

Summary
Optimal T cell activation is vital for the successful resolution of microbial infections. Programmed death‐1 (PD‐1) is a key immune check‐point receptor expressed by activated T cells. Aberrant/excessive inhibition mediated by PD‐1 may impair host immunity to Mycobacterium tuberculosis infection, leading to disseminated disease such as miliary tuberculosis (MTB). PD‐1 mediated inhibition of T cells in pulmonary tuberculosis and TB pleurisy is reported. However, their role in MTB, particularly at the pathological site, remains to be addressed. The objective of this study was to investigate the role of PD‐1–PD‐ligand 1 (PD‐L1) in T cell responses at the pathological site from patients of TB pleurisy and MTB as clinical models of contained and disseminated forms of tuberculosis, respectively. We examined the expression and function of PD‐1 and its ligands (PD‐L1–PD‐L2) on host immune cells among tuberculosis patients. Bronchoalveolar lavage‐derived CD3 T cells in MTB expressed PD‐1 (54·2 ± 27·4%, P ≥ 0·0009) with significantly higher PD‐1 ligand‐positive T cells (PD‐L1: 19·8 ± 11·8%; P ≥ 0·019, PD‐L2: 12·6 ± 6·2%; P ≥ 0·023), CD19+ B cells (PD‐L1: 14·4 ± 10·4%; P ≥ 0·042, PD‐L2: 2·6 ± 1·43%; not significant) and CD14+ monocytes (PD‐L1: 40·2 ± 20·1%; P ≥ 0·047, PD‐L2: 22·4 ± 15·6%; P ≥ 0·032) compared with peripheral blood (PB) of MTB and healthy controls. The expression of PD‐1 was associated with a diminished number of cells producing effector cytokines interferon (IFN)‐γ, tumour necrosis factor (TNF)‐α, interleukin (IL)−2 and elevated apoptosis. Locally accumulated T cells were predominantly PD‐1+–PD‐L1+, and blocking this pathway restores the protective T cell response. We conclude that M. tuberculosis exploits the PD‐1 pathway to evade the host immune response by altering the T helper type 1 (Th1) and Th2 balance at the pathological site of MTB, thereby favouring disease dissemination.
Keywords: bronchoalveolar lavage, miliary tuberculosis, programmed death‐1
Introduction
Protective immunity to mycobacteria involves T helper type 1 (Th1) cells, which are under the control of other T cell subsets 1, 2, 3, 4, 5, 6, 7. Polarized Th1 cells produce interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α in response to mycobacterial antigens, which are critical for macrophage activation and control of bacterial replication 8. Cross‐talk between various T cell subsets and antigen‐presenting cells (APC) is important for induction of the T cell response. Indeed, the interaction of co‐stimulatory and co‐inhibitory receptors [e.g. CD28, cytotoxic T lymphocyte antigen‐4 (CTLA‐4) and programmed death‐1 (PD‐1)] expressed on T cells with their respective ligands (e.g. B7‐1/B7‐2 and PD‐L1/PD‐L2) on APC influences the quality and magnitude of the antigen‐reactive T cell response 9. Diverse mechanisms are known to suppress host immunity among pulmonary tuberculosis (PTB) patients 10, 11. Previously, we have shown that interleukin (IL)−10 mediates suppression of the T cell response by regulatory T cells (Treg) among miliary tuberculosis (MTB) patients 12. Recent studies indicate the importance of the PD‐1–PD‐L pathway in infection, autoimmunity and transplant rejection 13, 14. Several studies indicate that pathogenic microbes exploit the PD‐1–PD‐L pathway as a strategy for immune evasion and persistent infection. For example, PD‐1 is expressed highly on functionally impaired (exhausted) T cells in chronic infection with HIV 15, 16, 17, 18 and SIV 19, 20. Helicobacter pylori suppresses the T cell response by up‐regulating PD‐L1 expression on gastric epithelial cells that are thought to act as APC 21, 22. Recently, we have demonstrated that the co‐inhibitory receptor PD‐1 and the PD‐1 ligands are expressed on a higher percentage of peripheral blood mononuclear cells (PBMCs) of patients with active tuberculosis. Of particular interest, live infection with the M. tuberculosis H37Rv strain up‐regulates PD‐L1 expression selectively on monocytes 23. PD‐1‐blocking in vitro enhances IFN‐γ and IL‐2 production by M. tuberculosis‐specific T cells via regulating their apoptosis and proliferation. In‐vitro blockade of the PD‐1 signalling enhances cytokine production and proliferation by T and natural killer (NK) T cells on M. tuberculosis antigen stimulation 23, 24, 25, 26, suggesting that the PD‐1–PD‐L pathway may play an important role in selective suppression of the Th1 response at the pathological site in MTB. This may result in a Th2‐dominant effector immune response, thus causing bacillary dissemination characterized by haematogenous spread of the bacilli, thus giving rise to progressive and disseminated form(s) of diseases such as MTB.
Previously we have shown that inhibiting the PD‐1 pathway rescues M. tuberculosis‐specific IFN‐γ‐producing T cells from apoptosis in PTB patients, and the number of PD‐1‐expressing T cells decreased significantly during therapy and correlated inversely with the IFN‐γ dominant T cell response against M. tuberculosis 23. In this study, we evaluated the regulation and immune function of PD‐1–PD‐L1 pathways at the pathological site of MTB and tuberculosis pleural effusion (TB‐PE). Perhaps, for the first time, we have demonstrated a profound increase in PD‐1+ T cells and PD‐L1/PD‐L2‐expressing monocytes at the pathological site of MTB patients. Of note, we found that the PD‐1 pathway selectively inhibits M. tuberculosis‐specific TNF‐α, IFN‐γ and IL‐2‐producing multi‐functional T cells at the pathological site of MTB with greater magnitude than that of peripheral blood (PB) and TB‐PE cells.
Material and methods
Subjects
The study included 27 patients with TB [13 MTB (mean age 33 ± 12·6 years, range 17–52; 10 males and three females) and 14 TB‐PE (mean age 29·9 ± 11·2 years, range 18–50; 11 males and three females)] attending the All India Institute of Medical Sciences Hospital, New Delhi, India for treatment. Twenty three healthy controls (HCs) (mean age 27·53 ± 8·69, range 23–48 with 15 males and eight females) with no diagnosed ailments, normal blood parameters and chest radiographs were also recruited. Written informed consent was obtained from all study subjects. The Institutional Ethics Committee (Ref. no. A‐60:/25.07.2007, dated 28 August 2008) approved the study. Diagnosis of MTB was made according to the previously described criteria 12, 27. Details of the diagnostic work‐up are provided in Table 1. Diagnosis of patients with TB‐PE was based on the parameters shown in Table 1, with presence of fluid in chest radiograph (postero‐anterior view). All subjects were HIV‐negative. PB (8–12 ml) was collected from all the subjects. Pleural effusion fluid (PEF) was collected from patients with TB‐PE, whereas bronchoalveolar lavage (BAL) fluid was collected from patients with MTB as local disease site specimens (LDSS), as per our previously described protocol 12 and Supporting information, Fig. S1. None of the patients was on anti‐TB treatment at the time of enrolment into the study.
Table 1.
Demographic and Clinical Characteristics of 27 Indian Patients With Tuberculosis Pleural effusion and Miliary TB
| Tuberculosis pleuraleffusion (n = 14) | Miliary tuberculosis (n = 13) | |
|---|---|---|
| Demographic characteristics | ||
| Age (mean ± s.d.), range | (29·9 ± 11·2), 18–50 | (33·0 ± 12·6), 17–52 |
| Sex (M/F) | 11/3 | 10/3 |
| Ethnicity | Indian | Indian |
| Diagnostic characteristics | ||
| Smear for Mycobacterium tuberculosis | ||
| PEF | 3 | – |
| BAL | – | 6 |
| Pus (cold abscess) | – | 1 † |
| Culture for Mycobacterium tuberculosis | ||
| L‐J medium | ||
| PEF | 4 | – |
| BAL | – | 3 |
| BACTEC (460) | ||
| PEF | 3 | – |
| BAL | – | 2 † |
| PCR for Mycobacterium tuberculosis | ||
| PEF | 2 | – |
| BAL | – | 2 † |
| Bone marrow | – | – |
| Histopathological diagnosis* | ||
| Bone marrow | – | 1 † |
| Bronchial biopsy | – | – |
| Clinical and radiological responseto anti‐TB treatment | 2 | 2 |
*On histology, a compatible diagnosis of tuberculosis. †Together refers to 13 patients with miliary tuberculosis; of these, four were found to show Mycobacterium tuberculosis from more than one site(s). PCR = polymerase chain reaction; PEF = pleural fluid; BAL = bronchoalveolar lavage; L‐J medium = Lowenstein–Jensen medium; TB = tuberculosis; s.d. = standard deviation; M/F = male/female; BACTEC (460): from Becton Dickinson, Franklin Lakes, NJ, USA.
Cell isolation and flow cytometry
PBMCs were isolated from heparinized blood by Ficoll Hypaque gradient centrifugation and suspended in complete RPMI‐1640 (Caisson Laboratories, Logan, UT, USA). Cells were harvested from PEF and BAL by methods described previously 12. The viability of the cells was greater than 98% (Trypan blue dye exclusion). Cells were used for surface phenotyping using α‐CD3, α‐CD14, α‐CD19 (BD Biosciences, San Jose, CA, USA), α‐PD‐1, α‐PD‐L1 and α‐PD‐L2 antibodies (eBiosciences, San Diego, CA, USA), and were run on a BD fluorescence activated cell sorter (FACS)Calibur (BD Biosciences) and analysed subsequently by Flow‐Jo software (TreeStar Inc., Ashland, OR, USA).
In‐vitro cell culture and PD‐1–PD‐L1 blocking
Freshly isolated PBMCs were incubated with or without blocking antibodies against PD‐1 (5 μg/ml, J116; eBiosciences) using α‐PD‐L1, α‐PD‐L2 (5 μg/ml, MIH1; eBiosciences), as described previously 23, 24 and Supporting information, Fig. S2, in the presence or absence of M. tuberculosis (20 μg/ml, whole cell lysate) for 72 h and brefeldin A (GolgiPLUG 10 mg/ml; Sigma, St Louis, MO, USA) for last 12 h of culture. Purified mouse immunoglobulin (Ig)G1 (final concentration of 10 μg/ml; eBiosciences) was used as an isotype control. Cultured cells were stained with the LIVE/DEAD fixable dead cell stain kit (Invitrogen, Carlsbad, CA, USA). Cells were stained subsequently with α‐CD3, α‐CD4 (BD Biosciences) followed by intracellular detection of IFN‐γ, TNF‐α, IL‐2 and IL‐4 using α‐IFN‐γ, α‐TNF‐α, α‐IL‐2 and α‐IL‐4 antibodies (BD Biosciences). Fixation/permeabilization buffers (eBioscience) were used for intracellular staining. In a separate experiment, the impact of PD‐1 blocking on T cells apoptosis (annexin V) was measured as described elsewhere 24, 28. Briefly, for apoptosis study of T cells, annexin V‐fluorescein isothiocyanate (FITC) (cat. no. 556420; BD Biosciences) staining was performed before fixation at room temperature, as per the manufacturer's instructions. Finally, cells were stained with α‐CD3 (BD Biosciences). Propidium iodide was added 15 min prior to acquisition of the sample. Stained cells were then acquired on a BD FACSCanto II equipped with Diva software and analysed using Flow‐Jo software (TreeStar Inc.). The gating strategy in this study is shown in Supporting information, Fig. S3).
Statistical analysis
Statistical analysis was performed using the non‐parametric Mann–Whitney test for unpaired samples and the Wilcoxon rank‐sum test for paired samples. Values of P ≤ 0·05 were considered significant, but * denotes P ≤ 0·05 and ** denotes P ≤ 0·005.
Results
PD‐1 and its ligand(s) (PD‐L1 and PD‐L2) expression on T cells of TB‐PE and miliary TB patients
We evaluated the expression of PD‐1 receptor and its ligand(s) on T and B cells and monocytes, both in immune‐reactive TB‐PE and MTB patients (disseminated form of tuberculosis). Flow cytometry was performed on MNCs of PB, PEF and BAL with the aim of evaluating the relevance of PD‐1 and its ligand(s) in suppression of the host effector T cell response at the pathological site. The frequency of PD‐1+ T cells was significantly higher (54·2 ± 27·4 versus 15·6 ± 9·2, P = 0·0002) at the disease site of MTB patients (BAL) compared to their peripheral compartment and with PB of HCs (9·74 ± 4·3, Fig. 1a,b). However, a similar enrichment of PD‐1+ T cells in the PE of TB‐PE patients relative to their PB was not observed, although it was increased significantly (28·8 ± 13·1 versus 18·2 ± 8·2, P = 0·0039). Our results concerning PD‐1 ligand expression on T cells show a significantly higher frequency of peripheral PD‐1 ligand(s)‐expressing T cells among both types of TB patients compared to that of HCs (Fig. 1c,d). We observed a substantial increase in the frequencies of these cells both in the PB and LDSS of TB‐PE and MTB patients. The frequency of PD‐1+ T cells was increased significantly in the PB of both TB‐PE and MTB patients compared to that of HCs, suggesting the possible role of PD‐1 expression of T cells in tuberculosis patients 23. However, their enrichment was profound in the BAL of MTB patients compared to that of PEF of TB‐PE patients. This further suggests the relevance of PD‐1 expression of T cells at the LDSS of MTB patients. A similar observation was noted with respect to PD‐L1 and PD‐L2 expression of the locally accumulated T cells among MTB patients. No difference was observed between ligand(s)‐expressing T cells in the MNCs of PB and PE of TB‐PE patients (Fig. 1c,d). Overall, our data highlight a possible role of the PD‐1–PD‐L1/2 pathway in the local immune response in MTB. Among all samples, the PD‐1 ligand(s)‐expressing T cells are significantly high in the MNCs from BAL. This is indicative of over‐expression of inhibitory receptor and ligand(s) on the surface of locally recruited T cells at the pathological site of MTB, and possibly suppresses the locally recruited T cells.
Figure 1.
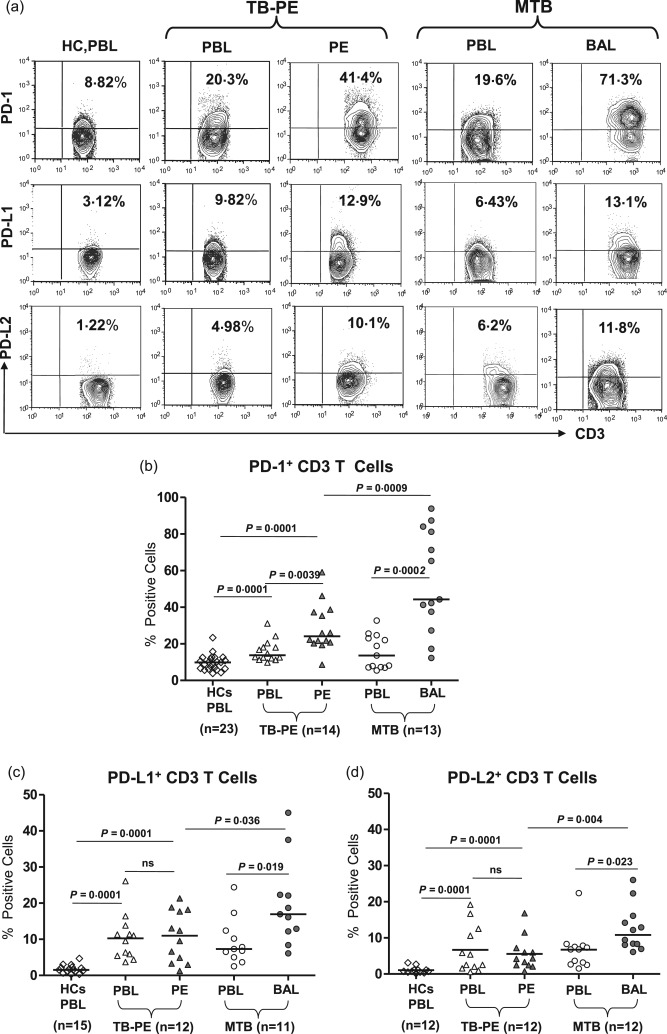
Profound increase in the frequency of programmed death‐1 (PD‐1) and its ligands (PD‐L1 and PD‐L2) expressing T cells from miliary tuberculosis (MTB): PD‐1 and its ligand expression was examined on total CD3+ T cells derived from healthy controls (HC) and various forms of tuberculosis (TB) patients [TB‐pleural effusion (PE) and MTB]. (a,b) Representative fluorescence activated cell sorter (FACS) plots show expression of PD‐1, PD‐L1 and PD‐L2 on gated CD3+ T cells of HC and in paired samples of TB‐PE [peripheral blood (PB) versus PE] and MTB [PB versus bronchoalveolar lavage (BAL)] patients. (b–d) Scatter diagram shows percentage of PD‐1‐, PD‐L1‐ and PD‐L2‐expressing CD3+ cells in paired samples of TB‐PE (PB versus PE) and MTB (PB versus BAL) patients. Analysis shows a significant increase in the frequency of PD‐1‐, PD‐L1‐ and PD‐L2‐positive T cells among patients compared to that of HC. Interestingly, the percentage of PD‐1 and its ligands expressing T cells from BAL of MTB were significantly higher than the PE of TB‐PE patients and the autologous mononuclear cells (MNC) of MTB. Each symbol represents a single individual. Horizontal line depicts the median value. Statistical analysis was based on the non‐parametric Mann–Whitney unpaired t‐test.
PD‐1 ligand(s) (PD‐L1 and PD‐L2) expression on CD14+ monocytes of TB‐PE and miliary TB patients
We analysed PD‐1 ligand(s) expression on CD14+ monocytes obtained from the PB and LDSS (PE and BAL) of TB‐PE and MTB patients, respectively, and compared this with the frequencies of PD‐1 ligand(s)‐expressing CD14+ cells of HCs. We found an almost ninefold increase in the frequency of PD‐L1‐expressing CD14+ monocytes in patients’ PB as well as PE of TB‐PE (PB; 37·7 ± 18·2, PE; 28·5 ± 9·7 versus 3 ± 2·3, P = 0·0001) and PB and BAL of MTB patients (PB; 28·5 ± 15·5, BAL; 40·2 ± 20·1 versus 3 ± 2·3, P = 0·0023). A similar observation was noted for PD‐L2, although the magnitude of expression was much lower (Fig. 2a,b and Supporting information, Fig. S4). No significant differences were observed in the frequencies of PD‐L2‐expressing CD14+ cells within all forms of tuberculosis patients, even though these cells were significantly higher than that of HCs.
Figure 2.
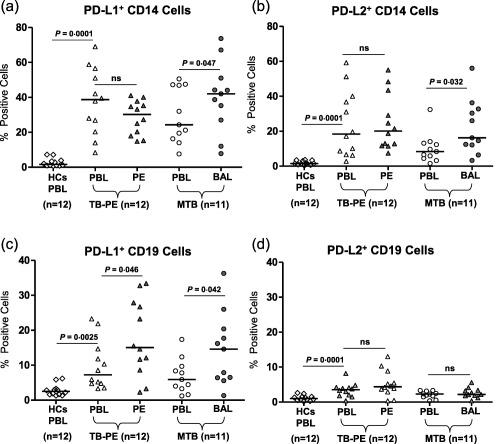
Comparative analysis of programmed death‐1 (PD‐1) ligands (PD‐L1 and PD‐L2) on CD14+ and CD19+ B cells in various forms of tuberculosis (TB) patients: PD‐1 ligand expression was examined on total CD14+ monocytes and CD19+ B cells in freshly isolated peripheral blood mononuclear cells (PBMCs) from healthy controls (HCs) and paired samples of tuberculosis‐pleural effusion (TB‐PE) and miliary tuberculosis (MTB) patients. Representative fluorescence activated cell sorter (FACS) plots show expression of PD‐L1 and PD‐L2 in CD14+ monocytes and CD19+ B cells among these patients and healthy controls (HCs) shown in Supporting information, Fig. S4. (a,b) Scatter diagram shows the percentage of PD‐L1‐ and PD‐L2‐expressing CD14+ monocyte cells (c,d) on CD19+ B cells in these study subjects. Analysis shows a significant increase in frequencies of PD‐1 ligands in paired samples of TB‐PE [peripheral blood (PB) versus PE] and MTB [PB versus bronchoalveolar lavage (BAL)] patients. Interestingly, the percentages of PD‐1 ligands expressing CD14 cells from BAL of MTB were significantly higher than the PB of HCs and the autologous mononuclear cells (MNC) of MTB. No significant difference was found in the paired sample of TB‐PE. Each symbol represents a single individual. Horizontal line depicts the median value. Statistical analysis was based on the non‐parametric Mann–Whitney unpaired t‐test.
PD‐1 ligand(s) (PD‐L1 and PD‐L2) expression on CD19+ B cells of TB‐PE and miliary TB patients
We analysed PD‐1 ligand(s) on CD19+ B cells among patients and in comparison with the frequency of HCs. We found an increased frequency of PD‐L1‐expressing B cells in the PB as well as MNCs of TB‐PE and MTB patients compared to HCs PB. PD‐L1+ B cells were increased significantly in PE of TB‐PE and BAL of MTB patients (Fig. 2c,d and Supporting information, Fig. S4), suggesting the possible role of PD‐L1‐expressing B cells on the accumulated effector T cell functions. Such interaction may be important in suppressing the local immune response, both in TB‐PE and MTB patients. However, due to much higher expression of PD‐1 on the T cells accumulated in the BAL of MTB, PD‐1–PD‐L1 interaction‐mediated suppression of the local immune response may be critical in the MTB pathological site. No significant differences were observed in the frequencies of PD‐L2‐expressing CD19+ B cells within all forms of tuberculosis patients (Fig. 2d).
Blocking PD‐1–PD‐L1 interaction augments M. tuberculosis‐specific IFN‐γ, TNF‐α and IL‐2 production by CD3+ T cells from TB patients
Previously, we have demonstrated that patients with TB‐PE had significantly higher IFN‐γ levels in their PF compared with that of PB of the same patients, thus exhibiting localization of a predominantly Th1‐type immunity in the PF. Conversely, patients with MTB had higher IFN‐γ levels in the PB compared to their BAL fluid, suggesting that the cytokine profile at the disease site is skewed towards a Th2‐like bias 29. To verify possible PD‐1‐mediated inhibition of Th1‐response antigen‐specific CD4+ T cells of the local disease site of MTB, we compared the ability of CD4+ T cells to produce IFN‐γ, TNF‐α, IL‐2 and IL‐4 in the presence or absence of PD‐1 blocking.
In‐vitro blocking of PD‐1 and PD‐L1/2 increased the frequency of M. tuberculosis‐specific IFN‐γ‐producing T cells both in PB as well as in LDSS, including TB‐PE and MTB. In general, blocking the PD‐1 pathway rescued M. tuberculosis‐specific IFN‐γ producers both in TB‐PE and MTB, substantiating our previous observation that inhibiting the PD‐1 pathway restores antigen‐specific IFN‐γ‐producing T and NK T cells among PTB patients 23, 24. However, the increase of antigen‐specific IFN‐γ‐producing T cells following PD‐1 blocking was maximal in BAL‐derived T cells from MTB patients (more than sixfold, or 600% rescue in BAL compared to 30% rescue in PB of MTB) compared to PB of TB‐PE and HCs, suggesting the highest degree of PD‐1‐mediated suppression of IFN‐γ‐producing effector T cells in the pathological site of MTB (Fig. 3a,b). Conversely, in the case of TB‐PE, the increase in antigen‐specific IFN‐γ+ producers in PE‐derived T cells after blocking the PD‐1–PD‐L1 pathway was just double, or 100% (Fig. 3a,b). Interestingly, PD‐1 pathway‐blocking failed to show any significant impact on the frequency of M. tuberculosis‐specific IL‐4‐producing T cells derived from both PB and LDSS of TB‐PE and MTB patients, respectively (Fig. 3c,d). Taken together, our results clearly suggest maximum inhibition of IFN‐γ producers in the pathological site sparing the IL‐4 producers, thus skewing the local immune response of MTB patients towards IL‐4 dominance. The recently appreciated important role of polyfunctional T cells producing multiple cytokines at single‐cell level prompted us to determine if PD‐1 blocked rescue of the antigen‐specific polyfunctional T cells in the BAL of MTB patients. We observed that, along with the increase in the single cytokine producers, PD‐1 inhibition increased the frequency of M. tuberculosis‐specific single or dual cytokine producers (IFN‐γ+TNF‐α+ and IL‐2+IFN‐γ+) and rescue of these polyfunctional T cells was significantly higher in the BAL of MTB patients (Fig. 4a,b,c). In addition to dual cytokine producers, PD‐1 blocking profoundly enhances the frequency of single TNF‐α‐producing CD4 T cells (approximately 600%) followed by single IFN‐γ+ CD4 T cells (approximately 400%) in BAL of MTB compared to its PB counterpart. This indicates that, in addition to suppressing the single IFN‐γ and TNF‐α producers, PD‐1 inhibits the polyfunctional T cells significantly more in the pathological site of MTB, which is believed to be critical for host immunity.
Figure 3.
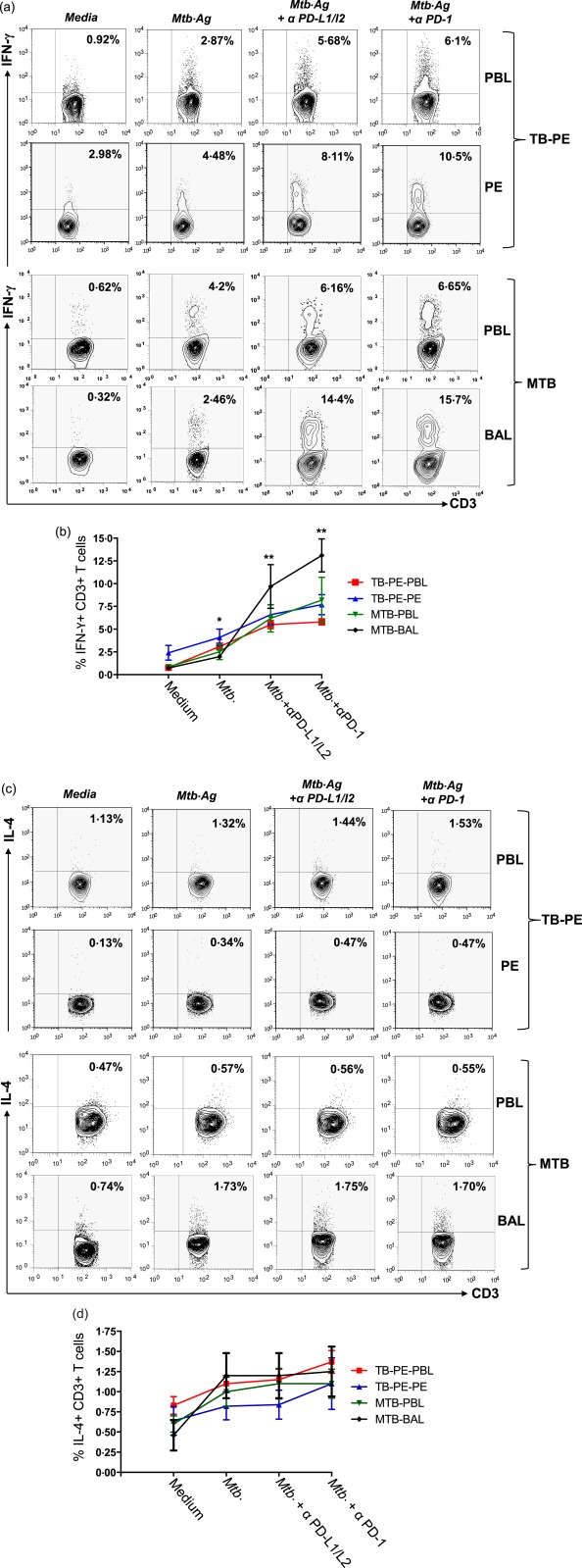
Mycobacterium tuberculosis stimulation combined with programmed death‐1 (PD‐1)–PD‐ligand (PD‐L2) blockade efficiently increases the frequency of T helper type 1 (Th‐1) cells, and sparing Th‐2 T cells. Mononuclear cells from peripheral blood (PB) and pleural fluids of tuberculosis‐pleural effusion (TB‐PE) patients (n = 4); PB and bronchoalveolar lavage of miliary tuberculosis (MTB) patients (n = 4) were blocked with α‐PD‐1 alone or α‐PD‐L1 and α‐PD‐L2 in combination with purified monoclonal antibodies (mAbs) blocking antibodies for 72 h with or without M. tuberculosis antigen (WCL = whole cell lysate). T cells from cultured cells were analysed for M. tuberculosis‐specific cytokine production by flow cytometry. Lymphocytes were gated based on their scatter profile and gated further on the basis of CD3 expression. Representative fluorescence activated cell sorter (FACS) plots show production of (a) interferon (IFN)‐γ and (c) interleukin (IL)−4 among study groups. Each point represents the mean percentage ± standard error of the mean from each group for (b) IFN‐γ and (d) IL‐4 production in various culture conditions. *P = 0·01; **P = 0·001. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 4.
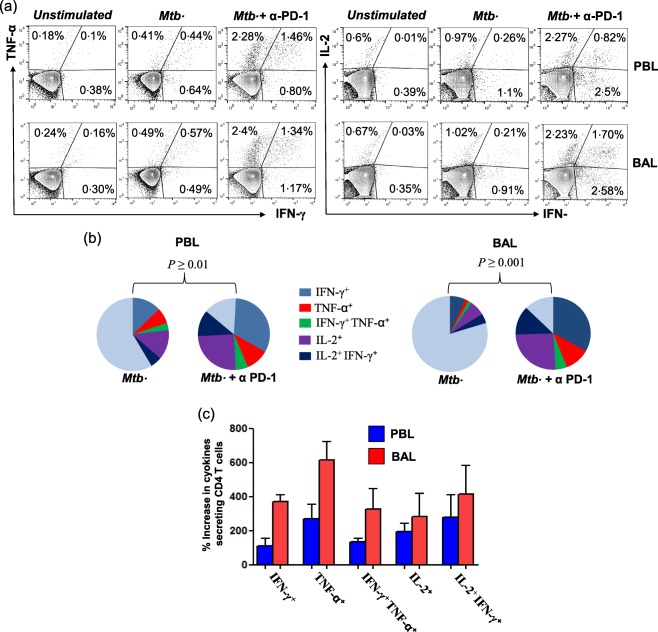
Restoration of polyfunctionality of Mycobacterium tuberculosis‐specific CD4+ T cell immune responses with programmed death 1 (PD‐1) blockade. Mononuclear cells (MNC) obtained from peripheral blood (PB) and bronchoalveolar lavage fluids from miliary tuberculosis (MTB) patients (n = 3) were cultured with M. tuberculosis antigen [whole cell lysate (WCL)] in the presence and absence of PD‐1 blockade for 48 h. Cultured cells were analysed for the presence of intracellular interferon (IFN)‐γ‐, tumour necrosis factor (TNF)‐α‐ and interleukin (IL‐2)‐producing CD4+ T cells by fluorescence activated cell sorter (FACS). (a) Representative flow cytometry plots show the percentages of M. tuberculosis‐specific cytokine‐secreting CD4+ T cells. (b) The pie‐charts show a significant increase in the percentages of CD4+ T cells producing one (IFN‐γ+, TNF‐α+ and IL‐2+) or two (IFN‐γ+TNF‐α+ and IL‐2+IFN‐γ+) cytokines upon PD‐1 blockade. The pie‐chart represents percentages of different M. tuberculosis‐specific combinations of cytokine‐secreting CD4+ T cells as a percentage of total CD4+ T cells. (c) Percentage increases in cytokine co‐expression subsets of total cytokine‐producing CD4+ T cells are shown. Each group consists of data from three MTB patients. The P‐values are calculated using one‐way analysis of variance (anova) followed by Bonferroni's non‐parametric post‐test. [Colour figure can be viewed at wileyonlinelibrary.com]
Inhibition of T cell apoptosis upon blocking PD‐1–PD‐L1/L2 interaction
Further, to test the effect of blocking PD‐1–PD‐L1/L2 interactions on T cell apoptosis, we evaluated M. tuberculosis antigen‐induced CD3+ T cell apoptosis using flow cytometry‐based annexin V and propidium iodide staining, as described elsewhere 24, 28. We observed a significant reduction of both early (annexin+ PI–) and late (annexin+ PI+) apoptotic M. tuberculosis‐specific T cells in PB as well as BAL of MTB patients after PD‐1 blocking. However, the reduction of apoptosis following blocked PD‐1 was significantly higher in the BAL‐derived T cells over that of PB (Fig. 5a,b). This indicates that over‐expression of PD‐1 in the local pathological milieu of tuberculosis leads to higher induction of apoptosis of effector T cells in the lung of MTB patients.
Figure 5.
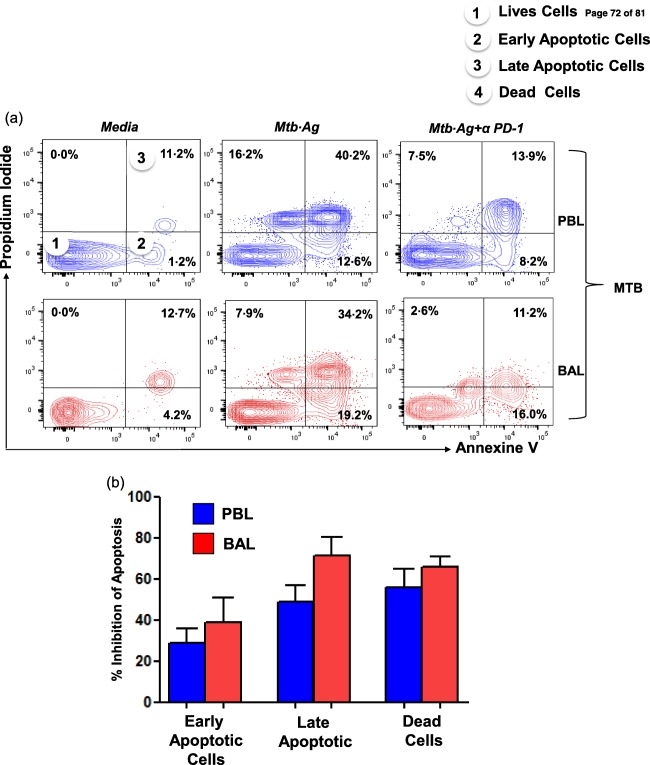
Programmed death‐1 (PD‐1) blockade rescues CD3+ T cells from Mycobacterium tuberculosis‐induced apoptosis. Mononuclear cells obtained from peripheral blood (PB) and bronchoalveolar lavage (BAL) fluids from miliary tuberculosis (MTB) patients (n = 3) were cultured with M. tuberculosis antigen in the presence and absence of PD‐1 blockade for 24 h. Cultured cells were analysed for co‐expression of annexin V and propidium iodide (PI) on gated CD3+ T cells by fluorescence activated cell sorter (FACS). (a) Representative flow cytometry plots show the distribution of M. tuberculosis‐specific early apoptotic (annexin V+ only), late apoptotic (annexin V, PI dual‐positive) and dead cell (PI+ only) proportions under each experimental condition. (b) Inhibition of apoptosis following blocked PD‐1 was significantly higher in the BAL‐derived T cells. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
M. tuberculosis infection is contained by intense granulomatous inflammation at the pathological site through orchestrated generation and localization of Th1 effector cells, as observed in TB‐PE, a localized form of disease. Conversely, the suppressed state of local immunity fails to contain the infection, leading to development of progressive and disseminated disease such as MTB 29. We and others have demonstrated that local immune response in tuberculosis is determined by the proportional presence of effector and Treg cells in the milieu and this, in turn, dictates the form of disease development 12, 30, 31, 32. The mechanism(s) underlying this delicate balance of local immune response still remains incompletely understood. Therefore, it is important to understand the regulatory mechanisms regulating the type and magnitude of immune response elicited at the pathological site of TB. Recent studies demonstrate that PD‐1 regulates T cell activation, peripheral tolerance and autoimmunity, principally as an inhibitory molecule 9, 33, 34. Several studies have demonstrated that the PD‐1 signalling pathway is activated during persistent infection with various microorganisms and contributes to impairment of protective immunity 25, 26. We have shown recently that in‐vitro blockade of PD‐1 signalling enhanced M. tuberculosis‐specific IFN‐γ production by T cells 23 and NK T cells of PTB patients 24, indicating that this inhibitory pathway also affects the T cell functions during mycobacterial infection. In the present study, we investigated the role of the PD‐1 pathway in dampening local effector T cells obtained from BAL of MTB patients. Here we report the numerical dominance of PD‐1 receptor and ligand(s) (PD‐L1 and PD‐L2)‐expressing T cells, CD19+ B cells and CD14+ monocytes in the PB and LDSS of all forms of tuberculosis compared to that of HCs. The frequency of PD‐1+ T cells in the LDSS of various forms of tuberculosis demonstrates a gradient of PD‐1‐expressing T cell representation among TB‐PE and MTB patients, suggesting that a proportional increase of locally accumulated PD‐1+ T cells (MTB > TB‐PE > PTB) possibly underlies the spectrum of local immunity which may result in either inadequate or efficient local immunity, and may therefore dictate the clinical manifestation of human tuberculosis. Interestingly, the frequency of PD‐1‐expressing T cells and PD‐1 ligand(s)‐expressing CD14+ monocytes and CD19+ B cells was more profound in BAL of MTB in comparison to their autologous counterpart and PE as well as PB of TB‐PE patients. This may be relevant for the observed local immune suppression leading to disease dissemination in MTB.
Mycobacterial infections are controlled mainly by an activation of macrophages through type 1 cytokine production by T cells, and IFN‐γ is central to this process 35. Therefore, production of IFN‐γ and a shift towards a Th1 cytokine profile are crucial mediators in protection against M. tuberculosis 1. Previously, we showed that patients with TB‐PE have a predominantly Th1 cytokine (IFN‐γ), whereas the lungs of patients with MTB have a relative enrichment with the Th2 cytokine (IL‐4) 29, 36. Considering the plausible involvement of PD‐1 pathway in dampening host immunity among MTB patients, in this study we evaluated the impact of the PD‐1 pathway on the effector T cells infiltrated in the lung of MTB patients. In our study, blocking the PD‐1 pathway resulted in the most profound rescue of IFN‐γ+ and TNF‐α T cells obtained from the BAL relative to the rescue of that obtained from PB of MTB patients. However, inhibition of the PD‐1 pathway failed to rescue IL‐4 producers T cells both in BAL and PB. These findings, coupled with increased expression of PD‐1 and its ligands, indicates strongly that in the local milieu of MTB, PD‐1 dampens the IFN‐γ+ and TNF‐α T cells dominantly and preferentially, while sparing the IL‐4 producers (Fig. 3a–d, Fig. 4a–c). We also show that the PD‐1 pathway induces apoptosis and death of T cells more profoundly in the lung of MTB patients.Thus, at the local disease site of MTB, PD‐1 selectively inhibits the protective M. tuberculosis‐specific T cells while sparing the suppressive IL‐4 producers, thereby shifting the local immune functions towards a more deficient state. This plausibly disrupts the protective Th1 response and skews the host immunity towards a suppressed state, and leads to the failure of eliciting an adequate level of effector immunity at the pathological site of MTB. Previously, we have demonstrated that Treg cells could suppress peripheral M. tuberculosis‐specific T cells via the PD‐1 pathway 23. In the present study, we report a similar but more profound role of this pathway in the lung of MTB patients. In other words, our results indicate the potential use of the PD‐1 pathway in rescuing the relevant specific host immunity in the pathological milieu of disseminated forms of tuberculosis such as MTB. Interestingly, we observed that inhibiting the PD‐1 pathway could significantly rescue polyfunctional T cells (IFNγ+TNF‐α+ and IFN‐γ+IL‐2+) in the BAL of MTB patients (Fig. 4a–c). Recent evidence suggests strongly the critical role of polyfunctional or ‘quality’ T cells in containing tuberculosis 37, 38. Studies have shown that the diverse cytokine production profile of T cells shifts towards dominant single cytokine production by M. tuberculosis‐specific T cells among TB patients 39. Interestingly, in our blocking experiment we found that single TNF‐α producer CD4 T cells are dominant over other single cytokines (TNF‐α+, IFN‐γ+ and IL‐2+) and dual cytokine (IFNγ+TNF‐α+ and IFN‐γ+IL‐2+) producers. Our results suggest that inhibiting the PD‐1 pathway not only restores single cytokine producers, but more importantly restored the polyfunctional effector T cells in the pathological site of MTB. We conclude that the interaction of PD‐1 and its ligands is a mechanism responsible for local immunosuppression leading to disease dissemination.
PD‐1 regulates effector T cell activity in peripheral tissues in response to infection or tumour progression 40. Several recent studies, including ours 15, 16, 17, 18, 19, 20, 21, 22, shows that during chronic infection PD‐1 is over‐expressed on exhausted T cells and blocking the PD‐1–PD‐L1 pathway restores proliferation, cytokine secretion and cytotoxicity, suggesting that PD‐1‐mediated suppression of host immunity plays an important role in tuberculosis 23, 24, 25, 26. However, studies on PD‐1 knock‐out mice demonstrated reduced survival of mice infected with M. tuberculosis 41, 42, 43, and a report by Lee et al. showed that tuberculosis reactivation in a patient receiving the anti‐programmed death‐1 (PD‐1) inhibitor used in Hodgkin's lymphoma might indicate homeostatically different roles of PD‐1 at different stages of tuberculosis 44. We believe that such discordance may be due to an aberrantly heightened inflammatory response with excessive neutrophilic infiltration and necrosis elicited in PD‐1 knock‐out animals, which may exert a lethal influence on their survival 41. In our opinion, these studies do not rule out the immunosuppressive role of PD‐1 in suppression of the host T cell response in humans. We propose that a basal level of PD‐1 expression is required to control the excessive inflammatory response following M. tuberculosis infection, as observed among PD‐1‐deficient mice. Conversely, over‐expression of PD‐1 on T cells inhibits the protective T cell response against M. tuberculosis; this might lead to failure to contain M. tuberculosis infection despite an exaggerated inflammatory response. Knocking out PD‐1 or the excessive use of PD‐1 inhibitors may disrupt the physiological homeostatic control of the elicitation of an appropriate T cell response necessary for protection and allowing reactivation of latent TB infection after PD‐1 blockade immunotherapy. Therefore, we propose that inhibiting the PD‐1 pathway may restore host immunity and thus help in disease containment in active tuberculosis, while the same may disrupt the homeostatic containment of latent tuberculosis, thereby possibly activating the disease.
In recent years, increasing animal and human studies have demonstrated that the PD‐1–PD‐1 pathway plays an important role in immune response during tuberculosis 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26. Several of these studies in humans have shown that blockade of this pathway can enhance the Mtb‐specific cytokine production, proliferation and lytic activity of CD8 T cells 23, 24, 25, 26. Moreover, blockade of the PD‐1–PD‐L1 pathway enhances lytic degranulation of NK cells and IFN‐γ production by NK 26 and NK T 24 cells significantly. These studies show that Mtb antigens induced an increase PD‐1 expression on peripheral T cells of active TB patients. Here, we have demonstrated significant up‐regulation of PD‐1 on T cells and its ligands on B cells and monocytes in both TB‐PE and miliary TB patients. To the best of our knowledge, the present study is the first to investigate the expression of PD‐Ls on B cells and monocytes in BAL of MTB patients and unravel the role of the PD‐1 pathway in the suppression of effector T cells, including polyfunctional (IFN‐γ+TNF‐α+ Τ cells) at the pathological site of MTB (lungs).
A decline in the number of PD‐1+ T cells during chemotherapy has been reported in viral diseases and tuberculosis 23, 24. A recent study demonstrated increased levels of soluble PD‐1, expression of PD‐1 and PD‐L1 on T cells as well as pleural mesothelial cells of pleura in TB‐PE patients. This is in concordance with our present findings in TB‐PE and MTB 45. In the present study we did not have an opportunity to check PD‐1 expression in TB‐PE and MTB during successful anti‐tubercular treatment. However, we have demonstrated previously that effective anti‐tubercular treatment results in a significant reduction of PD‐1+ T and NK T cells [23,24]. Similarly, another recent study has demonstrated such a decline in PD‐1, PD‐L1 and PD‐L2 gene expression during the intensive phase of TB treatment 46.
Overall, the present study demonstrates the potential role of the PD‐1 pathway in the impairment of protective immunity, particularly at the disease site, of more disseminated forms of tuberculosis, such as miliary tuberculosis. We propose that blockade of the co‐inhibitory PD‐1 pathway may be a key to the augmentation of protective T cells, including polyfunctional T cells, at the pathological site, thus abrogating disease dissemination in MTB. It also reinforces the possibility of immune restoration with the possible application of a novel therapy for human tuberculosis. Further studies will help in understanding the detailed inhibitory role of the PD‐1 pathway in tuberculosis and the potential application of PD‐1 in immunotherapy and vaccination for TB.
Author contributions
A. S. and D. K. M. designed the study. A. S. performed all the experiments, statistical analysis and graphic design, wrote the manuscript under the guidance of D. K. M.; A. M. and A. B. D. provided samples from subjects and helped in terms of clinical and intellectual inputs. As the lead author, D. K. M. supervised scientific aspects of the research work and wrote the manuscript. All authors have scrutinized the manuscript.
Disclosure
The authors declare no disclosures.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Standardization of cell isolation from bronchoalveolar lavage (BAL) fluid. Cell separation from BAL has been a challenging task due to presence of sticky debris/carbon particles/mucous/coagulum/fibres, etc., causing cell clumping which prevented the preparation of single cell suspensions required for good flow cytometric analysis. Moreover, it was observed that due to the presence of the above‐mentioned debris, the forward (FSC)‐ versus side‐scatter (SSC) profile of the acquired cells obtained from BAL was unconvincing. Therefore, the protocol was modified in the laboratory for improved cell yield. After collection of BAL fluid, it was centrifuged at 4500 g for 10 min followed by washing with phosphate‐buffered saline (PBS) and incomplete RPMI‐1640. Thereafter, the cells obtained were subjected to filtration through a 40 µm filter to harvest enriched lymphocytes for further fluorescence activated cell sorter (FACS)‐based analysis. Finally, cells were suspended in complete RPMI‐1640 until further experimentation. It was interesting to note that this additional step of filtration led to the improvement in CD3+ T cell yield (a,b). Improvement in the cell yield and staining of CD3+ lymphocytes with the use of the 40 µm filter from the BAL fluid. (a) Representative FACS plots showing improvement in the yield of lymphocytes as well as staining of CD3+ lymphocytes with the use of the 40 µm filter while isolating the cells from BAL of patients with tuberculosis. (b) Cumulative data plot showing significant improvement in the yield of lymphocytes and staining of CD3+ cells with the use of the 40 µm filter while isolating the cells from BAL of patients with tuberculosis as determined by flow cytometry. As evident by flow cytometry, without filtration, the cell yield of lymphocytes varied generally from 5 to 20%, whereas CD3+ ranged from 5 to 40% of the total acquired events. Flow cytometry of BAL‐derived cells revealed that staining was observed after the filtration improvement in yields of both lymphocytes (ranging from 15 to 40%) and CD3+ cells (20–60%).
Fig. S2. Optimization of dose of pure anti‐programmed death‐1 (PD‐1), α‐PD‐ligand 1 (PD‐L1) and α‐PD‐L2 antibodies for the in‐vitro PD‐1–PD‐L1/L2 blocking experiment. Freshly isolated peripheral blood mononuclear cells (PBMCs) were resuspended in complete RPMI‐1640 at a concentration of 2 × 106 cells/ml to each well of the U‐bottomed culture plate (Falcon, Austin, TX, USA). Varying doses of purified recombinant antibodies (PD‐1, PD‐L1 and PD‐L2), ranging from 0·5 μg, 1 μg, 2·5 μg and 5 μg to 10 μg of each per ml culture, were added in duplicate for each concentration. Cells were incubated at 4°C for 45–60 min. Cells were stained with immunoglobulin (Ig)Gk1 fluorescein isothiocyanate (FITC) secondary antibody to determine the percentage of pure PD‐1 binding. Fresh PBMCs were also stained directly with anti‐PD‐1 FITC antibody as control. Details of surface staining are described in the Materials and methods. With an increasing dose of pure PD‐1 antibodies, an increase in the percentage of PD‐1+CD4+ cells was observed. We found a maximum number of PD‐1+CD4+ cells at concentrations of 5 μg and 10 μg per ml of pure PD‐1 antibody, which is equal to the percentage of cells obtained from anti‐PD‐1 FITC antibody. (a,b) Similar results were obtained for pure α‐PD‐L1 (5 μg/ml) and α‐PD‐L2 (5 μg/ml) antibodies. Thus, for each pure antibody, a concentration of 5 μg/ml was used for blockade of PD‐1–PD‐L1/L2 interaction. Dose optimization of pure anti‐PD‐1 antibody for in‐vitro PD‐1–PD‐L1/L2 blocking experiment: 2 × 106/ml was taken in a U‐bottomed culture plate (Falcon, Austin, TX, USA). Varying doses of anti‐PD‐1 pure antibody (range 0·5 μg, 1 μg, 2·5 μg, 5 μg, 10 μg per ml culture) were added in duplicate for each concentration. Cells were incubated at 4°C for 45–60 min. Cells were stained with IgGk1 FITC secondary antibodies to determine the percentage of pure PD‐1 binding. Fresh PBMCs were also stained directly with anti‐PD‐1 FITC antibodies as control. (a) The percentage of PD‐1+CD4+ cells was analysed by flow cytometry. (b) Data depicting the optimal dose of pure α‐PD‐1 antibody.
Fig. S3. Flow gating strategy: exclusion of doublets, debris or dead cells. Briefly, flow gating strategy showing the exclusion of debris, doublets or dead cells in cultured peripheral blood mononuclear cells/peripheral blood (PBMCs/PB) and bronchoalveolar lavage (BAL) fluid from human tuberculosis patients. Mononuclear cells obtained from PB or BAL were cultured as per the protocol described in the Material and methods. Cultured cells were stained for CD3 or CD4, intracellular cytokines [interleukin (IL)−2, interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α] and for apoptosis markers [annexin V and propidium iodide (PI)], as per our described methods. Singlets were selected based on forward‐scatter (FSC)‐H versus FSC‐A and side‐scatter (SSC)‐H versus SSC‐A. Further lymphocytes were selected based on FSC and SSC from gated singlets. Live CD3 or CD4 T cells were selected by excluding dead cells based on fluorescent amine reactive dye (violet live dead dye; Molecular Probes/Invitrogen, Carlsbad, CA, USA). Using this approach we achieved 50–80% of viable lymphocytes from our cultured cells. Further, gated live CD4 T cell intracellular cytokines were determined in our blocking experiment and gated CD3 cells for apoptosis assay. Left and right panels show representative fluorescence activated cell sorter (FACS) plots from PB and BAL of miliary tuberculosis (MTB) patients.
Fig. S4. Flow cytometry analysis of programmed death‐1 (PD)−1 ligands (PD‐L1 and PD‐L2) on antigen‐presenting cells (APC) (CD14+ cells and CD19 B cells) in various forms of tuberculosis (TB) patients. PD‐1 ligand (PD‐L1 and PD‐L2) expression was examined on total CD14+ monocytes and CD19+ B cells in the freshly isolated PBMCs from healthy controls (HC), pulmonary tuberculosis (PTB) patients and paired samples of TB‐pleural effusion (PE) and miliary tuberculosis (MTB) patients. (a) Representative fluorescence activated cell sorter (FACS) plot showing expression of PD‐L1 and PD‐L2 in CD14+ cells in among these patients and HC. (b) FACS plots showing expression of PD‐L1 and PD‐L2 on CD19+ cells among these study subjects. FACS plots show a substantial increase in frequencies of PD‐1 ligands in the PTB and in paired samples of TB‐pleural effusion (PE) [peripheral blood (PB) versus PE] and MTB [PB versus bronchoalveolar lavage (BAL)] patients. The percentage of PD‐1 ligands expressing CD14 cells in the BAL of MTB were much higher than the PB of PTB patients and the autologous MNC of MTB. No such difference was found in the paired samples of TB‐PE.
Acknowledgements
We thank our study volunteers for providing samples and supporting this work, as well as the clinical staff for their dedication to this research. This work was supported by Department of Biotechnology and Indian Council of Medical Research Grant, Government of India. We also acknowledge the financial support of the All India Institute of Medical Sciences. We are grateful to Dr U. Sengupta for M. tuberculosis antigen. We thank Dr A. K. Rai (Department TII, AIIMS) and Dr A. Saxena (Department TII, AIIMS) for helping in analysis of the flow data.
References
- 1. Spellberg B, Edwards JE Jr. Type 1/type 2 immunity in infectious diseases. Clin Infect Dis 2001; 32:76–102. [DOI] [PubMed] [Google Scholar]
- 2. Flynn JL, Chan J, Triebold KJ et al An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med 1993; 178:2249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caruso AM, Serbina N, Klein E et al Mice deficient in CD4 T cells have only transiently diminished levels of IFN‐γ, yet succumb to tuberculosis. J Immunol 1999; 162:5407. [PubMed] [Google Scholar]
- 4. Desvignes L, Ernst JD. Interferon‐gamma‐responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis . Immunity 2009; 31:974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baldridge MT, King KY, Boles NC et al Quiescent haematopoietic stem cells are activated by IFN‐gamma in response to chronic infection. Nature 2010; 465:793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cooper AM, Dalton DK, Stewart TA et al Disseminated tuberculosis in interferon gamma gene‐disrupted mice. J Exp Med 1993; 178:2243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saunders BM, Frank AA, Orme IM et al CD4 is required for the development of a protective granulomatous response to pulmonary tuberculosis. Cell Immunol 2002; 216:65–72. [DOI] [PubMed] [Google Scholar]
- 8. Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol 2001; 19:93–129. [DOI] [PubMed] [Google Scholar]
- 9. Chen L. Co‐inhibitory molecules of the B7‐CD28 family in the control of T‐cell immunity. Nat Rev Immunol 2004; 4:336–47. [DOI] [PubMed] [Google Scholar]
- 10. O'Garra A, Vieira PL, Vieira P et al IL‐10‐producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest 2004; 114:1372–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol 2005; 6:338–44. [DOI] [PubMed] [Google Scholar]
- 12. Sharma PK, Saha PK, Singh A et al FoxP3+ regulatory T cells suppress effector T‐cell function at pathologic site in miliary tuberculosis. Am J Respir Crit Care Med 2009; 179:1061–70. [DOI] [PubMed] [Google Scholar]
- 13. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol 2005; 23:515. [DOI] [PubMed] [Google Scholar]
- 14. Keir ME, Butte MJ, Freeman GJ et al PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Day CL, Kaufmann DE, Kiepiela P et al PD‐1 expression on HIV‐specific T cells is associated with T‐cell exhaustion and disease progression. Nature 2006; 443:350–4. [DOI] [PubMed] [Google Scholar]
- 16. Barber DL, Wherry EJ, Masopust D et al Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006; 439:6828–7. [DOI] [PubMed] [Google Scholar]
- 17. Trautmann L, Janbazian L, Chomont N et al Upregulation of PD‐1 expression on HIV‐specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 2006; 12:1198–202. [DOI] [PubMed] [Google Scholar]
- 18. Urbani S, Amadei B, Tola D et al PD‐1 expression in acute hepatitis C virus (HCV) infection is associated with HCV‐specific CD8 exhaustion. J Virol 2006; 80:11398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petrovas C, Price DA, Mattapallil J et al SIV‐specific CD8+ T cells express high levels of PD1 and cytokines but have impaired proliferative capacity in acute and chronic SIVmac251 infection. Blood 2007; 110:928–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hong JJ, Amancha PK, Rogers K et al Re‐evaluation of PD‐1 expression by T cells as a marker for immune exhaustion during SIV infection. PLOS ONE 2013; 8:e60186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu YY, Lin CW, Cheng KS et al Increased programmed death‐ligand‐1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin Exp Immunol 2010; 161:551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Das S, Suarez G, Beswick EJ et al Expression of B7‐H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol 2006; 176:3000–9. [DOI] [PubMed] [Google Scholar]
- 23. Singh A, Mohan A, Dey AB et al Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis‐specific interferon γ‐producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis 2013; 208:603–15. [DOI] [PubMed] [Google Scholar]
- 24. Singh A, Dey AB, Mohan A et al Programmed death‐1 receptor suppresses γ‐IFN producing NKT cells in human tuberculosis. Tuberculosis 2014; 94:97–206. [DOI] [PubMed] [Google Scholar]
- 25. Jurado JO, Ivana B, Pasquinelli V et al Programmed death (PD)−1:PD‐ligand 1/PD‐ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol 2008; 181:116–25. [DOI] [PubMed] [Google Scholar]
- 26. Alvarez IB, Pasquinelli V, Jurado JO et al Role played by the PD‐1‐programmed death ligand pathway during innate immunity against Mycobacterium tuberculosis . J Infect Dis 2011; 202:524–32. [DOI] [PubMed] [Google Scholar]
- 27. Sharma SK, Mohan A, Pande JN et al Clinical profile, laboratory characteristics and outcome in miliary tuberculosis. QJM 1995; 88:29–37. [PubMed] [Google Scholar]
- 28. Vermes I, Haanen C, Steffens‐Nakken H et al A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995; 184:39–51. [DOI] [PubMed] [Google Scholar]
- 29. Sharma SK, Mitra DK, Balamurugan A et al Cytokine polarization in miliary and pleural tuberculosis. J Clin Immunol 2002; 22:345–52. [DOI] [PubMed] [Google Scholar]
- 30. Kursar M, Koch M, Mittrücker H et al Cutting edge: regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis . J Immunol 2007; 178:2661–5. [DOI] [PubMed] [Google Scholar]
- 31. Scott‐Browne JP, Shafiani S, Tucker‐Heard G et al Expansion and function of Foxp3‐expressing T regulatory cells during tuberculosis. J Exp Med 2007; 204:2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh A, Dey AB, Mohan A, Sharma PK, Mitra DK. Foxp3(+) regulatory T cells among tuberculosis patients: impact on prognosis and restoration of antigen specific IFN‐gamma producing T cells. PLOS ONE 2012; 7:e44728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharpe AH, Wherry EJ, Ahmed R et al The function of programmed cell death1 and its ligands in regulating autoimmunity and infection. Nat Immunol 2007; 8:239–45. [DOI] [PubMed] [Google Scholar]
- 34. Keir ME, Francisco LM, Sharpe AH. PD‐1 and its ligands in T‐cell immunity. Curr Opin Immunol 2007; 19:309–314. [DOI] [PubMed] [Google Scholar]
- 35. Pieters J. Mycobacterium tuberculosis and the macrophages: maintaining a balance. Cell Host Microbe 2008; 3:399–407. [DOI] [PubMed] [Google Scholar]
- 36. Mitra DK, Sharma SK, Dinda AK et al Polarized helper T cells in tubercular pleural effusion: phenotypic identity and selective recruitment. Eur J Immunol 2005; 35:2367–75. [DOI] [PubMed] [Google Scholar]
- 37. Katlin A, Wilkinson RJW. Polyfunctional T cells in tuberculosis. Eur J Immunol 2010; 40:2139–42. [DOI] [PubMed] [Google Scholar]
- 38. Qui Z, Zhang M, Zhu Y et al Multifunctional CD4 T cell responses in patients with active tuberculosis. Sci Rep 2012; 2:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harari A, Rozot V, Enders FB et al Dominant TNF‐α+ Mycobacterium tuberculosis‐specific CD4+ T cell responses discriminate between latent infection and active disease. Nat Med 2011; 17:372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He J, Hu Y, Hu M, Li B. Development of PD‐1/PD‐L1 pathway in tumor immune microenvironment and treatment for non‐small cell lung cancer. Sci Rep 2015; 5:13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lazar‐Molnar E, Chen B, Sweeney KA et al Programmed death‐1 (PD‐1)‐deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci USA 2010; 107:13402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barber DL, Mayer‐Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD‐1‐mediated inhibition. J Immunol 2011; 186:1598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tousif S, Singh Y, Prasad DV et al T cells from programmed death‐1 deficient mice respond poorly to Mycobacterium tuberculosis infection. PLOS ONE 2011; 6:e19864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee JJ, Chan A, Tang T. Tuberculosis reactivation in a patient receiving anti‐programmed death‐1 (PD‐1) inhibitor for relapsed Hodgkin's lymphoma. Acta Oncol 2016; 55:519–20. [DOI] [PubMed] [Google Scholar]
- 45. Yin W, Tong ZH, Cui A et al PD‐1/PD‐Ls pathways between CD4 (+) T cells and pleural mesothelial cells in human tuberculous pleurisy. Tuberculosis 2014; 94:131–9. [DOI] [PubMed] [Google Scholar]
- 46. Hassan SS, Akram M, King EC, Dockrell HM, Cliff JM. PD‐1, PD‐L1 and PD‐L2 gene expression on T‐cells and natural killer cells declines in conjunction with a reduction in PD‐1 protein during the intensive phase of tuberculosis treatment. PLOS ONE 2015; 10:e0137646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Standardization of cell isolation from bronchoalveolar lavage (BAL) fluid. Cell separation from BAL has been a challenging task due to presence of sticky debris/carbon particles/mucous/coagulum/fibres, etc., causing cell clumping which prevented the preparation of single cell suspensions required for good flow cytometric analysis. Moreover, it was observed that due to the presence of the above‐mentioned debris, the forward (FSC)‐ versus side‐scatter (SSC) profile of the acquired cells obtained from BAL was unconvincing. Therefore, the protocol was modified in the laboratory for improved cell yield. After collection of BAL fluid, it was centrifuged at 4500 g for 10 min followed by washing with phosphate‐buffered saline (PBS) and incomplete RPMI‐1640. Thereafter, the cells obtained were subjected to filtration through a 40 µm filter to harvest enriched lymphocytes for further fluorescence activated cell sorter (FACS)‐based analysis. Finally, cells were suspended in complete RPMI‐1640 until further experimentation. It was interesting to note that this additional step of filtration led to the improvement in CD3+ T cell yield (a,b). Improvement in the cell yield and staining of CD3+ lymphocytes with the use of the 40 µm filter from the BAL fluid. (a) Representative FACS plots showing improvement in the yield of lymphocytes as well as staining of CD3+ lymphocytes with the use of the 40 µm filter while isolating the cells from BAL of patients with tuberculosis. (b) Cumulative data plot showing significant improvement in the yield of lymphocytes and staining of CD3+ cells with the use of the 40 µm filter while isolating the cells from BAL of patients with tuberculosis as determined by flow cytometry. As evident by flow cytometry, without filtration, the cell yield of lymphocytes varied generally from 5 to 20%, whereas CD3+ ranged from 5 to 40% of the total acquired events. Flow cytometry of BAL‐derived cells revealed that staining was observed after the filtration improvement in yields of both lymphocytes (ranging from 15 to 40%) and CD3+ cells (20–60%).
Fig. S2. Optimization of dose of pure anti‐programmed death‐1 (PD‐1), α‐PD‐ligand 1 (PD‐L1) and α‐PD‐L2 antibodies for the in‐vitro PD‐1–PD‐L1/L2 blocking experiment. Freshly isolated peripheral blood mononuclear cells (PBMCs) were resuspended in complete RPMI‐1640 at a concentration of 2 × 106 cells/ml to each well of the U‐bottomed culture plate (Falcon, Austin, TX, USA). Varying doses of purified recombinant antibodies (PD‐1, PD‐L1 and PD‐L2), ranging from 0·5 μg, 1 μg, 2·5 μg and 5 μg to 10 μg of each per ml culture, were added in duplicate for each concentration. Cells were incubated at 4°C for 45–60 min. Cells were stained with immunoglobulin (Ig)Gk1 fluorescein isothiocyanate (FITC) secondary antibody to determine the percentage of pure PD‐1 binding. Fresh PBMCs were also stained directly with anti‐PD‐1 FITC antibody as control. Details of surface staining are described in the Materials and methods. With an increasing dose of pure PD‐1 antibodies, an increase in the percentage of PD‐1+CD4+ cells was observed. We found a maximum number of PD‐1+CD4+ cells at concentrations of 5 μg and 10 μg per ml of pure PD‐1 antibody, which is equal to the percentage of cells obtained from anti‐PD‐1 FITC antibody. (a,b) Similar results were obtained for pure α‐PD‐L1 (5 μg/ml) and α‐PD‐L2 (5 μg/ml) antibodies. Thus, for each pure antibody, a concentration of 5 μg/ml was used for blockade of PD‐1–PD‐L1/L2 interaction. Dose optimization of pure anti‐PD‐1 antibody for in‐vitro PD‐1–PD‐L1/L2 blocking experiment: 2 × 106/ml was taken in a U‐bottomed culture plate (Falcon, Austin, TX, USA). Varying doses of anti‐PD‐1 pure antibody (range 0·5 μg, 1 μg, 2·5 μg, 5 μg, 10 μg per ml culture) were added in duplicate for each concentration. Cells were incubated at 4°C for 45–60 min. Cells were stained with IgGk1 FITC secondary antibodies to determine the percentage of pure PD‐1 binding. Fresh PBMCs were also stained directly with anti‐PD‐1 FITC antibodies as control. (a) The percentage of PD‐1+CD4+ cells was analysed by flow cytometry. (b) Data depicting the optimal dose of pure α‐PD‐1 antibody.
Fig. S3. Flow gating strategy: exclusion of doublets, debris or dead cells. Briefly, flow gating strategy showing the exclusion of debris, doublets or dead cells in cultured peripheral blood mononuclear cells/peripheral blood (PBMCs/PB) and bronchoalveolar lavage (BAL) fluid from human tuberculosis patients. Mononuclear cells obtained from PB or BAL were cultured as per the protocol described in the Material and methods. Cultured cells were stained for CD3 or CD4, intracellular cytokines [interleukin (IL)−2, interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α] and for apoptosis markers [annexin V and propidium iodide (PI)], as per our described methods. Singlets were selected based on forward‐scatter (FSC)‐H versus FSC‐A and side‐scatter (SSC)‐H versus SSC‐A. Further lymphocytes were selected based on FSC and SSC from gated singlets. Live CD3 or CD4 T cells were selected by excluding dead cells based on fluorescent amine reactive dye (violet live dead dye; Molecular Probes/Invitrogen, Carlsbad, CA, USA). Using this approach we achieved 50–80% of viable lymphocytes from our cultured cells. Further, gated live CD4 T cell intracellular cytokines were determined in our blocking experiment and gated CD3 cells for apoptosis assay. Left and right panels show representative fluorescence activated cell sorter (FACS) plots from PB and BAL of miliary tuberculosis (MTB) patients.
Fig. S4. Flow cytometry analysis of programmed death‐1 (PD)−1 ligands (PD‐L1 and PD‐L2) on antigen‐presenting cells (APC) (CD14+ cells and CD19 B cells) in various forms of tuberculosis (TB) patients. PD‐1 ligand (PD‐L1 and PD‐L2) expression was examined on total CD14+ monocytes and CD19+ B cells in the freshly isolated PBMCs from healthy controls (HC), pulmonary tuberculosis (PTB) patients and paired samples of TB‐pleural effusion (PE) and miliary tuberculosis (MTB) patients. (a) Representative fluorescence activated cell sorter (FACS) plot showing expression of PD‐L1 and PD‐L2 in CD14+ cells in among these patients and HC. (b) FACS plots showing expression of PD‐L1 and PD‐L2 on CD19+ cells among these study subjects. FACS plots show a substantial increase in frequencies of PD‐1 ligands in the PTB and in paired samples of TB‐pleural effusion (PE) [peripheral blood (PB) versus PE] and MTB [PB versus bronchoalveolar lavage (BAL)] patients. The percentage of PD‐1 ligands expressing CD14 cells in the BAL of MTB were much higher than the PB of PTB patients and the autologous MNC of MTB. No such difference was found in the paired samples of TB‐PE.