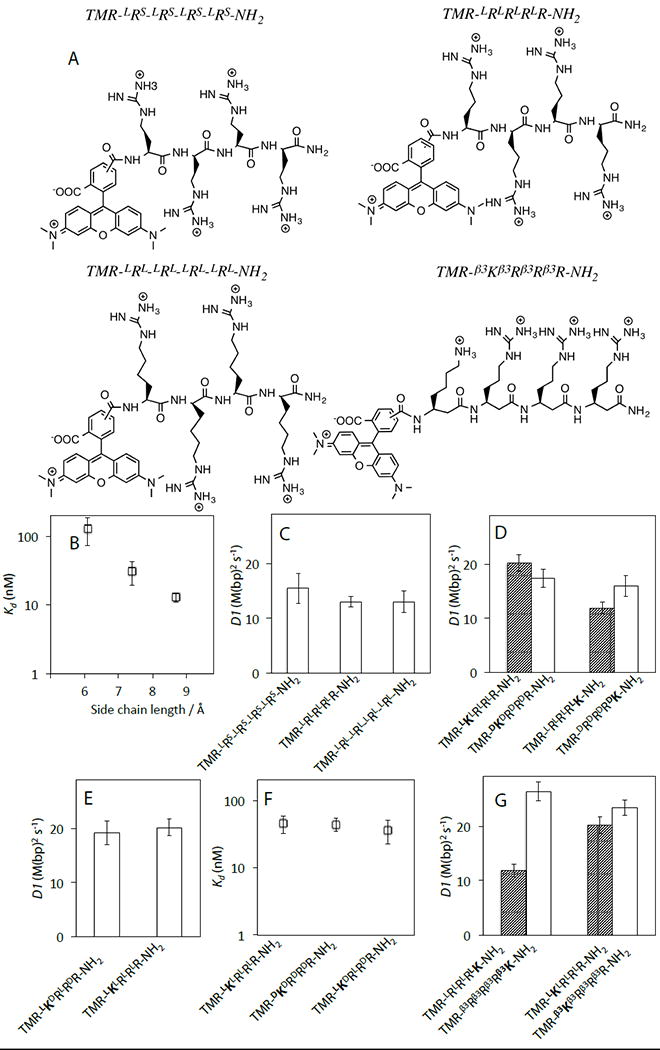
A). Selected chemical structures. B). Dependence of Kd values on guanadinium side chain length. The fully extended side chain lengths of LRS, LR and LRL are 6.1, 7.4 and 8.7 Å, respectively. C-D & F-G). D1 of peptide analogs in 2 mMNaCl buffer. E). The effect of the peptide backbone chirality on Kd. See Supplementary Table S4 for D1 and Kd values.
The data from the tetra-arginine-like series adds another example of relative independence between DNA binding affinity and D1. Previous work on NLS and cell penetrating peptide molecular sleds showed that DNA affinity and sliding activity do not always correlate.[4] Overall, our results suggest that chemical phenomena such as hydrogen bonding at the DNA/sled/solvent interfaces may be instrumental to fast sliding and explain the sensitivity ofD1 to terminal functionality. Deviations from a “friction”-based model for sliding were observed for the DNA repair protein, human oxoguanine DNA glycosylase (hOgg1), which may be affected byrelated chemical mechanisms[11].
DNA binding and sliding activity do not require specific stereochemistry
We next turned our attention to peptides with varied backbone stereochemistry, which interact differently with the right-handed B-DNA helix. The D1 and Kd values of TMR-KRRR-NH2 and TMR-RRRK-NH2 sleds are hardly affected by the backbone chirality, including alternating L- and D- amino acids (Fig. 2D & 2E, Table S2&S4). Inserting a flexible linker between TMR and the N-terminal amide does not impact the D1 or Kd values of D-peptides, indicating that the N-terminal TMR label interferes neither with sliding nor DNA binding properties (Fig. S6B, Table S4). From these observations we conclude that backbone stereochemistry is not an important constraint on molecular sled activity.
Non-canonical peptide backbone structure allows faster sliding activity with high DNA affinity
To further examine how backbone structure affects the activities of molecular sleds, we assayed β3-peptides, which have an additional methylene group in the backbone of each monomer. The D1 value ofTMR-β3Rβ3Rβ3Rβ3K-NH2 is twice that of TMR-LRLRLRLK-NH2 and that the D1 value of TMR-β3Kβ3Rβ3Rβ3R-NH2 is somewhat higher than that of TMR-LKLRLRLR-NH2(Fig. 2G). The affinities of β3-peptides for DNA are not reduced compared with the corresponding L-peptides, suggesting thatelectrostatic binding interactions with DNA are analogous to standard peptides. The faster sliding activity of β3-peptides might result from the more flexible backbone which allows these molecules to adopt conformations that better accommodate the geometric constraints of sliding transitionstate complexes.
A tiny linear small molecule slides on DNA
Based on the findings of our structure-function work with modified peptides, we set out to design a new minimal molecular sled unrelated to peptidesand smaller than known molecular sleds. Previous studies showed that polyamine-DNA bindingis predominantly electrostatic and DNA sequence nonspecific[13], characteristics that mirror those of known peptidylmolecular sleds. We selected PentaEthyleneHexAmine (PEHA, ~230 Da) as a test case based on its small size, the number and spacing of charges, and molecular symmetry that would yield a homogeneous population with a free terminal amine after mono-labeling with Cy3B-NHS. PEHA-Cy3B indeed binds DNA in line with the charge-affinity trends we have observed across peptides (Table S5)[4]. PEHA-Cy3B was found to be capable of fast 1D translocation on DNA with an estimated D1 value of 8.2 ± 0.5 M(bp)2s-1, ±SEM (Fig. 3B). Increasing the salt concentration does not increase the D1 value of PEHA further (Fig. 3B), indicating that PHEA predominantly translocates on DNA by sliding and almost certainly undergoes rotation-coupled helical sliding[11],[14]. These results suggest that a wide variety of other polyaminesand basic small molecules may slide on DNA. Polyamines are known to play important roles in DNA function such as DNA condensation, regulation of genome structure, and protection of DNA from external agents and radiation damage,[13a] but it remains to be discovered what role sliding plays in these biological processes.
Figure 3.
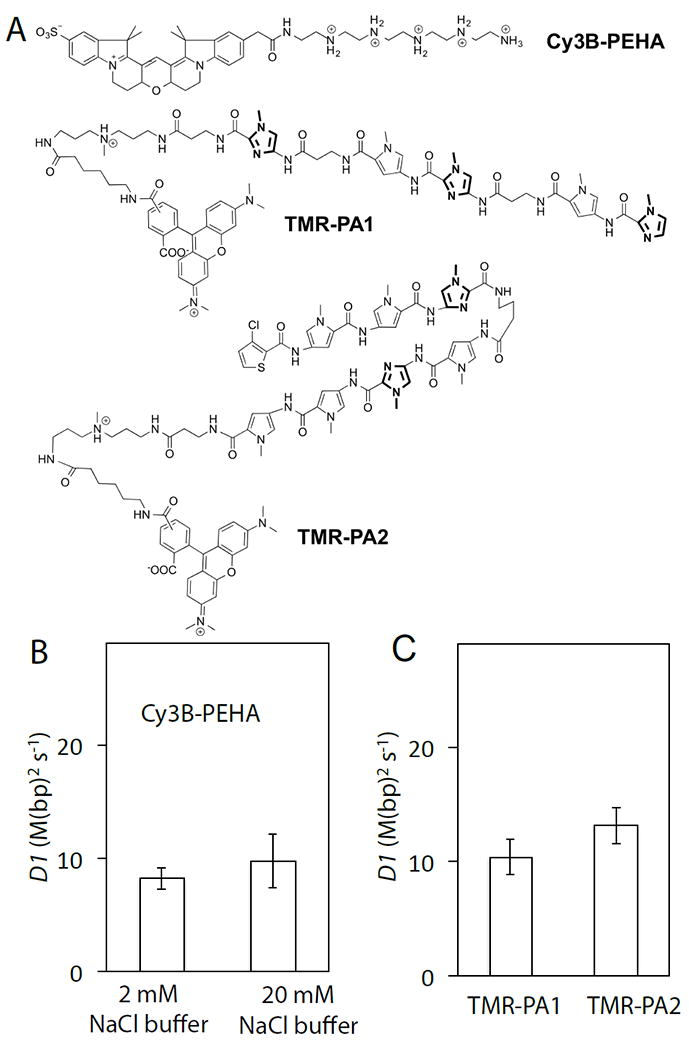
Molecular sled activity of synthetic small molecules. A). Chemicalstructures of Cy3B-PEHA, TMR-PA1 and TMR-PA2. DNA is colored by electrostatic potential. B). The D1 values of Cy3B-PEHA in 2 mMNaCl and 20 mMNaCl buffers. C). The D1 values of TMR-PA1 and TMR-PA2 in 2 mMNaCl buffer. See Supplementary Table S5 for D1 values.
N-methylpyrrole/N-methylimidazole polyamides slide along DNA
All known molecular sleds have a total net charge of ≥ +3 and bind DNA mainly through electrostatic interactions[3-4]. Likewise, essentially all proteins that slide on DNA have significant electrostatic interactions with nonspecific DNA. We were curious whether lower-charge molecules that achieve a preponderance of their interaction energy by a different means can also undergo 1D diffusion. Polyamides can be rationally designed to bind the DNA minor groove sequence-specifically through hydrogen bonding interactions and shape-selective recognition (Fig. S5) and are pursued as candidates for chemical regulation of gene expression.[8c, 10, 15],[16]
We observed that a linear polyamide[17], PA1, and a hairpin polyamide[18], PA2 (Figs. 3A, S5), each have a net charge of +1 and both exhibit fast 1D diffusion on DNA, with estimated D1 values of 10.4 ± 0.8 and 13.1 ± 0.8 M(bp)2 s-1, respectively (Figs. 3C, S3H&I). Despite their structural dissimilarity to peptides, both polyamides slide with rates similar to peptidyl sleds, raising the possibility of commonalities between their sequence non-specific binding modes and 1D diffusion mechanisms.
We were not able to differentiate a sliding vs. hopping mechanism for PA1 and PA2 by measuring the salt dependence of their D1 values as they bind DNA mainly through non-electrostatic interactions. An alternative approach to detect hopping is to test for drift of the sliding molecules in the direction of the buffer flow used to stretch the DNA templates. Molecules undergoing 1D diffusion may drift in the flow direction because of large size that presents significant flow drag[19] or because of hopping that enables the molecule to freely drift when not bound to DNA (see Estimation of the drift of molecules hopping on DNA in the supplementary text). Despite the high flow rate used in our measurements of PA1 and PA2, we detected no significant drift in the flow direction, consistent with a model where the diffusing molecule remains bound to DNA more than 99.99% of the time and very close to the DNA surface where the molecule is partially protected from the flow (Fig. S7). We estimated Kd values for PA1 and PA2 to be 363 ± 76 and 432 ± 87 nM and salt-independent (Table S6), in-line with previously reported values of PA1 and PA2 binding to non-cognate DNA sequences.[20] The average sliding lengths for PA1 and PA2 were calculated (from the apparent off-rate and D1; see Fig.S8) to be 2.2 kbp and 2.7 kbp, respectively. These findings lead us to predict that polyamides are capable of sliding hundreds to thousands of base pairs at physiological salt concentrations.
Polyamide sliding provides a biophysical mechanism for target site localization within the genome by RD to 1D and facilitated diffusion like DNA-binding proteins[1]. Sliding also helps explain the preference of polyamides for clustered sites in vivo[10] by providing a mechanism to couple the affinity of linked sites on the ~400 bp length scale where clustering effects are observed[10]. We imagine that polyamides become locally trapped in clusters of binding sites, leading to increased occupancy at these loci.
One limitation of polyamides is their poor uptake to cells and nuclei. Previous work showed improved cellular permeability of polyamides tagged with NLS sequences[21]. We recently reported that cell penetrating peptides exhibit molecular sled function, suggesting that conjugating protease-resistant peptide analogs with cell penetrating and nuclear uptake activity to polyamides might improve their efficacy.[4]
Our observations that small molecules with a wide range of chemical functionality slide on DNA, that specific modifications can be used to tune crucial properties like DNA affinity and sliding speed, and that polyamides with sequence-recognition capability exhibit sliding activity open the way for engineering facilitated diffusion. In addition to sequence-specific polyamides with improved properties, small molecule inhibitors with molecular sled activity could prove efficacious as therapeutics. For example, basic compounds with chromatin-proximal targets could be optimized for cell penetrating, nuclear localization, DNA-binding, and sliding activities to improve potency and reduce off-target activity outside the nucleus. Other compounds could be conjugated to minimal molecular sleds like PEHA to similarly target chromatin and suppress off-target interactions.
Supplementary Material
Acknowledgments
This work was supported by startup funding and a Career Award at the Scientific Interface (to PCB) from the Burroughs Wellcome Foundation and GM133508 (to A.Z.A.) from the National Institutes of Health. We also thank NihalKorkmaz at University of Wisconsin–Madison for help with structural modeling.
Contributor Information
Kan Xiong, Broad Institute of MIT and Harvard, Cambridge, MA, 02142, USA; Department of Biological Engineering, MIT, Cambridge, MA, 02142, USA.
Graham S. Erwin, Department of Biochemistry, University of Wisconsin–Madison, Madison, WI, 53706, USA
Prof. Aseem Z. Ansari, Department of Biochemistry, University of Wisconsin–Madison, Madison, WI, 53706, USA
Prof. Paul C. Blainey, Broad Institute of MIT and Harvard, Cambridge, MA, 02142, USA Department of Biological Engineering, MIT, Cambridge, MA, 02142, USA.
References
- 1.Berg OG, Winter RB, von Hippel PH. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 2.a Graziano V, Luo GB, Blainey PC, Perez-Berna AJ, Mcgrath WJ, Flint SJ, Martin CS, Xie XS, Mangel WF. J Biol Chem. 2013;288:2068–2080. doi: 10.1074/jbc.M112.407312. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Blainey PC, Graziano V, Perez-Berna AJ, McGrath WJ, Flint SJ, Martin CS, Xie XS, Mangel WF. J Biol Chem. 2013;288:2092–2102. doi: 10.1074/jbc.M112.407460. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Stratmann SA, Morrone SR, van Oijen AM, Sohn J. Elife. 2015;4 doi: 10.7554/eLife.11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mangel WF, McGrath WJ, Xiong K, Graziano V, Blainey PC. Nature Communications. 2016 doi: 10.1038/ncomms10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong K, Blainey PC. Nucleic Acids Res. 2016;44:2266–2273. doi: 10.1093/nar/gkw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turkin A, Zhang L, Marcozzi A, Mangel WF, Herrmann A, van Oijen AM. Chem Sci. 2016;7:916–920. doi: 10.1039/c5sc03063c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Ngo TA, Nakata E, Saimura M, Morii T. J Am Chem Soc. 2016 doi: 10.1021/jacs.5b10198. [DOI] [PubMed] [Google Scholar]; b Slomovic S, Collins JJ. Nat Methods. 2015;12:1085–1090. doi: 10.1038/nmeth.3585. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Liu DR. Angew Chem Int Ed. 2004;43:4848–4870. doi: 10.1002/anie.200400656. [DOI] [PubMed] [Google Scholar]
- 8.a Nishijima S, Shinohara K, Bando T, Minoshima M, Kashiwazaki G, Sugiyama H. Bioorg Med Chem. 2010;18:978–983. doi: 10.1016/j.bmc.2009.07.018. [DOI] [PubMed] [Google Scholar]; b Uil TG, Haisma HJ, Rots MG. Nucleic Acids Res. 2003;31:6064–6078. doi: 10.1093/nar/gkg815. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Carlson CD, Warren CL, Hauschild KE, Ozers MS, Qadir N, Bhimsaria D, Lee Y, Cerrina F, Ansari AZ. P Natl Acad Sci USA. 2010;107:4544–4549. doi: 10.1073/pnas.0914023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raskatov JA, Nickols NG, Hargrove AE, Marinov GK, Wold B, Dervan PB. P Natl Acad Sci USA. 2012;109:16041–16045. doi: 10.1073/pnas.1214267109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erwin GS, Bhimsaria D, Eguchi A, Ansari AZ. Angew Chem Int Edit. 2014;53:10124–10128. doi: 10.1002/anie.201405497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. P Natl Acad Sci USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vestergaard CL, Blainey PC, Flyvbjerg H. Physical review: E, Statistical, nonlinear, and soft matter physics. 2014;89:022726. doi: 10.1103/PhysRevE.89.022726. [DOI] [PubMed] [Google Scholar]
- 13.a Ouameur AA, Tajmir-Riahi HA. J Biol Chem. 2004;279:42041–42054. doi: 10.1074/jbc.M406053200. [DOI] [PubMed] [Google Scholar]; b Deng H, Bloomfield VA, Benevides JM, Thomas GJ., Jr Nucleic Acids Res. 2000;28:3379–3385. doi: 10.1093/nar/28.17.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blainey PC, Luo GB, Kou SC, Mangel WF, Verdine GL, Bagchi B, Xie XS. Nat Struct Mol Biol. 2009;16:1224–U1234. doi: 10.1038/nsmb.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang F, Nickols NG, Li BC, Marinov GK, Said JW, Dervan PB. P Natl Acad Sci USA. 2013;110:1863–1868. doi: 10.1073/pnas.1222035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilch DS, Poklar N, Baird EE, Dervan PB, Breslauer KJ. Biochemistry. 1999;38:2143–2151. doi: 10.1021/bi982628g. [DOI] [PubMed] [Google Scholar]
- 17.a Burnett R, Melander C, Puckett JW, Son LS, Wells RD, Dervan PB, Gottesfeld JM. Proc Natl Acad Sci U S A. 2006;103:11497–11502. doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Urbach AR, Love JJ, Ross SA, Dervan PB. J Mol Biol. 2002;320:55–71. doi: 10.1016/S0022-2836(02)00430-8. [DOI] [PubMed] [Google Scholar]
- 18.a Viger A, Dervan PB. Bioorg Med Chem. 2006;14:8539–8549. doi: 10.1016/j.bmc.2006.08.028. [DOI] [PubMed] [Google Scholar]; b Chenoweth DM, Dervan PB. J Am Chem Soc. 2010;132:14521–14529. doi: 10.1021/ja105068b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blainey PC. PhD thesis. Harvard University; 2007. [Google Scholar]
- 20.Puckett JW, Muzikar KA, Tietjen J, Warren CL, Ansari AZ, Dervan PB. Journal of the American Chemical Society. 2007;129:12310–12319. doi: 10.1021/ja0744899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Best TP. PhD thesis. California Institute of Technology; 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.