

Abstract
Our aims were to summarize the clinical pharmacokinetics and pharmacodynamics of the dipeptidyl‐peptidase‐4 inhibitor, linagliptin, and to consider how these characteristics influence its clinical utility. Differences between linagliptin and other dipeptidyl‐peptidase‐4 inhibitors were also considered, in addition to the influence of Asian race on the pharmacology of linagliptin. Linagliptin has a xanthine‐based structure, a difference that might account for some of the pharmacological differences observed with linagliptin versus other dipeptidyl‐peptidase‐4 inhibitors. The long terminal half‐life of linagliptin results from its strong binding to dipeptidyl‐peptidase‐4. Despite this, linagliptin shows a short accumulation half‐life, as a result of saturable, high‐affinity binding to dipeptidyl‐peptidase‐4. The pharmacokinetic characteristics of linagliptin make it suitable for once‐daily dosing in a broad range of patients with type 2 diabetes mellitus. Unlike most other dipeptidyl‐peptidase‐4 inhibitors, linagliptin has a largely non‐renal excretion route, and dose adjustment is not required in patients with renal impairment. Furthermore, linagliptin exposure is not substantially altered in patients with hepatic impairment, and dose adjustment is not necessary for these patients. The 5‐mg dose is also suitable for patients of Asian ethnicity. Linagliptin shows unique pharmacological features within the dipeptidyl‐peptidase‐4 inhibitor class. Although most clinical trials of linagliptin have involved largely Caucasian populations, data on the pharmacokinetic/pharmacodynamic properties of linagliptin show that these features are not substantially altered in Asian populations. The 5‐mg dose of linagliptin is suitable for patients with type 2 diabetes mellitus irrespective of their ethnicity or the presence of renal or hepatic impairment.
Keywords: Linagliptin, Pharmacodynamics, Pharmacokinetics
Introduction
The global burden of type 2 diabetes mellitus continues to grow1, and is becoming an increasingly urgent health issue across the world, particularly in low‐ and middle‐income countries; almost one‐fifth of people with diabetes live in Southeast Asia1. As a result of the growing burden of type 2 diabetes mellitus, there remains a need for effective, well‐tolerated therapies, and a range of treatment options is available for the management of hyperglycemia2. However, some of the commonly used therapies for type 2 diabetes mellitus have limitations as a result of troublesome side‐effects, such as risk of hypoglycemia and weight gain (e.g., sulfonylureas, thiazolidinediones, insulin), the possibility of gastrointestinal side‐effects (e.g., metformin, α‐glucosidase inhibitors), or are contraindicated in patients with moderate or severe renal impairment (e.g., metformin, sulfonylureas)2.
An important advance in the management of type 2 diabetes mellitus has been the development of incretin‐based therapies, including the dipeptidyl‐peptidase (DPP)‐4 inhibitors. These agents are being increasingly incorporated into clinical practice, and are listed as treatment options in the latest joint guidelines from the American Diabetes Association and the European Association for the Study of Diabetes2, and guidelines from the American Association of Clinical Endocrinologists3 for diabetes management. The mechanism of action of DPP‐4 inhibitors is distinct from that of other antidiabetes agents: their glucose‐lowering efficacy is based on an effect on the incretin hormones, active glucagon‐like peptide (GLP)‐1 and gastric inhibitory peptide (also known as glucose‐dependent insulinotropic polypeptide [GIP]), which are secreted from the intestine after a meal4. In the presence of hyperglycemia, these hormones are secreted in response to food intake, and exert a key role in the control of glucose levels by enhancing glucose‐dependent insulin release and reducing glucagon secretion. Both active GLP‐1 and GIP are rapidly inactivated through cleavage by DPP‐45, and thus, the antihyperglycemic activity of DPP‐4 inhibitors results from enhancement of the incretin effect. Importantly, DPP‐4 inhibitors have been shown to improve glycemic control with a low risk of hypoglycemia (when used without insulin secretagogues) or weight gain6.
Linagliptin is a selective and potent DPP‐4 inhibitor with a xanthine‐based molecular structure, and is indicated for the treatment of type 2 diabetes mellitus7. The efficacy of linagliptin has been shown in a range of clinical trials of patients with type 2 diabetes mellitus, both as monotherapy8, 9, 10 and in combination with other antidiabetes agents11, 12, 13, 14, 15, 16, 17, 18. The safety and tolerability of linagliptin has also been shown during its clinical development; a recent pooled analysis of 22 randomized, double‐blind trials of linagliptin showed that the frequency of adverse events was similar for linagliptin‐ and placebo‐treated patients across a wide range of patients, including elderly subjects and individuals with declining renal function19.
The aim of the present review is to provide a summary of the clinical pharmacokinetics (PK) and pharmacodynamics (PD) of linagliptin, and to show how the PK/PD profile of linagliptin influences its clinical utility. The features of linagliptin will be compared with other drugs in its class. In view of the high prevalence of type 2 diabetes mellitus in Asian populations, consideration will be given to how the PK/PD of linagliptin compare between Caucasian and Asian populations.
Methods
The Medline database was searched through PubMed to retrieve relevant references from the past 10 years. Search terms included: linagliptin, DPP‐4 inhibitors, PK, PD, Japanese, Chinese, Asian, renal, hepatic and interactions. Other relevant literature was obtained based on personal knowledge and experience. A narrative overview of the literature was then synthesized based on manual assessment of the retrieved literature.
Pharmacology of linagliptin
In contrast with other DPP‐4 inhibitors, linagliptin has a xanthine‐based chemical structure20, 21. This structural difference might account for some of the pharmacological differences observed with linagliptin compared with other drugs in its class22. Linagliptin is a potent and selective inhibitor of DPP‐4, with >10,000‐fold selectivity for DPP‐4 compared with the enzymes DPP‐8 and DPP‐922. In clinical studies, linagliptin administration has been shown to produce dose‐dependent DPP‐4 inhibition in healthy volunteers23, 24 and in patients with type 2 diabetes mellitus25, 26, 27. In healthy subjects, linagliptin doses of up to 600 mg (120 times higher than the 5‐mg dose) have been shown to be well tolerated24, and this wide therapeutic window might, at least in part, be related to the high selectivity of linagliptin for DPP‐422. For clinical use, linagliptin has an oral route of administration21, 22 and is indicated, as an adjunct to diet and exercise, to improve glycemic control in adults with type 2 diabetes mellitus, either alone or in combination with other oral antidiabetes agents7, 28.
Clinical PK
Absorption
After oral administration of linagliptin 5 mg, the drug is rapidly absorbed, and geometric mean values for the maximum plasma concentration are approximately 6–10 nmol/L after a single dose24, 27, and 11–12 nmol/L at steady state25, 27. The time taken to achieve maximum plasma concentration is approximately 1.5–2.0 h24, 25, 27. After multiple oral doses of linagliptin 1–10 mg, two studies have shown the mean area under the plasma concentration–time curve at steady state (AUCτ,ss) to be approximately 81.7–207 nmol h/L in patients with type 2 diabetes mellitus25, 27. The absolute bioavailability of linagliptin has been estimated to be approximately 30%29. Administration of linagliptin with food has been shown to have no clinically relevant effect on its absorption30.
Distribution
In a study of healthy men, the apparent volume of distribution at steady state after intravenous infusion of linagliptin 0.5–10 mg was shown to be 380–1,540 L29. After a single intravenous dose of 5 mg, the volume of distribution at steady state was 1,110 L29. This large apparent volume of distribution indicates extensive distribution of linagliptin in the tissues. In addition, linagliptin has been shown to bind extensively to plasma proteins (70–80%) in a concentration‐dependent manner31. This high‐affinity binding of linagliptin to DPP‐4 in the plasma and tissues contributes to its long terminal half‐life (>100 h)25, 27, short accumulation half‐life (approximately 10 h)25, 27 and non‐linear PK profile, as shown in both animal31, 32, 33 and human20 studies. Furthermore, the saturable binding of linagliptin to DPP‐4 results in less than dose‐proportional increases in exposure to linagliptin within the therapeutic dose range, and thus, a non‐linear relationship between linagliptin dose and drug exposure24, 25.
Metabolism
Metabolism is a minor contributor to the overall disposition and elimination of linagliptin, which is mainly eliminated unchanged through feces. Its main metabolite (CD 1790) accounts for approximately 18% of the molar linagliptin plasma exposure (AUC24) after a single oral 10‐mg dose of linagliptin, and is pharmacologically inactive34.
Elimination
Linagliptin has a mainly non‐renal route of excretion, with 84.7% of an orally administered 10‐mg dose being eliminated through bile and the gut, and 5.4% excreted in urine (Figure 1)34. Experiments in rats have shown that the bioavailability of orally administered linagliptin is enhanced by inhibition of intestinal P‐glycoprotein, indicating that this transport system can decrease the intestinal absorption of linagliptin. Although the potent, reversible binding of linagliptin to DPP‐4 in the plasma and tissues means that a proportion of the administered dose is not directly available for elimination, these studies showed that the systemically available linagliptin is mainly excreted with bile, with a minor proportion (12% of an intravenous dose) being secreted directly into the gut35. It is therefore possible that, in the presence of hepatic or renal impairment, the direct excretion of linagliptin into the gut could provide an alternative route of excretion of the drug.
Figure 1.
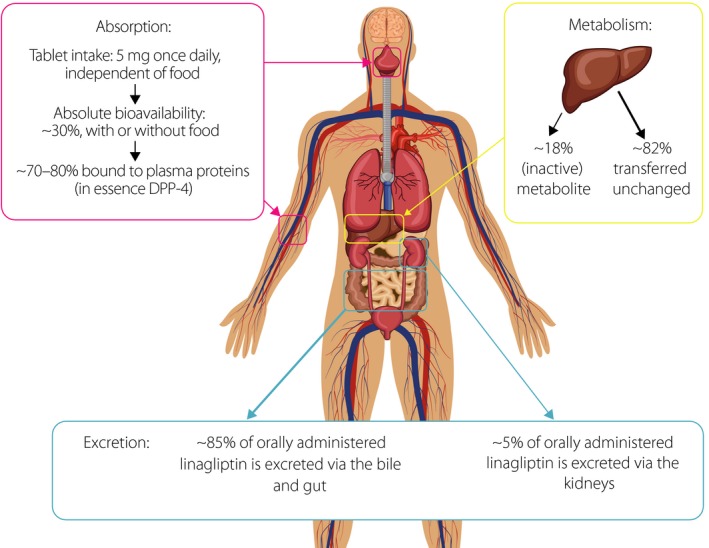
The absorption, metabolism and excretion of linagliptin after oral administration. The percentages shown for the excretion of linagliptin are based on data obtained up to 120 h after the oral administration of a 10‐mg dose of linagliptin34. DPP‐4, dipeptidyl‐peptidase‐4.
Clinical PD
DPP‐4 inhibition
The inhibition of DPP‐4 is an attractive strategy for the management of type 2 diabetes mellitus, in particular because the associated stimulation of insulin release is glucose‐dependent and, therefore, DPP‐4 inhibitors have a low risk of hypoglycemia36. Linagliptin provides sustained inhibition of DPP‐4 activity in a dose‐dependent manner. A once‐daily 5‐mg dose of linagliptin has been shown to achieve >80% inhibition of DPP‐4 in healthy volunteers23 and patients with type 2 diabetes mellitus25. This level of DPP‐4 inhibition is considered to be the threshold for glycemic control for DPP‐4 inhibitors, with maximum glucose‐lowering efficacy being achieved with DPP‐4 inhibitors that achieve at least 80% inhibition of DPP‐437. In a study of linagliptin doses of 2.5, 5 and 10 mg, inhibition of DPP‐4 was shown to be rapidly achieved; the mean maximum DPP‐4 inhibition ranged from 86% for the 2.5‐mg dose to 93% for linagliptin 10 mg, after a single dose27. At steady state, the mean maximum inhibition of DPP‐4 was 91–93% across all linagliptin doses. Therefore, it would be expected that maximum glucose‐lowering efficacy can be achieved with the evaluated linagliptin doses.
Effects on GLP‐1 and blood glucose
The antihyperglycemic effect of linagliptin arises from its effect on the incretin hormones, active GLP‐1 and GIP. In response to hyperglycemia, these hormones stimulate glucose‐dependent insulin secretion, and inhibit the secretion of glucagon38, 39, 40. DPP‐4 is the main enzyme involved in the breakdown of both GLP‐1 and GIP41, and DPP‐4 inhibition, therefore, prolongs the activity of GLP‐1 and potentiates its antihyperglycemic effects. For example, in a study of linagliptin administration to patients with type 2 diabetes mellitus, marked increases in plasma levels of GLP‐1 were observed after 28 days of linagliptin dosing27. After a meal tolerance test, carried out 24 h after the last linagliptin intake, there were statistically significant reductions in the AUC of the plasma glucose concentration–time graph27. Because both GIP and GLP‐1 promote glucose‐dependent insulin secretion42, 43, linagliptin therapy is associated with a low risk of hypoglycemia. This is supported by the findings of clinical trials of linagliptin, both alone9 and in combination with other non‐sulfonylurea oral antidiabetes agents11, 14, 18, and by an exploratory analysis of data from a 2‐year randomized, double‐blind study of linagliptin versus glimepiride in patients with type 2 diabetes mellitus and inadequate glycemic control despite metformin therapy44.
Special Populations
Renal impairment
The development of moderate‐to‐severe renal impairment (defined as an estimated glomerular filtration rate [eGFR] below 60 mL/min/1.73 m2) is a frequent complication of type 2 diabetes mellitus, and some degree of renal disease is estimated to be present in up to 40% of patients45, 46, 47. As a result, the impact of renal disease on antidiabetes therapies is an important consideration. The effect of various degrees of renal impairment on exposure to linagliptin has been evaluated under single‐dose and steady‐state conditions in subjects with or without type 2 diabetes mellitus, and mild, moderate or severe renal impairment, or end‐stage renal disease48. The findings showed that the renal excretion of unchanged linagliptin did not exceed 7%, regardless of renal function status. Although exposure to linagliptin was slightly increased (20–60%) among patients with renal impairment versus subjects with normal renal function, renal impairment was shown to have only a minor effect on the PK of linagliptin. These results were further confirmed in a pooled analysis of three randomized studies from the global phase III program for linagliptin; mean trough levels of linagliptin over time were similar for patients with normal renal function (eGFR ≥90 mL/min) and those with mild (eGFR 60 to <90 mL/min), moderate (eGFR 30 to <60 mL/min) or severe (eGFR <30 mL/min) renal impairment49. Therefore, no dose adjustment of linagliptin or drug‐related monitoring of eGFR is deemed necessary on the basis of renal function7, 28, 48.
Hepatic impairment
In addition to renal dysfunction, patients with type 2 diabetes mellitus frequently show evidence of hepatic disease, including non‐alcoholic fatty liver disease50 and cirrhosis51. Despite the largely hepatic route of elimination of linagliptin, the presence of hepatic impairment has been shown to have no clinically important effect on the PK, PD or tolerability of linagliptin52. In a study of subjects with mild, moderate or severe hepatic impairment, exposure to single or multiple doses of linagliptin 5 mg was not shown to be affected to a clinically relevant extent by the presence of hepatic impairment52. The degree of DPP‐4 inhibition was similar for all patient groups, with median DPP‐4 inhibition values of >80% for all patients regardless of the degree of hepatic impairment. These results show that dose adjustment is not required for patients with hepatic impairment7, 28.
Drug Interactions
Linagliptin is a weak‐to‐moderate inhibitor of cytochrome P450 enzymes7, 21. Because of the small proportion of linagliptin that is metabolized by these enzymes, changes in exposure to linagliptin by inhibition or induction of P450‐dependent pathways by concomitantly administered drugs are considered to be unlikely. Importantly, linagliptin has shown no clinically relevant PK interaction with commonly prescribed antidiabetes drugs, such as metformin53, pioglitazone54 and glyburide55.
Linagliptin is a P‐glycoprotein substrate, and full efficacy of linagliptin might not be achieved when administered in combination with strong inducers of P‐glycoprotein (such as rifampicin), particularly if these drugs are administered long term7, 28, 34, 56. As a consequence, alternative treatment is recommended in these circumstances.
PK/PD in Asian vs Caucasian Patients
The presentation of type 2 diabetes mellitus can differ between patients of Asian and Caucasian origin; in Asian patients, the condition generally starts at a younger age in individuals with a relatively low body mass index57. Asian individuals tend to show greater adiposity and a higher percentage of body fat for a given body mass index compared with Western populations57, 58. This feature is probably linked to the higher frequency of insulin resistance observed in Asian versus Caucasian populations58. Asian patients with type 2 diabetes mellitus are also at heightened risk of comorbidities, such as renal complications and cardiovascular disease59. In addition to these clinical factors, there is evidence to show that ethnic differences in dietary habits result in variations in glucose regulation between different Asian populations. For example, one study showed that Japanese subjects, with or without type 2 diabetes mellitus, demonstrated higher fasting insulin levels compared with Korean or Chinese participants60. These differences based on ethnicity could affect the PK and PD characteristics of antidiabetes therapies and, therefore, are an important consideration.
In a study of Japanese patients with type 2 diabetes mellitus, linagliptin showed a non‐linear PK profile, low accumulation and low (<7%) urinary excretion rate, all of which were consistent with findings in healthy Japanese subjects and Caucasian populations26. After 4‐week administration of multiple doses of linagliptin (0.5, 2.5, 10 mg), a long terminal half‐life (223–260 h) was reported, in contrast with a shorter accumulation half‐life (10.0–38.5 h), resulting in a moderate accumulation ratio of <2.9 that decreased with rising doses. As with other populations, this observation reflects the saturable high‐affinity binding of linagliptin to DPP‐4 at the evaluated doses, leading to slow dissociation of the drug from its target. Similar findings have been reported from another study of multiple doses of linagliptin (1, 2.5, 5, 10 mg) given to healthy Japanese men23. Although exposure to linagliptin at steady state is increased by approximately 30% in Japanese versus Caucasian subjects, this is not considered to be clinically relevant because of the wide therapeutic window of linagliptin24. Furthermore, data obtained from Japanese patients with type 2 diabetes mellitus have shown that the efficacy and safety of linagliptin is not substantially altered by the presence of renal impairment, indicating that, as in Western populations, dose adjustment in these patients is not required on the basis of renal function61. The 5‐mg and 10‐mg doses of linagliptin have been shown to inhibit DPP‐4 by >80% at 24‐h post‐dose in Japanese subjects23, 26, which is comparable with the efficacy that has been observed in Caucasian populations. The PK profile of linagliptin in healthy Chinese volunteers62, 63 has also been shown to be similar to that in other populations, including Japanese and Caucasian subjects (Table 1)23, 24, 25, 26, 27, 29, 34, 62, 63, 64.
Table 1.
Comparison of the main pharmacokinetic parameters of linagliptin (5 mg, unless otherwise indicated) in mixed and Asian patient populations
| Parameter | Estimate (mixed populations) | Estimate (Asian population) | |
|---|---|---|---|
| Japanese | Chinese | ||
| Cmax (nmol/L) |
5.724
8.325 9.627 |
9.023 |
6.863, †
10.462 |
| Tmax (h) |
1.524
1.825 2.027 |
1.526, ‡ | 4.063, † |
| AUC0–24 (nmol h/L) |
10024
11825 |
15923 | 15062 |
| T½ (h) |
69.724
12729 |
10523 |
58.0–75.663, †
82.462 |
| CL/F (mL/min) | 23123 | 163–20463, † | |
| Cmax,ss (nmol/L) |
11.125
12.327 |
5.0–44.026, ‡
11.923 |
14.162 |
| Tmax,ss (h) |
1.027
1.525 |
1.3–1.526, ‡ | – |
| AUCτ,ss (nmol h/L) | 14827 |
89.4–37326, ‡
19323 |
20462 |
| T½ ss (h) |
13125
19427 |
14323
223–26026, ‡ |
10362 |
| Accumulation T½ (h) |
9.527
11.425 |
10–1523
10.0–38.526, ‡ |
11.562 |
| CL/Fss (mL/min) |
1,19027
1,33064 |
197–94526, ‡ | – |
| Renal elimination (%) |
<124, 25
3.264 5.434 6.225 |
1.2–4.923, <726, ‡ | 1.9–7.962 |
Values are geometric mean. Superscript numbers refer to source references. †In the pharmacokinetic study by Pichereau et al.63, data are shown for subjects who received linagliptin 2.5 mg daily. ‡In the study of Japanese patients with type 2 diabetes mellitus by Horie et al.26, data shown are for subjects receiving linagliptin 0.5–10 mg daily. AUC0–24, area under the plasma concentration–time curve from zero to 24 h; AUCτ,ss area under the plasma concentration–time curve at steady state; CL/F, apparent total clearance; CL/Fss, apparent clearance at steady state; Cmax, maximum plasma concentration; Cmax,ss, maximum plasma concentration at steady state; PK, pharmacokinetic; T½, half‐life; T½ ss, half‐life at steady state; T2DM, type 2 diabetes mellitus; Tmax, time to reach maximum plasma concentration; Tmax,ss, time to reach maximum plasma concentration at steady state.
Data from two studies on the bioequivalence of linagliptin fixed‐dose combination treatments versus administration of the individual drugs can provide some insight into the comparative PK characteristics of linagliptin in Chinese and Caucasian populations. Although the total exposure to linagliptin (AUC0–72 and maximum plasma concentration) was approximately 40% higher among Chinese participants63 than previously reported in a similar study of Caucasian subjects65, this is in line with the findings reported above for Japanese subjects, and is not considered to be clinically relevant63.
Mean bodyweight in some Asian populations can be lower than in Caucasians. However, bodyweight has been shown to have no clinically meaningful impact on the PK or PD of linagliptin66, and so dose adjustment is not required on the basis of bodyweight7.
Comparison with Other DPP‐4 Inhibitors
Although the DPP‐4 inhibitors share a common mode of action, they are structurally heterogeneous, and linagliptin has a unique chemical structure and pharmacological profile compared with the other agents in its class (Table 2)7, 20, 22, 28, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83.
Table 2.
Main pharmacological differences between currently available dipeptidyl‐peptidase‐4 inhibitors
| Characteristic | Sitagliptin69, 82 | Vildagliptin70 | Saxagliptin71, 81 | Alogliptin72, 80 | Linagliptin7, 28 |
|---|---|---|---|---|---|
| Therapeutic dose (mg) | 100 | 50 | 5 | 25 | 5 |
| Relative (fold) in vitro selectivity for DPP‐4 vs DPP‐8 or DPP‐9 | >2,60073 | <30074 | <45075 | >10,00076 | >10,00022 |
| Fraction bound to plasma protein | Intermediate | Low | Low | Low | High |
| Renal excretion route | Major | Intermediate | Major | Major | Minor |
| Need for dose adjustment for renal impairment | Yes (moderate or severe) | May be required (limited experience) | Yes (moderate or severe) | Yes (moderate or severe) | No |
| Need for dose reduction with hepatic impairment (mild/moderate) | No (No experience in patients with severe hepatic impairment) | Not recommended for patients with hepatic impairment | No (Not recommended for patients with severe hepatic impairment) | No (No experience in patients with severe hepatic impairment) | No |
| Drug interaction potential | Low | Low | Intermediate | Low | Low |
| Efficacy – HbA1c lowering | Similar efficacy | Similar efficacy | Similar efficacy | Similar efficacy | Similar efficacy |
| Overall safety† |
Good |
Good
|
Good |
Good |
Good
|
†For all dipeptidyl‐peptidase‐4 (DPP‐4) inhibitors listed, hypoglycemia is reported more frequently with concomitant sulfonylurea (SU) or insulin therapy. ‡Most frequent adverse event (AEs) are those listed in prescribing information to occur in ≥5% of patients and more frequently than with placebo. §Common AEs defined as a frequency of ≥1/100 to <1/10. CI, confidence interval; HbA1c, glycated hemoglobin; HF, heart failure; HR, hazard ratio; URTI, upper respiratory tract infection; UTI, urinary tract infection.
In vitro studies of the inhibition of DPP‐4 activity have shown that the potency of linagliptin was higher than that of other DPP‐4 inhibitors (vildagliptin, sitagliptin, saxagliptin and alogliptin; based on half maximal inhibitory concentration values)22. Furthermore, the non‐linear PK profile of linagliptin is not shown by other DPP‐4 inhibitors. In addition, linagliptin shows a much higher binding to plasma proteins than other DPP‐4 inhibitors, with a very long terminal half‐life22, 68. From a clinical perspective, an important difference between linagliptin and other DPP‐4 inhibitors is its mainly non‐renal route of elimination35, which means that unlike several other DPP‐4 inhibitors, linagliptin does not require dose adjustment in the presence of renal impairment48.
Conclusions
Linagliptin has unique pharmacological properties within the DPP‐4 inhibitor class. The long terminal half‐life of linagliptin is related to its non‐linear PK profile that results from strong binding to its primary target, DPP‐4. Despite having a long terminal half‐life, linagliptin also exhibits a short accumulation half‐life, which can be attributed to the saturable, high‐affinity binding to DPP‐4. When DPP‐4 is saturated, unbound linagliptin is rapidly cleared from the body through bile and the gut. The PK characteristics of linagliptin have an impact on its clinical utility, such that an oral dose of 5 mg once daily is suitable for a broad range of patients with type 2 diabetes mellitus84. In contrast with most other DPP‐4 inhibitors, the largely non‐renal route of excretion of linagliptin allows treatment to be administered to patients with renal impairment, without the need for dose adjustment. Although linagliptin is largely metabolized in the liver, dose adjustment is not required for patients with hepatic impairment. This feature might be related to its wide therapeutic window and the fact that exposure to linagliptin is not substantially altered by the presence of hepatic impairment. The 5‐mg dose is also suitable for patients of Asian ethnicity; small changes in PK parameters observed when linagliptin is given to Japanese and Chinese patients have not been shown to have clinically relevant effects. Despite the fact that many clinical trials of linagliptin have been carried out in largely Caucasian populations, these findings provide reassurance that the PK/PD properties of linagliptin are not altered to a clinically relevant extent in patients of Asian ethnicity.
Disclosure
AC disclosed the following. Advisory board membership: AstraZeneca, Bayer Healthcare, Boehringer Ingelheim, Bristol‐Myers Squibb, Danone, DOC Generici, Eli Lilly, Janssen, Medtronic, Merck Sharp & Dohme, Novartis, Novo Nordisk, OM Pharma, Roche Diagnostics, Sanofi, Takeda and Unilever. Consultancy: Bayer Pharma, Lifescan, Mendor, Novartis and Roche Diagnostics. Lectures: AstraZeneca, Bayer Healthcare, Bayer Pharma, Boehringer Ingelheim, Bristol‐Myers Squibb, Eli Lilly, Merck Sharp & Dohme, Mitsubishi, Novartis, Novo Nordisk, Nutricia, Sanofi, Servier and Takeda. Research grants: Mitsubishi, Novartis and Novo Nordisk. NI has received clinical research grants from MSD, Eli Lilly Japan, Shiratori Pharmaceutical, Mitsubishi Tanabe Pharma and Roche Diagnostics; and scholarship grants from Nippon Boehringer Ingelheim, Kissei Pharmaceutical, Taisho Toyama Pharmaceutical, Sanofi, Pfizer Japan, Daiichi Sankyo, Mitsubishi Tanabe Pharma, Takeda Pharmaceutical, Japan Tobacco, Kyowa Hakko Kirin, Sumitomo Dainippon Pharma, Astellas Pharma, MSD, Sanwa Kagaku Kenkyusho, Japan Diabetes Foundation and Ono Pharmaceutical.
Acknowledgments
The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development and have approved the final version of the review, which reflects their interpretation and conclusions. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Jennifer Edwards, MB, BS, of Envision Scientific Solutions, during the preparation of this review. Boehringer Ingelheim was given the opportunity to check the data used in the manuscript for factual accuracy only.
J Diabetes Investig 2017; 8: 19–28
References
- 1. International Diabetes Federation . IDF Diabetes Atlas, 6th edn, 2014 Update. Brussels, Belgium. 2014. Available from: http://www.idf.org/sites/default/files/EN_6E_Atlas_Full_0.pdf. Accessed August 20, 2015.
- 2. Inzucchi SE, Bergenstal RM, Buse JB, et al Management of hyperglycemia in type 2 diabetes, 2015: A patient‐centered approach: Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38: 140–149. [DOI] [PubMed] [Google Scholar]
- 3. Garber AJ, Abrahamson MJ, Barzilay JI, et al AACE/ACE comprehensive diabetes management algorithm 2015. Endocr Pract 2015; 21: 438–447. [DOI] [PubMed] [Google Scholar]
- 4. Chahal H, Chowdhury TA. Gliptins: A new class of oral hypoglycaemic agent. QJM 2007; 100: 671–677. [DOI] [PubMed] [Google Scholar]
- 5. Deacon CF, Nauck MA, Toft‐Nielsen M, et al Both subcutaneously and intravenously administered glucagon‐like peptide I are rapidly degraded from the NH2‐terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44: 1126–1131. [DOI] [PubMed] [Google Scholar]
- 6. Scheen AJ. DPP‐4 inhibitors in the management of type 2 diabetes: A critical review of head‐to‐head trials. Diabetes Metab 2012; 38: 89–101. [DOI] [PubMed] [Google Scholar]
- 7. Boehringer Ingelheim Pharmaceuticals . Highlights of prescribing information. Tradjenta (linagliptin) tablets. 2015; Available from: http://bidocs.boehringer-ingelheim.com/BIWebAccess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing+Information/PIs/Tradjenta/Tradjenta.pdf. Accessed August 20, 2015.
- 8. Barnett AH, Patel S, Harper R, et al Linagliptin monotherapy in type 2 diabetes patients for whom metformin is inappropriate: An 18‐week randomized, double‐blind, placebo‐controlled phase III trial with a 34‐week active‐controlled extension. Diabetes Obes Metab 2012; 14: 1145–1154. [DOI] [PubMed] [Google Scholar]
- 9. Del Prato S, Barnett AH, Huisman H, et al Effect of linagliptin monotherapy on glycaemic control and markers of beta‐cell function in patients with inadequately controlled type 2 diabetes: A randomized controlled trial. Diabetes Obes Metab 2011; 13: 258–267. [DOI] [PubMed] [Google Scholar]
- 10. Kawamori R, Inagaki N, Araki E, et al Linagliptin monotherapy provides superior glycaemic control versus placebo or voglibose with comparable safety in Japanese patients with type 2 diabetes: A randomized, placebo and active comparator‐controlled, double‐blind study. Diabetes Obes Metab 2012; 14: 348–357. [DOI] [PubMed] [Google Scholar]
- 11. Gomis R, Espadero RM, Jones R, et al Efficacy and safety of initial combination therapy with linagliptin and pioglitazone in patients with inadequately controlled type 2 diabetes: A randomized, double‐blind, placebo‐controlled study. Diabetes Obes Metab 2011; 13: 653–661. [DOI] [PubMed] [Google Scholar]
- 12. Haak T, Meinicke T, Jones R, et al Initial combination of linagliptin and metformin improves glycaemic control in type 2 diabetes: A randomized, double‐blind, placebo‐controlled study. Diabetes Obes Metab 2012; 14: 565–574. [DOI] [PubMed] [Google Scholar]
- 13. Forst T, Uhlig‐Laske B, Ring A, et al Linagliptin (BI 1356), a potent and selective DPP‐4 inhibitor, is safe and efficacious in combination with metformin in patients with inadequately controlled Type 2 diabetes. Diabet Med 2010; 27: 1409–1419. [DOI] [PubMed] [Google Scholar]
- 14. Taskinen MR, Rosenstock J, Tamminen I, et al Safety and efficacy of linagliptin as add‐on therapy to metformin in patients with type 2 diabetes: A randomized, double‐blind, placebo‐controlled study. Diabetes Obes Metab 2011; 13: 65–74. [DOI] [PubMed] [Google Scholar]
- 15. Gallwitz B, Rosenstock J, Rauch T, et al 2‐year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: A randomised, double‐blind, non‐inferiority trial. Lancet 2012; 380: 475–483. [DOI] [PubMed] [Google Scholar]
- 16. Ross SA, Rafeiro E, Meinicke T, et al Efficacy and safety of linagliptin 2.5 mg twice daily versus 5 mg once daily in patients with type 2 diabetes inadequately controlled on metformin: A randomised, double‐blind, placebo‐controlled trial. Curr Med Res Opin 2012; 28: 1465–1474. [DOI] [PubMed] [Google Scholar]
- 17. Lewin AJ, Arvay L, Liu D, et al Efficacy and tolerability of linagliptin added to a sulfonylurea regimen in patients with inadequately controlled type 2 diabetes mellitus: An 18‐week, multicenter, randomized, double‐blind, placebo‐controlled trial. Clin Ther 2012; 34: 1909–1919 e1915. [DOI] [PubMed] [Google Scholar]
- 18. Owens DR, Swallow R, Dugi KA, et al Efficacy and safety of linagliptin in persons with type 2 diabetes inadequately controlled by a combination of metformin and sulphonylurea: A 24‐week randomized study. Diabet Med 2011; 28: 1352–1361. [DOI] [PubMed] [Google Scholar]
- 19. Lehrke M, Marx N, Patel S, et al Safety and tolerability of linagliptin in patients with type 2 diabetes: A comprehensive pooled analysis of 22 placebo‐controlled studies. Clin Ther 2014; 36: 1130–1146. [DOI] [PubMed] [Google Scholar]
- 20. Deacon CF, Holst JJ. Linagliptin, a xanthine‐based dipeptidyl peptidase‐4 inhibitor with an unusual profile for the treatment of type 2 diabetes. Expert Opin Investig Drugs 2010; 19: 133–140. [DOI] [PubMed] [Google Scholar]
- 21. Eckhardt M, Langkopf E, Mark M, et al 8‐(3‐(R)‐aminopiperidin‐1‐yl)‐7‐but‐2‐ynyl‐3‐methyl‐1‐(4‐methyl‐quinazolin‐2‐ylmethyl)‐3,7‐dihydropurine‐2,6‐dione (BI 1356), a highly potent, selective, long‐acting, and orally bioavailable DPP‐4 inhibitor for the treatment of type 2 diabetes. J Med Chem 2007; 50: 6450–6453. [DOI] [PubMed] [Google Scholar]
- 22. Thomas L, Eckhardt M, Langkopf E, et al (R)‐8‐(3‐amino‐piperidin‐1‐yl)‐7‐but‐2‐ynyl‐3‐methyl‐1‐(4‐methyl‐quinazolin‐2‐ylmethyl)‐3,7‐dihydro‐purine‐2,6‐dione (BI 1356), a novel xanthine‐based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase‐4 inhibitors. J Pharmacol Exp Ther 2008; 325: 175–182. [DOI] [PubMed] [Google Scholar]
- 23. Sarashina A, Sesoko S, Nakashima M, et al Linagliptin, a dipeptidyl peptidase‐4 inhibitor in development for the treatment of type 2 diabetes mellitus: A Phase I, randomized, double‐blind, placebo‐controlled trial of single and multiple escalating doses in healthy adult male Japanese subjects. Clin Ther 2010; 32: 1188–1204. [DOI] [PubMed] [Google Scholar]
- 24. Hüttner S, Graefe‐Mody EU, Withopf B, et al Safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of BI 1356, an inhibitor of dipeptidyl peptidase 4, in healthy male volunteers. J Clin Pharmacol 2008; 48: 1171–1178. [DOI] [PubMed] [Google Scholar]
- 25. Heise T, Graefe‐Mody EU, Hüttner S, et al Pharmacokinetics, pharmacodynamics and tolerability of multiple oral doses of linagliptin, a dipeptidyl peptidase‐4 inhibitor in male type 2 diabetes patients. Diabetes Obes Metab 2009; 11: 786–794. [DOI] [PubMed] [Google Scholar]
- 26. Horie Y, Kanada S, Watada H, et al Pharmacokinetic, pharmacodynamic, and tolerability profiles of the dipeptidyl peptidase‐4 inhibitor linagliptin: A 4‐week multicenter, randomized, double‐blind, placebo‐controlled phase IIa study in Japanese type 2 diabetes patients. Clin Ther 2011; 33: 973–989. [DOI] [PubMed] [Google Scholar]
- 27. Forst T, Uhlig‐Laske B, Ring A, et al The oral DPP‐4 inhibitor linagliptin significantly lowers HbA1c after 4 weeks of treatment in patients with type 2 diabetes mellitus. Diabetes Obes Metab 2011; 13: 542–550. [DOI] [PubMed] [Google Scholar]
- 28. European Medicines Agency . Trajenta (linagliptin). Summary of product characteristics. 2013; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002110/human_med_001482.jsp&mid=WC0b01ac058001d124. Accessed July 8 2015.
- 29. Retlich S, Duval V, Ring A, et al Pharmacokinetics and pharmacodynamics of single rising intravenous doses (0.5 mg‐10 mg) and determination of absolute bioavailability of the dipeptidyl peptidase‐4 inhibitor linagliptin (BI 1356) in healthy male subjects. Clin Pharmacokinet 2010; 49: 829–840. [DOI] [PubMed] [Google Scholar]
- 30. Graefe‐Mody U, Giessmann T, Ring A, et al A randomized, open‐label, crossover study evaluating the effect of food on the relative bioavailability of linagliptin in healthy subjects. Clin Ther 2011; 33: 1096–1103. [DOI] [PubMed] [Google Scholar]
- 31. Fuchs H, Tillement JP, Urien S, et al Concentration‐dependent plasma protein binding of the novel dipeptidyl peptidase 4 inhibitor BI 1356 due to saturable binding to its target in plasma of mice, rats and humans. J Pharm Pharmacol 2009; 61: 55–62. [DOI] [PubMed] [Google Scholar]
- 32. Fuchs H, Binder R, Greischel A. Tissue distribution of the novel DPP‐4 inhibitor BI 1356 is dominated by saturable binding to its target in rats. Biopharm Drug Dispos 2009; 30: 229–240. [DOI] [PubMed] [Google Scholar]
- 33. Retlich S, Withopf B, Greischel A, et al Binding to dipeptidyl peptidase‐4 determines the disposition of linagliptin (BI 1356)–investigations in DPP‐4 deficient and wildtype rats. Biopharm Drug Dispos 2009; 30: 422–436. [DOI] [PubMed] [Google Scholar]
- 34. Blech S, Ludwig‐Schwellinger E, Grafe‐Mody EU, et al The metabolism and disposition of the oral dipeptidyl peptidase‐4 inhibitor, linagliptin, in humans. Drug Metab Dispos 2010; 38: 667–678. [DOI] [PubMed] [Google Scholar]
- 35. Fuchs H, Runge F, Held HD. Excretion of the dipeptidyl peptidase‐4 inhibitor linagliptin in rats is primarily by biliary excretion and P‐gp‐mediated efflux. Eur J Pharm Sci 2012; 45: 533–538. [DOI] [PubMed] [Google Scholar]
- 36. Deacon CF. Therapeutic strategies based on glucagon‐like peptide 1. Diabetes 2004; 53: 2181–2189. [DOI] [PubMed] [Google Scholar]
- 37. Roy RSWJ, Eiermann G, Lyons K, et al Plasma DPP‐4 inhibition by sitagliptin and other DPP‐4 inhibitors correlates with and predicts glucose lowering efficacy. Diabetes 2009; 58: A612. [Google Scholar]
- 38. Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology 2007; 132: 2131–2157. [DOI] [PubMed] [Google Scholar]
- 39. Dupre J, Ross SA, Watson D, et al Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 1973; 37: 826–828. [DOI] [PubMed] [Google Scholar]
- 40. Kreymann B, Williams G, Ghatei MA, et al Glucagon‐like peptide‐1 7‐36: A physiological incretin in man. Lancet 1987; 2: 1300–1304. [DOI] [PubMed] [Google Scholar]
- 41. Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl‐peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon‐like peptide‐1(7‐36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 1993; 214: 829–835. [DOI] [PubMed] [Google Scholar]
- 42. Nauck MA, Bartels E, Orskov C, et al Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon‐like peptide‐1‐(7‐36) amide infused at near‐physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab 1993; 76: 912–917. [DOI] [PubMed] [Google Scholar]
- 43. Schmidt WE, Siegel EG, Creutzfeldt W. Glucagon‐like peptide‐1 but not glucagon‐like peptide‐2 stimulates insulin release from isolated rat pancreatic islets. Diabetologia 1985; 28: 704–707. [DOI] [PubMed] [Google Scholar]
- 44. Gallwitz B, Rosenstock J, Patel S, et al Regardless of the degree of glycaemic control, linagliptin has lower hypoglycaemia risk than all doses of glimepiride, at all time points, over the course of a 2‐year trial. Diabetes Obes Metab 2015; 17: 276–284. [DOI] [PubMed] [Google Scholar]
- 45. Koro CE, Lee BH, Bowlin SJ. Antidiabetic medication use and prevalence of chronic kidney disease among patients with type 2 diabetes mellitus in the United States. Clin Ther 2009; 31: 2608–2617. [DOI] [PubMed] [Google Scholar]
- 46. Meyers JL, Candrilli SD, Kovacs B. Type 2 diabetes mellitus and renal impairment in a large outpatient electronic medical records database: Rates of diagnosis and antihyperglycemic medication dose adjustment. Postgrad Med 2011; 123: 133–143. [DOI] [PubMed] [Google Scholar]
- 47. Plantinga LC, Crews DC, Coresh J, et al Prevalence of chronic kidney disease in US adults with undiagnosed diabetes or prediabetes. Clin J Am Soc Nephrol 2010; 5: 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Graefe‐Mody U, Friedrich C, Port A, et al Effect of renal impairment on the pharmacokinetics of the dipeptidyl peptidase‐4 inhibitor linagliptin. Diabetes Obes Metab 2011; 13: 939–946. [DOI] [PubMed] [Google Scholar]
- 49. Friedrich C, Emser A, Woerle HJ, et al Renal impairment has no clinically relevant effect on the long‐term exposure of linagliptin in patients with type 2 diabetes. Am J Ther 2013; 20: 618–621. [DOI] [PubMed] [Google Scholar]
- 50. Cusi K. Nonalcoholic fatty liver disease in type 2 diabetes mellitus. Curr Opin Endocrinol Diabetes Obes 2009; 16: 141–149. [DOI] [PubMed] [Google Scholar]
- 51. Garcia‐Compean D, Jaquez‐Quintana JO, Gonzalez‐Gonzalez JA, et al Liver cirrhosis and diabetes: Risk factors, pathophysiology, clinical implications and management. World J Gastroenterol 2009; 15: 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Graefe‐Mody U, Rose P, Retlich S, et al Pharmacokinetics of linagliptin in subjects with hepatic impairment. Br J Clin Pharmacol 2012; 74: 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Scheen AJ. Linagliptin plus metformin: A pharmacokinetic and pharmacodynamic evaluation. Expert Opin Drug Metab Toxicol 2013; 9: 363–377. [DOI] [PubMed] [Google Scholar]
- 54. Graefe‐Mody EU, Jungnik A, Ring A, et al Evaluation of the pharmacokinetic interaction between the dipeptidyl peptidase‐4 inhibitor linagliptin and pioglitazone in healthy volunteers. Int J Clin Pharmacol Ther 2010; 48: 652–661. [DOI] [PubMed] [Google Scholar]
- 55. Graefe‐Mody U, Rose P, Ring A, et al Assessment of the pharmacokinetic interaction between the novel DPP‐4 inhibitor linagliptin and a sulfonylurea, glyburide, in healthy subjects. Drug Metab Pharmacokinet 2011; 26: 123–129. [DOI] [PubMed] [Google Scholar]
- 56. Matheny CJ, Lamb MW, Brouwer KR, et al Pharmacokinetic and pharmacodynamic implications of P‐glycoprotein modulation. Pharmacotherapy 2001; 21: 778–796. [DOI] [PubMed] [Google Scholar]
- 57. Chan JC, Malik V, Jia W, et al Diabetes in Asia: Epidemiology, risk factors, and pathophysiology. JAMA 2009; 301: 2129–2140. [DOI] [PubMed] [Google Scholar]
- 58. Ramachandran A, Snehalatha C, Vijay V. Low risk threshold for acquired diabetogenic factors in Asian Indians. Diabetes Res Clin Pract 2004; 65: 189–195. [DOI] [PubMed] [Google Scholar]
- 59. Ma RC, Chan JC. Type 2 diabetes in East Asians: Similarities and differences with populations in Europe and the United States. Ann N Y Acad Sci 2013; 1281: 64–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takeuchi M, Okamoto K, Takagi T, et al Ethnic difference in inter‐East Asian subjects with normal glucose tolerance and impaired glucose regulation: A systematic review and meta‐analysis focusing on fasting serum insulin. Diabetes Res Clin Pract 2008; 82: 383–390. [DOI] [PubMed] [Google Scholar]
- 61. Ito H, Abe M, Antoku S, et al Comparison of the antidiabetic effects of linagliptin among groups with a normal renal function and a mild or severe renal impairment – retrospective observation study of Japanese patients with type 2 diabetes mellitus. Expert Opin Pharmacother 2015; 16: 289–296. [DOI] [PubMed] [Google Scholar]
- 62. Friedrich C, Shi X, Zeng P, et al Pharmacokinetics of single and multiple oral doses of 5 mg linagliptin in healthy Chinese volunteers. Int J Clin Pharmacol Ther 2012; 50: 889–895. [DOI] [PubMed] [Google Scholar]
- 63. Pichereau S, Zhao X, Cui Y, et al Relative bioavailability study of linagliptin/ metformin tablets in healthy Chinese subjects. Int J Clin Pharmacol Ther 2015; 53: 582–593. [DOI] [PubMed] [Google Scholar]
- 64. Friedrich C, Jungnik A, Retlich S, et al Bioequivalence of linagliptin 5 mg once daily and 2.5 mg twice daily: Pharmacokinetics and pharmacodynamics in an open‐label crossover trial. Drug Res (Stuttg) 2014; 64: 269–275. [DOI] [PubMed] [Google Scholar]
- 65. Buschke S, Ring A, Friedrich C, et al Linagliptin fixed‐dose combination with metformin is bioequivalent to co‐administration of linagliptin and metformin as individual tablets. Int J Clin Pharmacol Ther 2014; 52: 537–548. [DOI] [PubMed] [Google Scholar]
- 66. Retlich S, Duval V, Graefe‐Mody U, et al Population pharmacokinetics and pharmacodynamics of linagliptin in patients with type 2 diabetes mellitus. Clin Pharmacokinet 2015; 54: 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deacon CF. Dipeptidyl peptidase‐4 inhibitors in the treatment of type 2 diabetes: A comparative review. Diabetes Obes Metab 2011; 13: 7–18. [DOI] [PubMed] [Google Scholar]
- 68. Scheen AJ. Pharmacokinetics of dipeptidylpeptidase‐4 inhibitors. Diabetes Obes Metab 2010; 12: 648–658. [DOI] [PubMed] [Google Scholar]
- 69. Merck and Co., Inc. Januvia . Highlights of prescribing information. 2015; Available from: http://www.merck.com/product/usa/pi_circulars/j/januvia/januvia_pi.pdf. Accessed September 8, 2015.
- 70. Galvus® (Vildagliptin) Prescribing information. 2014; Available from: http://www.novartis.com.au/PI_PDF/gal.pdf. Accessed September 8, 2015.
- 71. AstraZeneca Pharmaceuticals LP . Onglyza® (saxagliptin). Highlights of prescribing information. 2015; Available from: http://www.azpicentral.com/onglyza/pi_onglyza.pdf#page=1. Accessed September 8, 2015.
- 72. Takeda Pharmaceuticals America, Inc. Nesina (alogliptin) . Highlights of prescribing information. 2013; Available from: http://general.takedapharm.com/content/file.aspx?FileTypeCode=KAZANOPI&cacheRandomizer=24332267-a1d1-46e5-b27a-bbe91544d02e. Accessed September 8, 2015.
- 73. Kim D, Wang L, Beconi M, et al (2R)‐4‐oxo‐4‐[3‐(trifluoromethyl)‐5,6‐dihydro[1,2,4]triazolo[4,3‐a]pyrazin‐7(8H)‐yl]‐1‐(2,4,5‐trifluorophenyl)butan‐2‐amine: A potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem 2005; 48: 141–151. [DOI] [PubMed] [Google Scholar]
- 74. Burkey BF, Hoffmann PK, Hassiepen U, et al Adverse effects of dipeptidyl peptidases 8 and 9 inhibition in rodents revisited. Diabetes Obes Metab 2008; 10: 1057–1061. [DOI] [PubMed] [Google Scholar]
- 75. Kirby MSDC, Wang A, Weigelt C, et al In vitro enzymologic characteristics of saxagliptin, a highly potent and selective DPP4 inhibitor with “slow binding” characteristic. (Abstract). Clin Chem Lab Med 2008; 46: A29. [Google Scholar]
- 76. Feng J, Zhang Z, Wallace MB, et al Discovery of alogliptin: A potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J Med Chem 2007; 50: 2297–2300. [DOI] [PubMed] [Google Scholar]
- 77. Green JB, Bethel MA, Armstrong PW, et al Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 373: 232–242. [DOI] [PubMed] [Google Scholar]
- 78. Scirica BM, Bhatt DL, Braunwald E, et al Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 79. Zannad F, Cannon CP, Cushman WC, et al Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: A multicentre, randomised, double‐blind trial. Lancet 2015; 385: 2067–2076. [DOI] [PubMed] [Google Scholar]
- 80. Vipidia . Summary of product characteristics. 2013; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002182/WC500152271.pdf. Accessed November 10, 2015.
- 81. Onglyza . Summary of product characteristics. 2014; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001039/WC500044316.pdf. Accessed November 10, 2015.
- 82. Januvia . Summary of product characteristics. 2015; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000722/WC500039054.pdf. Accessed November 10, 2015.
- 83. Rosenstock J, Marx N, Neubacher D, et al Cardiovascular safety of linagliptin in type 2 diabetes: A comprehensive patient‐level pooled analysis of prospectively adjudicated cardiovascular events. Cardiovasc Diabetol 2015; 14: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Forst T, Pfützner A. Linagliptin, a dipeptidyl peptidase‐4 inhibitor with a unique pharmacological profile, and efficacy in a broad range of patients with type 2 diabetes. Expert Opin Pharmacother 2012; 13: 101–110. [DOI] [PubMed] [Google Scholar]