

Abstract
Aims/Introduction
Omarigliptin is a novel, potent, long‐acting oral dipeptidyl peptidase‐4 inhibitor being developed as a once‐weekly (q.w.) treatment for type 2 diabetes mellitus patients, with 25 mg and 12.5 mg tablets recently being approved as market formulations in Japan.
Materials and Methods
This was a two‐part, double‐blind, randomized, placebo‐controlled study in healthy Japanese men to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of omarigliptin after single dose (5–100 mg) and multiple dose (1–50 mg q.w. for 3 weeks) administration.
Results
Omarigliptin was rapidly absorbed with a time to maximum concentration of 0.5–4 h. The pharmacokinetic profile was biphasic with a long terminal half‐life >100 h. The area under the concentration–time curve from 0 to 168 h, maximum concentration and the concentration at 168 h post‐dose increased dose‐dependently after 3 weeks of once‐weekly dosing for doses ranging 1–50 mg, with accumulation ratios ranging 1.03–1.35 and 0.87–1.36 for the area under the concentration–time curve from 0 to 168 h and maximum concentration, respectively. Plasma dipeptidyl peptidase‐4 inhibition levels 1 week post‐dose increased with dose, ranging 79.2–94.0% after 5–100 mg single dose administration and 51.3–90.2% after 1–50 mg multiple once‐weekly dose administration. Administration with food did not meaningfully alter the pharmacokinetics of omarigliptin. Omarigliptin was generally well tolerated, with no hypoglycemia being reported.
Conclusion
The results of the present study in healthy Japanese men showed that omarigliptin was well tolerated and had a pharmacokinetic and dipeptidyl peptidase‐4 inhibition profile that supports once‐weekly dosing in Japanese patients with type 2 diabetes mellitus.
Keywords: Dipeptidyl peptidase‐4 inhibitors, Omarigliptin, Once‐weekly
Introduction
Type 2 diabetes mellitus affects more than 415 million people worldwide, including more than 7 million people in Japan1. Despite the availability of several new oral antihyperglycemic agents (AHAs), many treated type 2 diabetes mellitus patients still fail to achieve glycemic control goals. A key reason for this is poor medication adherence2. Adherence to AHAs has been shown to decrease as the number of medications increases3. A prospective study of type 2 diabetes mellitus patients reported a mean adherence of 79% for a once‐daily AHA regimen, 65% for a twice‐daily AHA regimen and 38% for a thrice‐daily AHA regimen4. Furthermore, type 2 diabetes mellitus patients initiating once‐weekly (q.w.) exenatide had significantly higher adjusted odds of adherence compared with patients initiating twice‐daily exenatide or once‐daily liraglutide5. Thus, an efficacious, well‐tolerated and safe AHA that can be taken q.w. has the potential to reduce medication burden and increase treatment adherence in type 2 diabetes mellitus patients, which in turn might improve long‐term outcomes in these patients.
Dipeptidyl peptidase‐4 (DPP‐4) inhibitors are incretin enhancers that act by inhibiting the inactivation of incretin peptides, especially glucagon‐like peptide‐1 and glucose‐dependent insulinotropic polypeptide. DPP‐4 inhibitors have been shown to be an effective and generally well tolerated class of AHAs for the treatment of type 2 diabetes mellitus, with a low risk of hypoglycemia and neutral weight gain, and have anticipated long‐term beneficial effects on β‐cell function and mass6, 7, 8. Interestingly, it is suggested that the glucose‐lowering efficacy of DPP‐4 inhibitors is greater in the Asian population9. The favorable efficacy and safety profiles of DPP‐4 inhibitors make these therapies important treatment options for type 2 diabetes mellitus management worldwide, especially in an Asian population. To date, a q.w. dosing DPP‐4 inhibitor is not yet widely available.
Omarigliptin (MK‐3102) is a structurally distinct and potent oral DPP‐4 inhibitor, which has a longer plasma half‐life that supports q.w. dosing10. Inhibition of DPP‐4 by omarigliptin was shown to enhance post‐meal active glucagon‐like peptide‐1 by approximately twofold in healthy males. In a 12‐week, dose‐range finding study of omarigliptin, the dose‐dependent 2‐h post‐meal glucose reduction, fasting plasma glucose reduction, glycated hemoglobin reduction and safety profile were similar to that of once‐daily sitagliptin12. To date, the tolerability, safety, pharmacokinetics and pharmacodynamics of omarigliptin in Japanese people have not been reported. Therefore, the present study evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics of omarigliptin after single (5–100 mg) and multiple (1–50 mg q.w. for 3 weeks) doses in healthy Japanese men.
Materials and Methods
Participants
Healthy Japanese male volunteers aged ≥18 to ≤45 years with a body mass index of ≥18 to ≤30 kg/m2 at the prestudy (screening) visit were recruited to participate in the present study. Participants were excluded if they had a history of clinically significant endocrine, gastrointestinal, cardiovascular, hematological, hepatic, immunological, renal, respiratory, neurological or genitourinary abnormalities or diseases. Participants were also excluded if they had a history of neoplastic disease, hypoglycemia or glucose intolerance, type 1 diabetes, or type 2 diabetes mellitus. Participants with an estimated creatinine clearance of ≤80 mL/min based on the Cockcroft–Gault equation13 were also excluded.
All participants provided written informed consent before the initiation of any study procedures. The study protocol was approved by the institutional review board (Aspire IRB LLC, LA Mesa, CA, USA). The study was conducted in accordance with the guidelines on good clinical practices and with ethical standards for human experimentation established by the Declaration of Helsinki.
Study design
This was a two‐part, double‐blind, randomized, placebo‐controlled study (Protocol 005). The study was conducted at one study site.
In part 1 of the study, two panels (panels A and B) of eight different participants each (six on omarigliptin, two on placebo) alternately received single rising doses of omarigliptin or placebo in three treatment periods (periods 1, 2 and 3). In panel A, participants received single doses of 5, 25 or 100 mg omarigliptin or matching placebo in the fasted state. In periods 1 and 2 of panel B, participants received single doses of 10 and 50 mg omarigliptin or matching placebo in the fasted state. Participants fasted from all food and drink (except water) for a minimum of 10 h before dosing. In panel B, period 3, participants received 10 mg omarigliptin or matching placebo after consuming a Japanese standard breakfast before study drug administration. The participants were allocated to receive the same treatment, omarigliptin or a placebo, in both period 1 and period 3 for panel B. There was a minimum of 2 weeks washout between administration of the study drug within each treatment panel for all periods (summary of the study design is provided in Supplementary Material; Table S1).
In part 2 of the study, four serial panels (panels A, B, C and D) of eight participants each (six on omarigliptin, two on placebo) received multiple doses of omarigliptin (1, 10, 25 or 50 mg) or a placebo q.w. under the fasted state for 3 weeks (a total of three doses to be given on days 1, 8 and 15).
In part 1 and on days 1 and 15 of part 2, a standard meal was provided in each period 4 h post‐dose, immediately after blood draws, which was required to be consumed entirely with 240 mL of water within 10 min. Meals at 10 and 24 h post‐dose, and snacks at 7.5 and 13 h post‐dose were required to be consumed entirely, and had the same caloric content and composition when administered across participants and periods. In panel B period 3 of part 1, the Japanese standard breakfast was provided 30 min before dosing, and the contents of the entire meal were required to be consumed by 10 min before dosing. The Japanese standard breakfast totaled 411 kcal, with a nutrient breakdown of 53.4 g carbohydrates, 27.1 g protein and 9.0 g fat.
Pharmacokinetic assessments
Blood samples for the determination of plasma omarigliptin concentrations were collected from each participant at predose and at specified time‐points after the administration of omarigliptin in part 1 and part 2 (days 1 and 15) in dipotassium ethylenediaminetetraacetic acid‐containing tubes and centrifuged for 10 min at 1000–1300 g. The plasma samples were then frozen at −20°C. The omarigliptin plasma pharmacokinetic parameters evaluated were area under the concentration–time curve from 0 to 168 h (AUC0–168h) and 0 to infinity (AUC0–∞), the maximum concentration (Cmax), time to Cmax (Tmax), the concentration at 168 h post‐dose (C168h) and apparent terminal half‐life (t½) after single or multiple doses.
Urine samples for the determination of urine omarigliptin concentrations were collected at predose on day 1 and at specified time intervals up to 168 h after the administration of omarigliptin at 50 mg on day 15 in part 2. Tween 20 was added to each urinary sample to give a 0.2% Tween 20 final concentration and then stored at −20°C. Urine omarigliptin pharmacokinetic parameters included: the cumulative amount of unchanged drug excreted in urine determined by the sum of the product of urine concentration and the urine volume per collection interval over 168 h after the last dose of omarigliptin; the fraction of omarigliptin dose that was excreted unchanged in urine over the collection interval up to 168 h, calculated as the ratio of the cumulative amount of unchanged drug excreted in urine and dose; and the renal clearance, calculated by the ratio of the cumulative amount of unchanged drug excreted in urine and AUC0–168h.
Omarigliptin was assayed in plasma and urine using a high‐turbulence liquid chromatography online extraction method with a lower limit of quantification of 1.0 ng/mL for plasma and 40 ng/mL for urine.
Pharmacodynamic assessments
Blood samples for determination of plasma DPP‐4 activity were collected from each participant at predose and at specified time‐points after the administration of omarigliptin in part 1 and part 2 (days 1 and 15). At the 4‐ and 24‐h time‐points, blood samples were obtained before administration of the standardized meals. Plasma samples collected for DPP‐4 activity measurement were analyzed by the PPD Global Central Labs (Highland Height, KY, USA). Enzymatic activity of DPP‐4 was determined by incubating ethylenediaminetetraaceticacid plasma with the substrate glycyl‐prolylparanitraniline, and measuring the release of peptide nucleic acid by an increase in absorbance at 390 nm (in mOD/min) from 4 to 14 min. The assay's limit of quantitation was 0.6 mOD/min.
Safety assessments
Adverse events were assessed throughout the study. All adverse events were evaluated in terms of intensity, duration, severity, outcome and relationship to study medication. Other safety parameters, including vital signs, physical examinations, neurological examinations, 12‐lead electrocardiograms (ECGs), standard laboratory safety tests and glucometer measurements, were assessed predose at various post‐dose time‐points, and post‐study.
Statistical analysis
Pharmacokinetics
In part 1, pharmacokinetic parameters AUC0–∞, AUC0–168h, Cmax and C168h were natural log transformed and analyzed based on a linear mixed effects model containing a fixed effect for treatment and a random effect for participant. Exponentiating the least‐squares means (mean differences) and upper and lower limits of confidence interval yielded estimates for the population geometric means (population geometric mean ratios) and corresponding confidence intervals on the original scale. To assess the effect of food for those parameters, 90% confidence intervals for population geometric mean ratios were constructed. In part 2, pharmacokinetic parameters were analyzed based on a linear mixed effects model with fixed effects for treatment, day, and treatment‐by‐day interaction and a random effect for participant. The estimates for the population geometric mean ratios of day 15/day 1 to assess the accumulation with corresponding confidence intervals on the original scale were provided, exponentiating the relevant statistics generated on the log scale. For both parts, median, minimum and maximum were reported for Tmax, whereas geometric mean and percent coefficient of variation were reported for t1/2.
Pharmacodynamics
For part 1, baseline DPP‐4 activity was defined as the measurements taken at pretreatment in period 1. The effect of omarigliptin on percent inhibition from baseline of DPP‐4 activity at 168 h post‐dose was analyzed using a linear mixed effects model containing a fixed effect for treatment and a random effect for participant. All analyses were conducted on the log scale, and final results were back‐transformed for reporting purposes. The point estimates and corresponding confidence intervals were generated for the difference in percent inhibition (omarigliptin – placebo) of DPP‐4 activity at 168 h post‐dose. For part 2, baseline DPP‐4 activity was defined as the measurements taken at pretreatment. The effects of omarigliptin were analyzed using a linear mixed effects model with fixed effects for treatment, day and treatment‐by‐day interaction, and a random effect for participant on the log scale, and results were back‐transformed for reporting. For each participant, the percentage inhibition of plasma DPP‐4 activity was plotted against the plasma omarigliptin concentration, and a population‐based three‐parameter maximum effect (Emax) model with parameters of Emax, 50% effective concentration, and Hill slope was fitted to the DPP‐4 inhibition data to determine the 80% effective concentration.
Safety
All adverse events were tabulated for each dose of omarigliptin and for placebo. Summary statistics were generated for the change from baseline values in vital signs, ECG parameters and selected laboratory safety parameters.
Results
Baseline characteristics
A total of 16 Japanese men participated in part 1, and 32 Japanese men participated in part 2. All participants were in good general health according to medical history, physical examination, vital signs and laboratory data.
Pharmacokinetics
Table 1 summarizes the pharmacokinetic parameters after single and multiple dose administration of omarigliptin, respectively. Figure 1 shows the mean plasma concentration–time profiles of omarigliptin after once‐weekly multiple doses.
Table 1.
Pharmacokinetic parameters after single and multiple dose administration of omarigliptin in healthy Japanese men
| Single dose (panel A) | ||||
|---|---|---|---|---|
| Parameters | 5 mg | 25 mg | 100 mg | |
| (n = 6) | (n = 6) ‡‡ | (n = 6) | ||
| AUC0–∞ (μmol/L·h) † | 6.27 (5.70–6.90) | 25.13 (22.85–27.64) | 98.87 (89.89–108.75) | |
| AUC0–168h (μmol/L·h) † | 5.34 (4.85–5.88) | 23.81 (21.63–26.21) | 94.63 (85.96–104.18) | |
| Cmax (nmol/L) † | 141.51 (120.18–166.62) | 749.54 (636.57–882.56) | 2709.59 (2301.20–3190.46) | |
| C168h (nmol/L) † | 7.55 (5.86–9.74) | 19.98 (15.29–26.11) | 54.49 (42.28–70.24) | |
| Tmax (h) § | 1.50 (0.50–2.00) | 1.00 (0.50–4.00) | 2.00 (1.00–4.00) | |
| t½ (h) †† | 66.65 (34.10) | 38.89 (25.78) | 43.41 (33.73) | |
| Single dose (panel B) | ||||
| Parameters | 10 mg | 50 mg | 10 mg (Fed) | |
| (n = 6) | (n = 6) | (n = 6) | ||
| AUC0–∞ (μmol/L·h) † | 9.78 (8.87–10.78) | 50.54 (45.94–55.60) | 10.07 (9.14–11.10) | |
| AUC0–168h (μmol/L·h) † | 9.07 (8.22–10.00) | 48.66 (44.19–53.58) | 8.75 (7.94–9.65) | |
| Cmax (nmol/L) † | 297.84 (252.17–351.78) | 1712.32 (1453.67–2016.99) | 223.03 (188.83–263.43) | |
| C168h (nmol/L) † | 8.10 (6.26–10.49) | 23.62 (18.32–30.46) | 9.16 (7.08–11.86) | |
| Tmax (h) § | 1.00 (1.00–2.00) | 1.00 (0.50–4.00) | 4.00 (2.00–6.00) | |
| t½ (h) †† | 49.91 (32.62) | 33.43 (9.99) | 89.45 (45.07) | |
| Multiple dose | ||||
| Parameters | 1 mg | 10 mg | 25 mg | 50 mg |
| (n = 6) ‡‡ | (n = 6) | (n = 6) | (n = 6) ‡‡ | |
| Day 1 | ||||
| AUC0–168h (μmol/L·h) † | 1.03 (0.91–1.17) | 8.92 (7.85–10.13) | 21.19 (18.66–24.07) | 40.32 (35.50–45.79) |
| Cmax (nmol/L) † | 15.66 (12.76–19.22) | 236.65 (192.77–290.50) | 802.60 (653.81–985.26) | 1083.59 (882.71–1330.20) |
| Ctrough (nmol/L) † | 3.68 (2.80–4.83) | 10.36 (7.90–13.60) | 14.75 (11.24–19.36) | 27.09 (20.64–35.55) |
| Tmax (h) § | 2.00 (1.00–4.00) | 1.50 (1.00–6.00) | 0.50 (0.50–2.00) | 1.58 (1.00–4.00) |
| Day 15 | ||||
| AUC0–168h (μmol/L·h) † | 1.40 (1.23–1.60) | 9.75 (8.59–11.08) | 22.31 (19.65–25.34) | 41.56 (36.50–47.31) |
| Cmax (nmol/L) † | 21.25 (16.78–26.91) | 278.21 (226.64– 341.53) | 700.63 (570.74–860.08) | 1305.97 (1050.40–1623.72) |
| Ctrough (nmol/L) † | 4.56 (3.42–6.08) | 10.06 (7.67–13.20) | 16.40 (12.49–21.52) | 27.85 (21.09–36.79) |
| Tmax (h) § | 2.00 (2.00–8.00) | 1.00 (1.00–2.00) | 1.50 (0.33–2.17) | 1.00 (0.50–4.00) |
| t1/2 (h) † † | 144.88 (44.42) | 143.52 (39.28) | 82.48 (53.27) | 73.73 (34.73) |
| fe (%) ¶ | – | – | – | 74.4 (10.0) |
| CLr (L/h) ¶ | – | – | – | 2.3 (0.4) |
| CrCL (L/h) ¶ | – | – | – | 6.9 (1.3) |
| Accumulation ratio (day 15/day 1) ‡ | ||||
| AUC0–168h (μmol/L·h) | 1.35 (1.27–1.44) | 1.09 (1.04–1.15) | 1.05 (1.00–1.11) | 1.03 (0.97–1.09) |
| Cmax (nmol/L) | 1.36 (1.13–1.63) | 1.18 (1.00–1.38) | 0.87 (0.75–1.02) | 1.21 (1.02–1.43) |
| Ctrough (nmol/L) | 1.24 (1.06–1.44) | 0.97 (0.86–1.10) | 1.11 (0.98–1.26) | 1.03 (0.90–1.18) |
†(‡)Back‐transformed least squares mean and 95% (90%) confidence interval from a linear mixed effects model performed on natural log‐transformed values. §Median (minimum, maximum). ¶Mean (standard deviation). ††Geometric mean with percent coefficient of variation. ‡‡ n = 5 for concentration at 168 h post‐dose (C168h) for single dose of 25 mg, and n = 4 (multiple dose of 1 mg) and n = 5 (multiple dose of 50 mg) on day 15. –, Not calculated; AUC0–168h, area under the concentration–time curve from 0 to 168 h; AUC0–∞, area under the concentration–time curve from 0 to infinity; CLr, renal clearance; Cmax, maximum concentration; Ctrough, trough plasma concentration; CrCL, creatinine clearance; fe, fraction of dose excreted in urine; t½, terminal half‐life; Tmax, time to maximum concentration.
Figure 1.
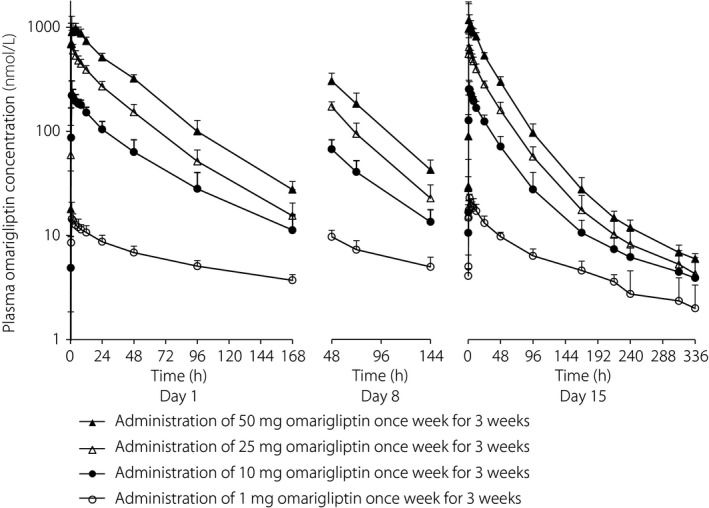
Mean plasma concentration–time profiles of omarigliptin after multiple dose administration of omarigliptin in healthy Japanese men (mean + standard deviation, n = 4−6).
Omarigliptin was rapidly absorbed after a single dose administration, with observed median Tmax values ranging 1.00–4.00 h (Table 1). The plasma pharmacokinetics of omarigliptin after single dose administration were biphasic, with a long t½ of >100 h. The single dose pharmacokinetic parameters of omarigliptin (AUC0–∞, AUC0–168h and Cmax) appeared to increase in an approximately dose‐proportional manner, whereas C168h appeared to increase in a less than dose‐proportional manner over the dose range of 5–100 mg.
No difference in mean plasma concentration–time plot was observed after administration of a single dose of omarigliptin 10 mg under fasted or fed conditions (data provided in Supplementary Material; Figure S1/Table 1). Omarigliptin 10 mg was absorbed with a median Tmax value of 1.00 h in the fasted state and 4.00 h in the fed state. The observed geometric mean ratios (fed/fasted; 90% CI) for AUC0–∞, AUC0–168h, Cmax and C168h were 1.03 (0.96–1.11), 0.97 (0.90–1.03), 0.75 (0.65–0.86) and 1.13 (0.93–1.37), respectively (data provided in Supplementary Material; Figure S1/Table S2).
Omarigliptin was rapidly absorbed after multiple dose administration (Figure 1). The observed median Tmax values ranged 0.50–2.00 h (Table 1). As observed with a single dose administration, the pharmacokinetic profile of omarigliptin after multiple dose administration was biphasic with a long t½ (Figure 1 and Table 1). Cmax appeared to increase in a dose‐proportional manner, whereas AUC0–168h and trough plasma concentration appeared to increase in a less than dose‐proportional manner across the omarigliptin 1–50 mg q.w. dose range. Accumulation of omarigliptin with multiple dose administration was minimal. The observed geometric mean accumulation ratios (day 15/day 1) after omarigliptin 1–50 mg for 3 weeks of q.w. administration ranged 1.03–1.35 for AUC0–168h, 0.87–1.36 for Cmax and 0.97–1.24 for trough plasma concentration. All participants had reached steady state after the first dose for all doses tested (Table 1).
Over 168‐h collection intervals, at steady state on day 15, 74.4% of the administered omarigliptin 50 mg q.w. dose was recovered as unchanged parent in urine (Table 1). The mean renal clearance of omarigliptin 50 mg q.w., calculated from the 0–168‐h urinary excretion data, was 2.3 L/h (Table 1). The renal clearance of omarigliptin 50 mg q.w. was lower than creatinine clearance (mean creatinine clearance over 168 h: 6.9 L/h; Table 1).
Pharmacodynamics
Administration of omarigliptin q.w. for 3 weeks produced sustained dose‐dependent inhibition of DPP‐4 activity for 1 week post‐dose compared with placebo (Figure 2). The mean and 95% CIs for percent inhibition of DPP‐4 activity from baseline at 168 h after single dosing of omarigliptin ranged between 79.2% (76.2–81.9) and 94.0% (93.0–94.9) over the 5–100 mg dose range (Table 2). The mean and 95% CIs for percent inhibition of DPP‐4 activity from baseline at 168 h after 3 weeks dosing of omarigliptin ranged between 51.3% (29.7–66.3) and 90.2% (86.6–92.9) over the 1–50 mg q.w. dose range (Table2). Greater than 80% inhibition from baseline of DPP‐4 activity was rapidly reached after the first dosing, and maintained for 168 h after single and multiple q.w. administration of omarigliptin at a dose of 25 mg or higher (Figure 2). The pharmacokinetic and pharmacodynamic relationship followed an Emax model, with an Emax value of 96.5% inhibition and an estimated 80% effective concentration of 11.4 nmol (data provided in Supplementary Material; Figure S2/Table S3).
Figure 2.
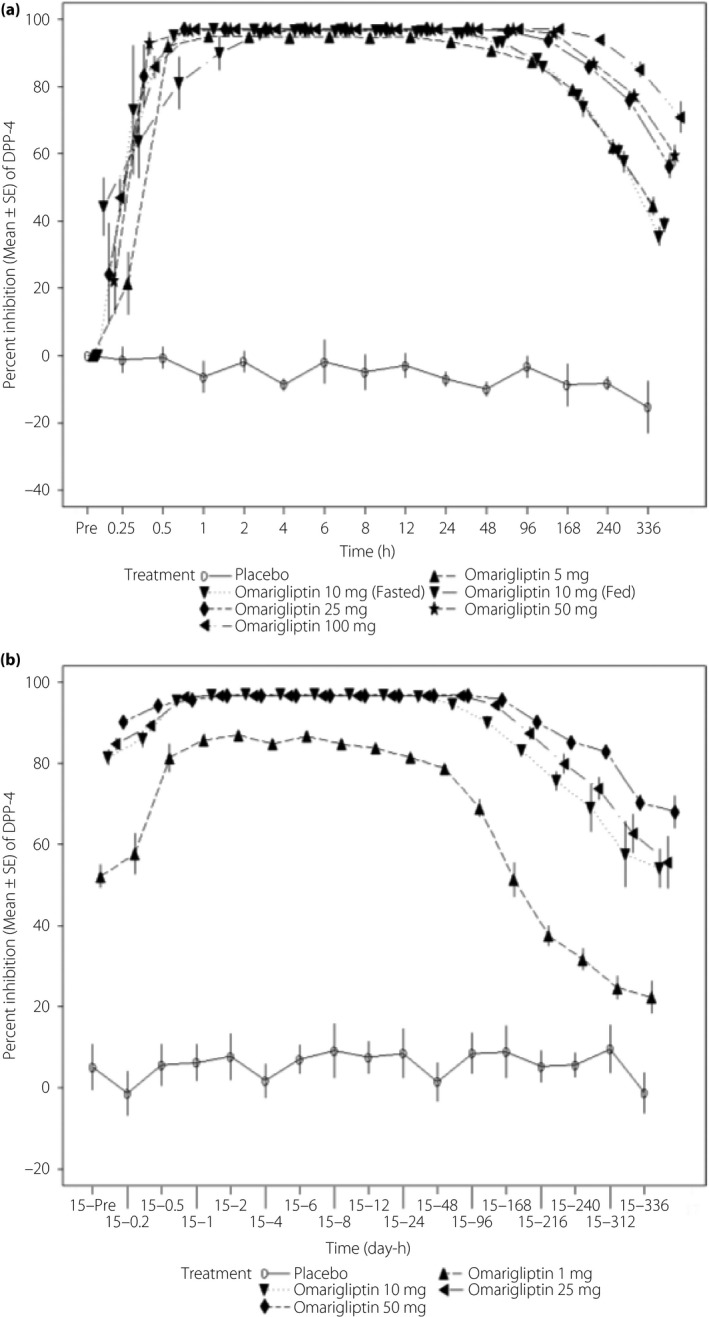
Percent inhibition of plasma dipeptidyl peptidase‐4 (DPP‐4) activity from baseline after (a) single and (b) multiple dose administration of omarigliptin or placebo in healthy Japanese men. Data are shown as mean ± standard error percent inhibition from baseline (day 1 predose). The time‐scale shown in the x‐axis was modified to present plots clearly without overlap.
Table 2.
Dipeptidyl peptidase‐4 percent inhibition from baseline and percent inhibition from baseline difference at 168‐h post‐dose by treatment after single and multiple dose administration of omarigliptin or placebo in healthy Japanese men
| Treatment | n | Percent inhibition from baseline (%) | Difference from placebo (%) |
|---|---|---|---|
| LS mean (95% CI) † | difference in LS means (90% CI) ‡ | ||
| Single dose | |||
| Placebo § | 4 | −8.69 (−28.82–8.29) | |
| Omarigliptin 5 mg | 6 | 79.20 (76.17–81.85) | 87.89 (80.96–95.09) |
| Omarigliptin 10 mg (fasted) | 6 | 77.39 (74.14–80.24) | 86.09 (79.05–93.38) |
| Omarigliptin 10 mg (fed) | 6 | 73.90 (70.14–77.18) | 82.59 (75.36–90.06) |
| Omarigliptin 25 mg | 5 | 85.97 (83.66–87.95) | 94.66 (88.11–101.49) |
| Omarigliptin 50 mg | 5 | 86.76 (84.59–88.63) | 95.45 (88.95–102.25) |
| Omarigliptin 100 mg | 5 | 94.01 (93.03–94.85) | 102.70 (96.60–109.11) |
| Multiple dose (day 15) | |||
| Placebo | 7 | 8.85 (−17.71–29.42) | |
| Omarigliptin 1 mg | 4 | 51.33 (29.67–66.32) | 42.48 (31.06–54.19) |
| Omarigliptin 10 mg | 5 | 83.26 (77.03–87.80) | 74.41 (65.73–83.59) |
| Omarigliptin 25 mg | 6 | 87.52 (83.55–90.53) | 78.67 (70.36–87.51) |
| Omarigliptin 50 mg | 5 | 90.23 (86.59–92.88) | 81.37 (73.30–90.00) |
†Back transformed least square (LS) means and confidence intervals (CI) obtained from a linear mixed effects model performed on natural log transformed values. ‡Difference of least square means. §Placebo data after period 1 in single dose part were excluded because of carryover effect.
Safety
There were no serious clinical or laboratory adverse events, and no participants were discontinued from the study because of an adverse event. There were no adverse events of hypoglycemia. In part 1, six participants reported a total of eight clinical adverse events after single dosing of omarigliptin or placebo. Six of these eight clinical adverse events were considered drug‐related (five after omarigliptin and one after placebo). In part 2, nine participants reported a total of 13 clinical adverse events after multiple dosing of omarigliptin. Eight of these 13 adverse events were considered drug‐related. The most common drug‐related clinical adverse events reported in the study were dizziness (two participants per each study part). All adverse events were of mild or moderate intensity, and resolved by the end of the study. There were no clinically significant, consistent treatment‐related or dose‐related changes from baseline in the laboratory, vital signs, glucose monitoring or ECG parameters, and no changes were noted in the neurological examination results after administration of the study drug.
Discussion
Omarigliptin is a structurally distinct and potent DPP‐4 inhibitor that is being developed as a q.w. treatment regimen for type 2 diabetes mellitus. Weekly dosing of an efficacious, well‐tolerated, safe, oral agent that reduces medication burden has the potential to increase adherence to AHA therapy, which in turn might improve long‐term outcomes. The present study assessed the safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple doses of omarigliptin in healthy Japanese men.
Absorption of omarigliptin was rapid after single and multiple oral dose administration, with median Tmax ranging 0.50–4.00 h across the doses studied. Omarigliptin had a biphasic plasma pharmacokinetic profile, with long t½ >100 h (Table 1). The apparent t½ values with omarigliptin at steady state supported q.w. dosing. Accumulation of omarigliptin on multiple q.w. dosing was negligible. After multiple dosing, all participants reached steady state after the first dose for all doses tested, resulting in at least the week 3 (day 15) pharmacokinetic parameters being representative of steady‐state exposure (Table 1).
Administration of a single dose of omarigliptin 10 mg with a Japanese standard breakfast resulted in a slight decrease in Cmax and a slight delay in Tmax. The difference of t½ under fed and fasted states after administration of 10 mg omarigliptin is due to the longer blood sampling time for the fed state (up to 336 h for fed condition, vs 240 h for fasted condition), which captured more of the longer beta phase (data provided in Supplementary Material: Figure S1). However, the differences in t½ between the fed and fasted states were not considered clinically meaningful. There was no difference in overall extent of absorption of omarigliptin 10 mg in the fed state compared with the fasted state, as assessed by AUC (data provided in Supplementary Material: Table S2). These findings support the administration of omarigliptin without regard to food.
Omarigliptin was primarily eliminated in the urine as parent drug over 168 h after q.w. multiple dose administration of omarigliptin 50 mg, approximately 74% of the dose was excreted unchanged in the urine (Table 1). The renal clearance of omarigliptin 50 mg q.w. was low at 2.3 L/h. Considering the free fraction of omarigliptin in humans spans a range of 56–76% for the concentration range for 50 mg q.w. dosing (unpubl. data) and converting to mL/min, the unbound renal clearance would then be approximately 50–68 mL/min. This unbound clearance is below the typical glomerular filtration rates of approximately 90 mL/min for healthy subjects, and is also below the creatinine clearance in the present study of 115 mL/min (6.9 L/h), suggesting that omarigliptin is cleared by filtration with net reabsorption. Omarigliptin shows high permeability and is not a substrate of drug transporters, such as organic anion transporters (OAT1/OAT3) and organic cation transporters (OCT2) expressed in the basolateral side of renal tubular cells. For renally excreted compounds whose unbound renal clearance is lower than typical glomerular filtration rates, reabsorption generally occurs by a passive process14. Thus, it is highly likely that passive permeation mediates the renal reabsorption of omarigliptin, consistent with its high membrane permeability. It is noted that other DPP‐4 inhibitors; for example, sitagliptin, alogliptin and trelagliptin, which are also primarily cleared through renal excretion of intact drug and show higher renal clearance than typical glomerular filtration rates, likely undergo active secretion in the kidney15, 16, 17, whereas omarigliptin undergoes net‐reabsorption in the kidney, consistent with the substantially longer elimination half‐life of the compound.
Plasma DPP‐4 activity is considered a valid biomarker for predicting the magnitude of clinically significant glucose‐lowering efficacy with a DPP‐4 inhibitor. Eighty percent of DPP‐4 inhibition has been reported to correlate with maximal glycemic efficacy11. Administration of omarigliptin q.w. for 3 weeks resulted in sustained, dose‐dependent inhibition of DPP‐4 activity for 1 week post‐dose compared with placebo. Greater than 80% inhibition from baseline of DPP‐4 activity was reached rapidly after the first administration, and maintained for approximately 168 h after single and q.w. administration of omarigliptin at a dose of 25 mg or higher (Figure 2).
Administration of omarigliptin at single doses of 5–100 mg and multiple doses of 1–50 mg q.w. was generally well tolerated in healthy Japanese men. No deaths, serious adverse events or laboratory adverse events were reported, and no participant discontinued from the study because of an adverse events. There were no adverse events of hypoglycemia. The majority of adverse events were of mild or moderate intensity, and all resolved by the end of the study. The most common drug‐related clinical adverse events reported in the study were dizziness. There were no consistent treatment‐related or dose‐related changes in the laboratory, vital signs, orthostatic vital signs, glucose monitoring or ECG safety parameters, and no changes were noted in neurological examination results after administration of the study drug.
In conclusion, the present study in healthy Japanese men showed that omarigliptin was well tolerated, and had a pharmacokinetic and DPP‐4 inhibition profile that supports once‐weekly dosing.
Disclosure
Saori Tsuchiya, Akira Wakana, Yuki Matsumoto and Hideyo Suzuki are employees of MSD K.K., Tokyo, Japan. Evan Friedman, Carol Addy, Daniel Tatosian and Eunkyung Kauh are or were employees of Merck & Co., Inc., Kenilworth, NJ, USA, and might hold stock in the company.
Supporting information
Figure S1 | Mean plasma concentration–time profiles of omarigliptin after single dose administration of omarigliptin under fasted and fed state in healthy Japanese men
Figure S2 | Omarigliptin individual DPP‐4 percent inhibition versus plasma concentrations after single dose administration of omarigliptin in healthy Japanese men
Table S1 | Summary of study design
Table S2 | Summary of pharmacokinetic parameters after single dose administration of omarigliptin under the fasted and fed state in healthy Japanese men
Table S3 | Emax model parameter estimates after single dose administration of omarigliptin in healthy Japanese men
Acknowledgments
The authors thank Alan Meehan, Kristen Lewis and Sheila Erespe (Merck & Co., Inc., Kenilworth, NJ, USA) for their assistance in preparing this article for publication. Funding for this study was provided by MSD K.K., Tokyo, Japan, a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, USA. The material presented in this article was presented in poster format at the Japan Diabetes Society (Osaka, Japan).
J Diabetes Investig 2017; 8: 84–92
References
- 1. IDF Diabetes Atlas. 7th ed Brussels, Belgium: International Diabetes Federation; http://www.diabetesatlas.org/component/attachments/ [Google Scholar]
- 2. Cramer JA. A systematic review of adherence with medications for diabetes. Diabetes Care 2004; 27: 1218–1224. [DOI] [PubMed] [Google Scholar]
- 3. Cheong C, Barner JC, Lawson KA, et al Patient adherence and reimbursement amount for antidiabetic fixed‐dose combination products compared with dual therapy among Texas Medicaid recipients. Clin Ther 2008; 30: 1893–1907. [DOI] [PubMed] [Google Scholar]
- 4. Paes AH, Bakker A, Soe‐Agnie CJ. Impact of dosage frequency on patient compliance. Diabetes Care 1997; 20: 1512–1517. [DOI] [PubMed] [Google Scholar]
- 5. Johnston SS, Nguyen H, Felber E, et al Retrospective study of adherence to glucagon‐like peptide‐1 receptor agonist therapy in patients with type 2 diabetes mellitus in the United States. Adv Ther 2014; 31: 1119–1133. [DOI] [PubMed] [Google Scholar]
- 6. Brubaker PL. Minireview: update on incretin biology: focus on glucagon‐like peptide‐1. Endocrinology 2010; 151: 1984–1989. [DOI] [PubMed] [Google Scholar]
- 7. Holst JJ, Vilsboll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol 2009; 297: 127–136. [DOI] [PubMed] [Google Scholar]
- 8. Xu L, Man CD, Charbonnel B, et al Effect of sitagliptin, a dipeptidyl peptidase‐4 inhibitor, on beta‐cell function in patients with type 2 diabetes: a model‐based approach. Diabetes Obes Metab 2008; 10: 1212–1220. [DOI] [PubMed] [Google Scholar]
- 9. Cho YM. Incretin physiology and pathophysiology from an Asian perspective. J Diabetes Investig 2015; 6: 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Biftu T, Sinha‐Roy R, Chen P, et al Omarigliptin (MK‐3102): a novel long‐acting DPP‐4 inhibitor for once‐weekly treatment of type 2 diabetes. J Med Chem 2014; 57: 3205–3212. [DOI] [PubMed] [Google Scholar]
- 11. Krishna R, Addy C, Tatosian D, et al Pharmacokinetics and pharmacodynamics of omarigliptin, a once‐weekly dipeptidyl peptidase‐4 (DPP‐4) inhibitor, after single and multiple doses in healthy subjects. J Clin Pharmacol 2016; 56: 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sheu WH, Gantz I, Chen M, et al Safety and efficacy of omarigliptin (MK‐3102), a novel once‐weekly DPP‐4 inhibitor for the treatment of patients with type 2 diabetes. Diabetes Care 2015; 38: 2106–2114. [DOI] [PubMed] [Google Scholar]
- 13. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16: 31–41. [DOI] [PubMed] [Google Scholar]
- 14. Rowland M, Tozer T. Clinical Pharmacokinetics Concepts and Applications, 3rd edn Lippincott William & Wikins, Philadelphia, 2004; p173. [Google Scholar]
- 15. Sitagliptin [product label]. Tokyo, Japan: MSD K.K., 2015. Available from: http://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/170050_3969010F1034_2_24 Accessed March 25, 2016 (Japanese). [Google Scholar]
- 16. Alogliptin [product label]. Tokyo, Japan: Takeda, 2014. Available from: http://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/400256_3969012F1025_1_14 Accessed March 25, 2016 (Japanese). [Google Scholar]
- 17. Trelagliptin [product interview form]. Tokyo, Japan: Takeda, 2015. Available from: http://www.pmda.go.jp/PmdaSearch/iyakuSearch/ Accessed March 25, 2016 (Japanese). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 | Mean plasma concentration–time profiles of omarigliptin after single dose administration of omarigliptin under fasted and fed state in healthy Japanese men
Figure S2 | Omarigliptin individual DPP‐4 percent inhibition versus plasma concentrations after single dose administration of omarigliptin in healthy Japanese men
Table S1 | Summary of study design
Table S2 | Summary of pharmacokinetic parameters after single dose administration of omarigliptin under the fasted and fed state in healthy Japanese men
Table S3 | Emax model parameter estimates after single dose administration of omarigliptin in healthy Japanese men