

Abstract
Aims/Introduction
Moderate elevation of glucose level has been shown to effectively promote β‐cell replication in various models in vitro and in normal rodents. Here, we aimed to test the effect of moderately elevated glucose on β‐cell mass expansion and islet function recovery in diabetic animal models.
Materials and Methods
A single high dose of streptozotocin was given to induce insulin‐deficient diabetes in adult male Sprague–Dawley rats. Then, 48 h after streptozotocin injection, newly diabetic rats were randomly divided into three groups: (i) no treatment to maintain hyperglycemia; (ii) daily exogenous long‐acting human insulin analog injection that maintained mild hyperglycemia (15 mmol/L < blood glucose < 18 mmol/L); (iii) daily exogenous long‐acting human insulin analog injection to restore normoglycemia (blood glucose <8 mmol/L) as a control. Islet function, β‐cell regeneration and β‐cell replication were monitored during the entire analysis period.
Results
A single high dose of streptozotocin induced massive loss of β‐cells, resulting in irreversible hyperglycemia. Mild hyperglycemia markedly promoted β‐cell proliferation, leading to robust β‐cell regeneration. Importantly, rats that maintained mild hyperglycemia showed nearly normal glucose‐stimulated insulin secretion, glucose disposal and random blood glucose levels, suggesting almost full restoration of the islet function. Normalization of blood glucose levels profoundly blunted β‐cell replication, regeneration and islet function recovery observed in mild hyperglycemia.
Conclusions
Our research provides a feasible approach to stimulate in situ β‐cell regeneration in diabetic rats, offering new perspectives for diabetes therapy.
Keywords: β‐Cell proliferation, Islet function recovery, Mild hyperglycemia
Introduction
In the pancreas, the endocrine cells are organized in defined islet structures, which function as key regulators of carbohydrate metabolism. Type 1 diabetes is a disease caused by an autoimmune attack on insulin‐secreting β‐cells in the islets, resulting in a lack of insulin production and hyperglycemia1. The most commonly used treatment for type 1 diabetes is insulin injection, but the degree of glycemic control with this approach does not compare with functional pancreatic β‐cells. Regenerative β‐cell treatments in diabetic patients would potentially give rise to long‐term restoration of normal glycemic control. A feasible approach to stimulate β‐cell regeneration might represent a curative therapy for type 1 diabetes2.
Glucose has long been considered one of the key regulators in β‐cell proliferation3. Moderate glucose elevation effectively stimulates β‐cell proliferation. For instance, the most effective glucose concentration range on INS1 cell proliferation occurred between 15 and 18 mmol/L glucose, and the proliferation rate plummeted above this glucose concentration4. Moderate elevated glucose concentration also markedly increases β‐cell proliferation in fetal and adult rat islets, mouse islets, and importantly, human β‐cells in vitro 5, 6, 7, 8. In vivo, consistent with earlier rodent infusion studies9, 10, 11, Alonso et al. showed that a 4‐day intravenous infusion of 50% glucose into normal mice modestly increases blood glucose (BG) concentrations, leading to markedly increased β‐cell proliferation as determined by bromodeoxyuridine incorporation12. Therefore, we wondered whether moderately elevated glucose levels could stimulate β‐cell proliferation in diabetic models, and whether islet function would be restored by such regeneration.
Streptozotocin (STZ) preferentially accumulates in pancreatic β‐cells through the Glut2 glucose transporter, fragments deoxyribonucleic acid (DNA) and therefore specifically destroys β‐cells in the pancreas. A single high dose of STZ destroys the majority of β‐cells as a result of direct toxic effects without autoimmune attack, and thus has been widely used to study β‐cell regeneration13, 14. Here, we used the single high‐dose STZ‐induced insulin‐deficient diabetic model. Diabetic rats were randomized into three groups: (i) no treatment to maintain hyperglycemia (HG); daily exogenous long‐acting human insulin analog injection to maintain mild hyperglycemia (mHG; 15 mmol/L < BG < 18 mmol/L) and daily exogenous long‐acting human insulin analog injection to restore normoglycemia (NG; BG <8 mmol/L), as insulin injection itself might generate a beneficial effect for β‐cell regeneration and function. We showed that mild hyperglycemia triggered β‐cell proliferation in single high‐dose STZ‐induced diabetic rats, leading to robust β‐cell regeneration and almost full islet function recovery.
Materials and Methods
Cell culture
INS1 cells were cultured in RPMI 1640 containing 11.1 mol/L glucose, with 10 mmol/L HEPES, 10% fetal bovine serum, 1 mmol/L sodium pyruvate, 50 μL/L β‐mercaptoethanol, 100 IU/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidified 5% CO2 atmosphere. Cells were initially starved overnight in RPMI 1640 containing 2.8 mmol/L glucose and 0.2% bovine serum albumin before treatment. Subsequently, the cells were incubated with 11.1 mmol/L, 15 mmol/L and 35 mmol/L for 72 h for apoptosis assay. For cell proliferation assay, INS1 cells were cultured with 11.1 mmol/L glucose, 15 mmol/L glucose and a combination of 15 mmol/L glucose with a PI3K inhibitor (Ly294002, 10 mol/L) for 24, 48 and 72 h, respectively. For western blot, INS1 cells were cultured with 11.1 mmol/L glucose, 15 mmol/L glucose and a combination of 15 mmol/L glucose with a PI3K inhibitor (Ly294002, 10 mol/L) for 24 h.
Apoptosis assay
INS1 cells were cultured, and then cells were trypsinized and collected. An annexin V‐FITC apoptosis detection kit (BD Biosciences, San Diego, CA, USA) was used according to the manufacturer's instructions. Cells were stained with annexin V‐FITC antibody for 15 min and propidium iodide for 5 min at room temperature in the dark, and stained cells were immediately analyzed using a FACSCalibur flow cytometer (BD Biosciences) and FlowJo software (Tomy Digital Biology, Tokyo, Japan).
Cell proliferation assay
Briefly, 4 × 103 INS‐1 cells were seeded into 100 μL RPMI‐1640 medium in 96‐well plates. Cell proliferation was determined 24 h, 48 h and 72 h after seeding using the CCK‐8 kit. CCK‐8 (10 μL/well, 96‐well plate) was added to the medium, and the cells were incubated for 3 h at 37°C. The absorbance was determined at 450 nm with an enzyme‐linked immunosorbent assay analyzer.
Animals and treatment
Eight‐week‐old male Sprague–Dawley rats (Laboratory Animal Research Center of the Academy of Military Medical Science, Beijing, China) weighing 240–260 g were injected intraperitoneally with a single dose of STZ (65 mg/kg; Sigma, St. Louis, MO, USA). STZ was dissolved in 0.05 mol/L citrate buffer (pH 4.5), and injected within 15 min of preparation. At 48 h after STZ treatment, rats with a BG level more than 25 mmol/L were included for further experiments. Included rats were randomly divided into tree groups: HG (n = 6); daily exogenous injection of long‐acting human insulin analog (glargine insulin) for the mHG group (15 mmol/L < BG < 18 mmol/L, n = 15); or to restore NG (BG <8 mmol/L, n = 8) as a control. Six normal rats were used as a normal control. The random BG levels were carefully monitored every day at 15.00 h, and we adjusted insulin doses the next morning according to the random BG levels. For example, four units of glargine insulin were given to one mHG rat in the morning. If the random BG level decreased to 12 mmol/L in the afternoon, lower than the target value, we adjusted the insulin dose to three units the next morning to keep the random BG level between 15 and 18 mmol/L. If the BG level was maintained between 15 and 18 mmol/L, we would maintain that insulin dose the next day. All animal procedures were approved by the Institutional Animal Care and Use Committee of the Chinese PLA General Hospital, and were carried out in accordance with the guidelines of the China Council on Animal Care and Use.
Preparation of tissue samples
The rats were killed at indicated time‐points. Eight to 12 rats were examined for each time‐point. For immunohistochemistry assay, the rats were injected intraperitoneally with 1% pentobarbital sodium (50 mg/kg), and then perfused through the left ventricle with 100 mL phosphate‐buffered saline, followed with 500 mL 4% paraformaldehyde. When the perfusion finished, the pancreata were isolated and incubated in 30% sucrose/phosphate buffer overnight. The tissues were then embedded (Tissue‐Tek OCT Compound; Sakura Finetek, Torrance, CA, USA) and frozen at −80°C for long‐term storage.
Immunohistochemistry
For the immunofluorescence analysis, the frozen sections were incubated for 14 h at 4°C with antisera specific for insulin (1/150, guinea pig; Sigma), glucagon (1/2,000, mouse; Sigma), glucagon (1/100, rabbit; Cell Signaling Technology, Danvers, MA, USA), Glut2 (1/100, mouse; Abcam, San Francisco, CA, USA), v‐maf musculopeoneurotic fiberosarcoma oncogene homologue A (MafA; 1/200, rabbit; Bethyl Laboratories, Montgomery, TX, USA), Pdx1 (1/50, goat; R&D System, Minneapolis, MN, USA), Ngn3 (1/200, rabbit; Millipore, St. Louis, MO, USA), E‐cadherin (1/100, rabbit; Abcam) and Ki67 (1/50, mouse; BD, Biosciences). The slides were then incubated for 2 h at room temperature with species‐specific secondary antibodies (1:500, Alexa‐594 or Alexa‐488; Invitrogen, Basel, Switzerland). β‐Cell apoptosis was determined using In Situ Cell Death Detection Kit (Roche, Basel, Switzerland). The nuclei were visualized with 40′,6‐diamidino‐2‐phenylindole (Sigma). Images were captured with a Fluoview FV1000 camera (Olympus, Tokyo, Japan) and recorded on a computer using the Olympus Fluoview Ver.1.7a viewer.
Islet isolation
In brief, islets were isolated from rats by distending the pancreas by injection of collagenase (Sigma) into the pancreatic duct followed by digestion at 37°C after removing the organ from the rat. The islets were separated from exocrine tissue by centrifugation on Histopaque 1077 (Sigma).
Quantitative reverse transcription polymerase chain reaction
Total ribonucleic acid extraction, complementary DNA synthesis and quantitative polymerase chain reaction (PCR). Adult pancreata from three rats for each group (normal, STZ‐2d, HG‐3 m, mHG‐3 m, NG‐3 m) were harvested. Adult islets were pooled as aforementioned from five rats for each group (normal, STZ‐2d, HG‐15d, mHG‐15d, NG‐15d). Ribonucleic acid samples were extracted from tissues or isolated cells using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Single‐stranded complementary DNA was synthesized using SuperScript II reverse transcriptase and oligo (dT; Invitrogen). Real‐time PCR analysis was carried out using Power SYBR Green RT–PCR Reagent (Applied Biosystems, Carlsbad, CA, USA) on ABI Prism thermal cycler model StepOnePlus (Applied Biosystems). The thermal cycling program was 50°C for 2 min, followed by 95°C for 10 min for one cycle, then 95°C for 30 s, followed by 60°C for 1 min for 40 cycles. Melting curve analysis was carried out to ensure the specificity of primers. β‐Actin was used as a reference gene in each reaction. The PCR primers are listed in Table S1.
Western blotting
The concentration of proteins in isolated islets and cell lysates was quantitated by bicinchoninic acid protein assay (Pierce, Rockford, IL, USA). Protein samples (30 μg) were separated by electrophoresis through 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel and transferred to a poly‐vinylidene fluoride membrane, followed by immunoblotting according to the protocol outlined by Cell Signaling Technology. The following antibodies were used: rabbit anti‐PI3k (1:1,000; Cell Signaling), rabbit anti‐Akt (1:1,000; Cell Signaling), rabbit anti‐phospho‐Akt (Ser473; 1:1000; Cell Signaling), mouse anti‐PCNA (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and mouse anti‐actin (1:5,000; Santa Cruz Biotechnology). The blots were analyzed with densitometry using ImageJ software (NIH, Bethesda, MD, USA).
Morphological analysis and quantifications
The middle and tail of the pancreas of each rat were harvested and separated into three or four samples. The pancreas samples were sectioned at 7 μm. A total of 50 consecutive sections were carried out from each pancreas sample. After the immunofluorescence staining of serial sections, the number of different cell types in each islet was manually counted in the islet microscopy images. For each cell type, 80–100 representative islets from 6 to 10 rats were counted.
Statistical analysis
All data are presented as mean ± standard deviation. The statistical analysis was carried out using the SPSS v.14.0.1 software for Windows (SPSS, Chicago, IL, USA). Differences between means were assessed using a Student's t‐test, Mann–Whitney test, χ2‐test or by one‐way anova when required. Group differences at the level of P < 0.05 were considered statistically significant.
Results
Mild hyperglycemia incubation reversed hyperglycemia and restored islet function in insulin‐deficient diabetic rats
To evaluate the effect of mild hyperglycemia in restoring islet function of diabetic rats, 8‐week‐old male Sprague–Dawley rats were treated with a single high dose of STZ (65 mg/kg), and 48 h post‐STZ injection newly diabetic rats (BG >25 mmol/L) were randomly treated with: no treatment to maintain hyperglycemia (referred to as HG, n = 6), daily exogenous injection of long‐acting human insulin analog (glargine insulin) to maintain mild hyperglycemia (15 mmol/L < BG < 18 mmol/L, referred to as mHG, n = 15), and daily exogenous long‐acting human insulin analog injection to restore normoglycemia (BG <8 mmol/L, referred to as NG), as insulin injection itself might generate beneficial effects for β‐cell regeneration and function. Six normal rats were used as the normal control (Control). Random BG levels, glucose disposal and insulin‐release during intraperitoneal glucose tolerance tests, and fasting serum rat insulin levels were monitored at indicated time‐points for up to 90 days in different groups.
HG developed severe and irreversible hyperglycemia afterwards. Insulin dosage in NG gradually increased during the analysis period, the BG level rebounded back to approximately 25 mmol/L after insulin withdrawal at day 80 and islet function slightly improved as shown in intraperitoneal glucose tolerance tests at the end of the observation (Figures 1a–d and S1a,b). Remarkably, insulin treatment was no longer required after 1 month of insulin injection in mHG. At the end of the observation, the mHG islets were almost functionally indistinguishable from the normal control. Rats in the mHG group achieved near normal glucose clearance, insulin secretion and random BG levels (Figures 1a–d and S1a,b).
Figure 1.
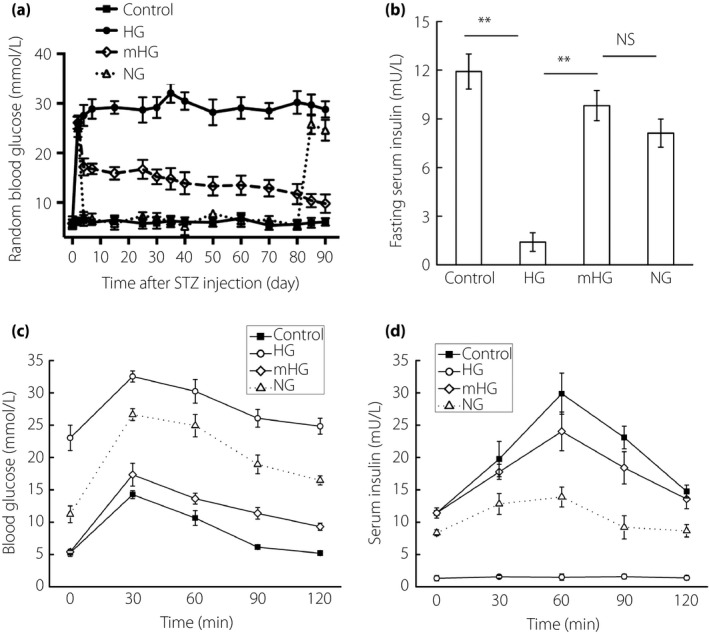
Chronic mild hyperglycemia incubation reversed diabetes in single high‐dose streptozotocin (STZ)‐treated rats. Eight‐week‐old male SD rats were treated with a single high dose of STZ (65 mg/kg), and 48 h post‐STZ injection newly diabetic rats (blood glucose >25 mmol/L) were randomly treated with: no treatment (HG; n = 6), daily exogenous long‐acting human insulin analog injection that restored normoglycemia (NG; blood glucose <8 mmol/L, n = 8), or maintained mild hyperglycemia (mHG; 15 mmol/L < blood glucose < 18 mmol/L, n = 15). Six normal rats were used as normal controls. Insulin withdrawal was executed within 30 days in the mHG group and at day 80 in the NG group. (a) Random blood glucose levels were monitored for 90 days. (b) Fasting serum insulin levels were detected at the end of observation. (c,d) Blood glucose levels and insulin levels during intraperitoneal glucose tolerance tests at the end of observation. The results are presented as the mean ± standard deviation. **P < 0.01. NS, not significant.
In summary, the present data suggest that mild hyperglycemia facilitated reversal of hyperglycemia in insulin‐deficient diabetic rats.
Mild hyperglycemia induced robust β‐cell regeneration in insulin‐deficient diabetic rats
Next, the pancreata from all the groups were collected for histological processing. At 48 h post‐STZ injection, when insulin treatment started, the residual insulin+ cell number was 211 per islet, accounting for approximately 14.2% of the normal control level, and decreased to less than 3% at the end of the observation in the HG group. Using insulin to restore normoglycemia appeared to stabilize the β‐cell number, which accounted for approximately 13.7% of the normal control at the end of the observation. As anticipated, mild hyperglycemia resulted in a dramatic increase in β‐cell mass (Figure 2a). The number of insulin‐stained cells in each islet increased on average by a factor of 3 (from 211 ± 0.8 to 806 ± 14.8), corresponding to 50% of the normal β‐cell number (Figure 2b). The distribution of islet sizes also almost returned to normal, except that oversized islets with a diameter larger than 250 m, which were readily seen in the normal controls, remained rare in mHG at the end of the observation (Figure S2a).
Figure 2.
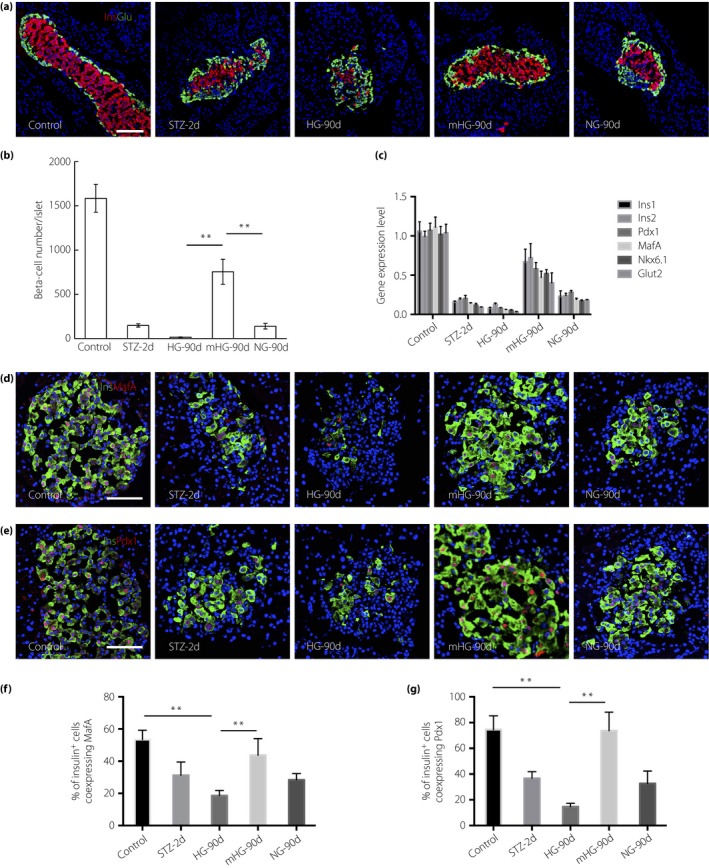
Mild hyperglycemia resulted in robust β‐cell regeneration. (a) Representative islets stained with antibodies against insulin (red) and glucagon (green) of normal rats (Control; n = 6), streptozotocin (STZ)‐treated rats at day 2 (STZ‐2d; n = 6) and rats at day 90 after no treatment (HG‐90d; n = 6), mild hyperglycemia incubation (mGH‐90d; n = 8) and euglycemia incubation (NG‐90d; n = 8). Nuclei were labeled with 40′,6‐diamidino‐2‐phenylindole. Scale bars, 100 μm. (b) Quantification of β‐cell number. The results are presented as the mean ± standard deviation. **P < 0.01. (c) Expression of β‐cell enriched genes in isolated islets (pooled from five rats). Gene expression levels were shown as fold change to the levels of normal rats. The results are presented as the mean ± standard deviation. (d) Photomicrographs of islets double stained with anti‐insulin (green) and anti‐v‐maf musculopeoneurotic fiberosarcoma oncogene homologue A (MafA; red) antibodies of Control, STZ‐2d and HG‐90d, mGH‐90d and NG‐90d. Nuclei were labeled with 40′,6‐diamidino‐2‐phenylindole. Scale bars, 50 μm. (e) Photomicrographs of islets double stained with anti‐insulin (green) and anti‐Pdx1 (red) antibodies. Nuclei were labeled with 40′,6‐diamidino‐2‐phenylindole. Scale bars, 50 μm. (f) The percentage of insulin+ cells co‐expressing MafA (n = 5) was quantified. The results are presented as the mean ± standard deviation. **P < 0.01. (g) The percentage of insulin+ cells co‐expressing Pdx1 (n = 5) was quantified. The results are presented as the mean ± standard deviation. **P < 0.01.
Glucose is known to activate nutrient signaling pathways that increase cell size15, 16. Measurement of β‐cell size by manually tracing cell borders showed no evidence of increased β‐cell size in mHG (Figure S2b). Thus, mild hyperglycemia for less than a month in rats does not induce β‐cell hypertrophy.
Gene expression quantification showed that several β‐cell markers were markedly downregulated in HG, but were significantly unregulated in mHG, consistent with the dramatic β‐cell regeneration observed. Limited improvement was detected in NG (Figure 2c). We further examined the expression of β‐cell‐enriched transcription factor, MafA, at the protein level in β‐cells in all the groups. We found that in control rats, not all of, but just half of the β‐cells showed MafA expression, which showed that the efficiency of the antibody is not very high. Long‐term hyperglycemia significantly deteriorated MafA expression, whereas the percentage of regenerated β‐cells co‐expressing MafA in mHG rats was comparable with the normal control (Figure 2d,f). Similar results were obtained when we stained another β‐cell marker, Pdx1, in normal rats and experimental groups (Figure 2e,g).
Taken together, the data suggested that mild hyperglycemia induced robust in situ β‐cell regeneration in the STZ‐induced insulin‐deficient diabetic rats.
Mild hyperglycemia stimulated β‐cell proliferation in insulin‐deficient diabetic rats
Consistent with previous reports, the present results showed that 15 mmol/L glucose induced INS1 cell proliferation in a PI3K/Akt‐dependent manner in vitro (Figure 3a–c). So we wondered if mild hyperglycemia promoted β‐cell regeneration through β‐cell replication in vivo. To accomplish that, we stained the insulin‐positive cells with Ki67, a protein expressed by cells in replicative phases, to show dividing β‐cells. Early adulthood β‐cells in rats had a very slow turnover with a replication rate of ~2%. It is well known that hyperglycemia deteriorates β‐cell proliferation. Indeed, after STZ treatment, the proliferation rates maintained at this low level at 48 h, and even had the tendency to further decrease afterwards in HG rats. However, a wave of β‐cell replication occurred in mHG; β‐cell proliferation rate increased four‐ to fivefold soon after glucose regulation. Proliferation of β‐cells persisted at this high level before normoglycemia was restored (Figure 3d,e). The increased β‐cell proliferation observed in mHG was profoundly blunted in NG rats, which was maintained at a level slightly higher than the control rats. The expression of the PI3K/Akt pathway protein in isolated islets was also tested in all the groups. The expression levels of PI3k and pAkt markedly increased in the islets of mHG (Figure 3f).
Figure 3.
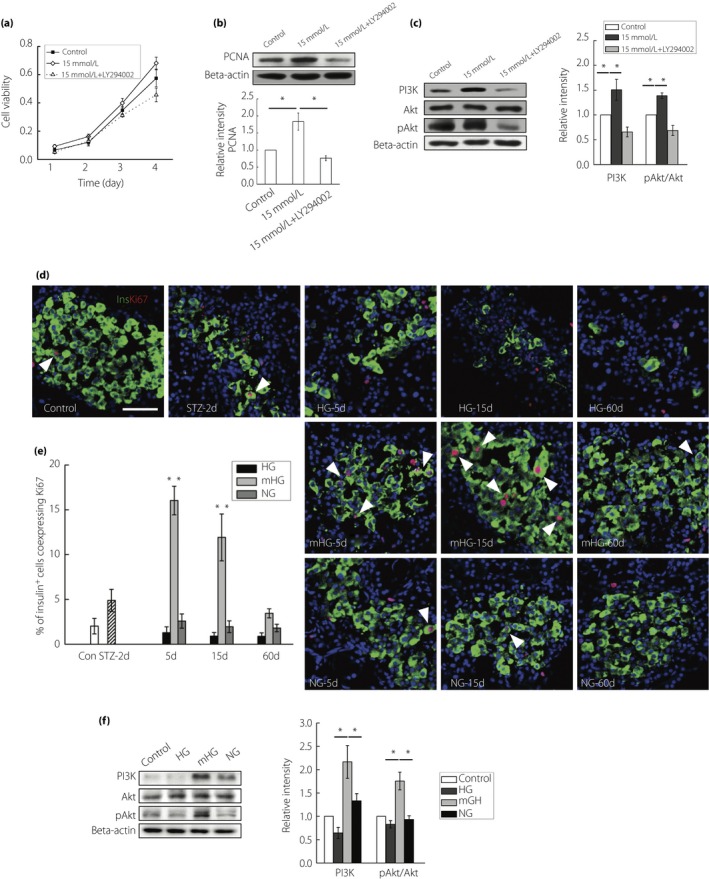
Mild hyperglycemia enhanced β‐cell replication. (a) INS1 cells were treated with 11.1 mmol/L glucose, 15 mmol/L glucose or a combination of 15 mmol/L glucose and a PI3K inhibitor (Ly294002, 10 mol/L). Cell viability of these cells was detected at indicated time‐points. Values are mean ± standard deviation of three individual experiments. (b) Western blot detection of proliferating cell nuclear antigen (PCNA) in INS1 cells exposed to 11.1 mmol/L glucose, 15 mmol/L glucose or a combination of 15 mmol/L glucose and 10 mol/L Ly294002, with densitometry analysis. (c) Western blot detection of PI3K, Akt and pAkt in INS1 cells exposed to 11.1 mmol/L glucose, 15 mmol/L glucose or a combination of 15 mmol/l glucose and 10 mol/L Ly294002, with densitometry analysis. (d) Representative islets stained with antibodies against insulin (green) and Ki67 (red) of normal rats (Control; n = 5), streptozotocin (STZ)‐treated rats at day 2 (STZ‐2d; n = 5), and rats at day 5, day 15 and day 60 with no treatment (HG‐5d, HG‐15d, HG‐60d; n = 4), mild hyperglycemia incubation (mHG‐5d, mHG‐15d, mHG‐60d; n = 4) and euglycemia incubation (NG‐5d, NG‐15d, NG‐60d; n = 4). The arrows: insulin+ cells showing Ki67 expression. Nuclei were labeled with 40′,6‐diamidino‐2‐phenylindole. Scale bars, 50 μm. (e) Quantification of insulin+ cells co‐expressing Ki67 was shown. The results are presented as the mean ± standard deviation. (f) Western blot detection of PI3K, Akt and pAkt in isolated islets of normal rats, and rats at day 15 with no treatment (HG; n = 3), hyperglycemia incubation (mHG; n = 3) and euglycemia incubation (NG; n = 3), with densitometry analysis. The results of western blot are presented as the mean ± standard deviation from three independent experiments. *P < 0.05, **P < 0.01.
We also assayed for putative pancreatic progenitor cells during β‐cell regeneration17, 18. Immunostaining and reverse transcription PCR showed no upregulation of the embryonic endocrine progenitor cell marker neurogenin‐3 in regenerating pancreata (Figure S3b,c), which was readily detected in the embryonic pancreas (Figure S3a). We also searched for cells co‐expressing insulin and glucagon, proposed by some reports to represent β‐cell progenitors after near‐total β‐cell ablation19, 20. The frequency of insulin+/glucagon+ cells increased from 1:5,500 β‐cells in normal control rats to 1:800 β‐cells in the NG group and 1:1,000 in the mHG group (Figure S4). If these double hormone‐positive cells are progenitor cells, their number in this context appeared too low to make a significant contribution to β‐cell regeneration.
Thus, the present data suggested that chronic residual β‐cell proliferation was markedly enhanced after chronic mild hyperglycemia incubation and could account for the regeneration observed in our model.
Mild hyperglycemia did not induce β‐cell apoptosis in insulin‐deficient diabetic rats
It is well accepted that prolonged exposure to elevated glucose concentrations both in vitro 21, 22, 23 and in vivo exerts deleterious or toxic effects on the β‐cell phenotype24, 25. Therefore, we next sought to investigate the impact of moderately elevated glucose levels on β‐cell survival. Consistent with previous reports, exposure of INS1 cells to 35 mmol/L glucose increased apoptosis, whereas 15 mmol/L glucose had no such effects (Figure 4a). In vivo, the proportion of insulin+ cells expressing terminal dexynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL)‐positive was determined 5 days post‐insulin treatment. Pancreas sections were incubated with DNase I recombinant for 10 min to induce DNA strand breaks before TUNEL labeling as a positive control (Figure 4b). TUNEL staining showed a slight increase in the number of apoptotic cells within diabetic islets from HG, compared with islets from control. When normoglycemia was restored by exogenous insulin, the proportion of apoptotic β‐cells decreased to a level comparable with that of the normal control. Importantly, mild hyperglycemia did not induce β‐cell apoptosis (Figure 4c,d).
Figure 4.
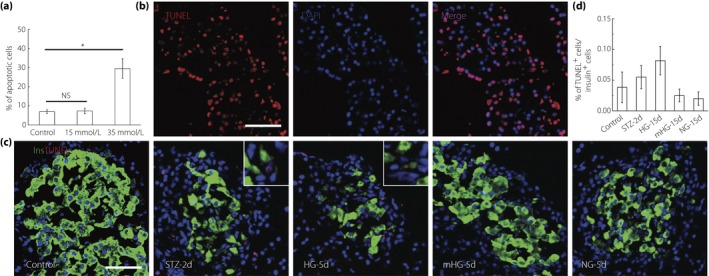
Mild hyperglycemia did not induce β‐cell apoptosis. (a) Apoptosis quantification in INS1 cells after 72‐h culture in 11.1 mmol/L, 15 mmol/L and 35 mmol/L glucose. Values are mean ± standard deviation (n = 6) of three individual experiments. *P < 0.05. (b) Pancreas sections were incubated with DNase I recombinant for 10 min to induce deoxyribonucleic acid strand breaks before terminal dexynucleotidyl transferase‐mediated dUTP nick end labeling labeling (red) as positive control. Nuclei were labeled with 40′,6‐diamidino‐2‐phenylindole. Scale bars, 50 μm. (c) Representative islets labeling with insulin (green) and terminal dexynucleotidyl transferase‐mediated dUTP nick end labeling (red) of normal rats (Control; n = 6), streptozotocin (STZ)‐treated rats at day 2 (STZ‐2d; n = 6), and rats at day 5 with no treatment (HG‐5d; n = 6), mild hyperglycemia incubation (mHG‐5d; n = 6) and euglycemia incubation (NG‐5d; n = 6). Nuclei were labeled with 40′,6‐diamidino‐2‐phenylindole. Scale bars, 50 μm. (d) Quantification of apoptosis. The results are presented as the mean ± standard deviation.
Mild hyperglycemia slightly increased α‐cell proliferation in insulin‐deficient diabetic rats
The α‐cell in pancreatic islets plays an essential counterpart and regulatory role to the insulin‐producing β‐cell in the regulation of BG homeostasis26. It has become increasingly evident that glucagon excess accounts for many diabetic manifestations27, 28, 29. In addition, previous studies have noted the increase of α‐cell composition in diabetes patients and self‐duplication in diabetic animal models30, 31, 32, 33. Therefore, we assessed α‐cell number and proliferation dynamically as well as glucagon secretion at indicated time‐points.
Quantification of the α‐cells in each islet showed a subtle increase in α‐cell number at day 5 in HG and a decrease afterwards, with the number even lower than that in normal islets, which might be explained by the general toxicity related to the high BG levels at these points. A slight, statistically significant, increase in α‐cell mass was noted in mHG, but not in NG (Figure 5c). Quantification of the percentage of Ki67+ α‐cells showed that α‐cell proliferation rate increased one‐ to twofold at day 5 after glucose regulation, and maintained at this high level before normoglycemia was restored. Increased α‐cell proliferation was not observed in NG (Figure 5a,b). Enzyme‐linked immunosorbent assay showed that serum glucagon level reached approximately twofold of the normal control at day 5, and further increased in the HG group afterwards despite the decreased α‐cell number, whereas serum glucagon in the mHG group was maintained at a level comparable with that of the normal controls despite the increased α‐cell number (Figure 5d).
Figure 5.
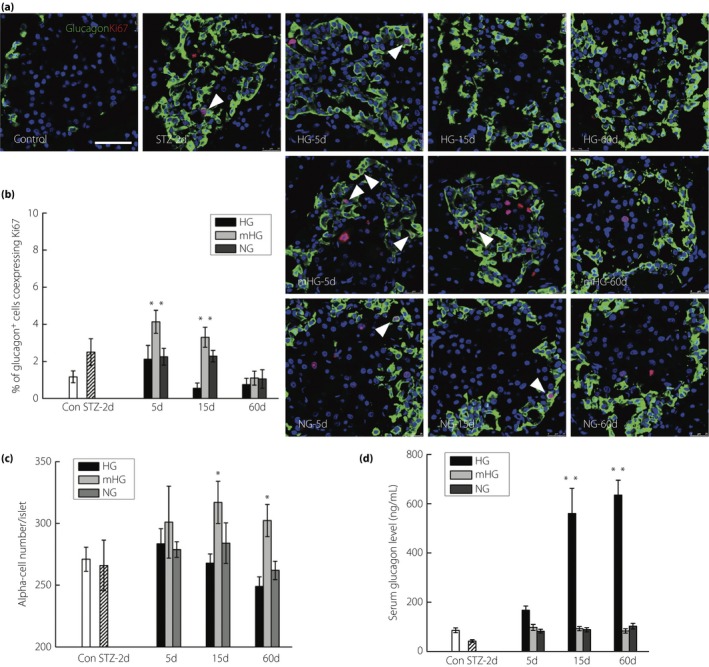
Chronic mild hyperglycemia slightly enhanced α‐cell replication with no excess glucagon secretion. (a) Representative islets stained with antibodies against glucagon (green) and Ki67 (red) of normal rats (Control; n = 5), streptozotocin (STZ)‐treated rats at day 2 (STZ‐2d; n = 5), and rats at day 5, day 15 and day 60 with no treatment (HG‐5d, HG‐15d, HG‐60d; n = 4), mild hyperglycemia incubation (mHG‐5d, mHG‐15d, mHG‐60d; n = 4) and euglycemia incubation (NG‐5d, NG‐15d, NG‐60d; n = 4). The arrows: glucagon+ cells showing Ki67 expression. Nuclei were labeled with 40′,6‐diamidino‐2‐phenylindole. Scale bars, 50 μm. (b) Quantification of glucagon+ cells co‐expressing Ki67. The results are presented as the mean ± standard deviation. *P < 0.05, **P < 0.01. Quantification of (c) α‐cell number was shown in and (d) serum glucagon level. The results are presented as the mean ± standard deviation. *P < 0.05, **P < 0.01.
In conclusion, mild hyperglycemia incubation slightly increased α‐cell proliferation and α‐cell mass, but glucagon excess did not occur in the experimental rats.
Discussion
The present study showed that mild hyperglycemia triggered β‐cell proliferation in single high‐dose STZ‐induced diabetic rats, leading to robust β‐cell regeneration, and almost full islet function recovery without excess glucagon secretion and β‐cell apoptosis. A principal goal in diabetes research is to develop viable therapeutic approaches to induce adult human β‐cell replication/expansion for regeneration therapies in patients with type 1 diabetes and/or type 2 diabetes34. In the current context, adult human β‐cells do not replicate in response to the long list of growth factors, nutrients and maneuvers that induce rodent β‐cells to replicate35. The natural mitogen glucose, however, has been shown to stimulate human adult β‐cells both in vitro and in vivo 7, 36, showing that it is possible that our procedure could be duplicated in humans and open a new door for diabetes therapy.
However, several issues need to be addressed before translation of these experimental results from bench to bedside. The most effective glucose concentration range of human β‐cell proliferation is unclear. An in vivo model might be necessary to acquire accurate data. A varying number of human islets could be transplanted under the kidney capsule of STZ‐induced diabetic severe combined immunodeficiency mice to investigate the effect of varying glucose levels on human β‐cell proliferation. In addition, in both human diabetes patients and diabetes rat models, long‐term chronic mild hyperglycemia inevitably resulted in diabetic complications, such as blindness and kidney failure. In the present study, mild hyperglycemia lasted less than a month in the diabetic rat model, and the β‐cell mass and function greatly recovered. Therefore, short‐term mild hyperglycemia incubation might be more appropriate to stimulate β‐cell regeneration and to avoid diabetic complications in clinical use.
STZ preferentially accumulates in pancreatic β‐cells through the Glut2 glucose transporter, and therefore specifically destroys β‐cells in the pancreas. A single high dose of STZ induced acute and massive β‐cell injury, leading to pure insulin‐deficient diabetes without autoimmunity attack as in type 1 diabetes, or insulin resistance, obesity and inflammation as in type 2 diabetes. The discrepancy between our rodent model and human diabetes would generate some issues when applying our procedure in clinic. In type 1 diabetes37, the regenerated β‐cells using our protocol theoretically could not escape from the autoimmune attack. Stem cell educator therapy has been reported to reverse autoimmunity in type 1 diabetes patients38. The combination of stem cell educator therapy and mild hyperglycemia incubation might result in greater clinical improvement in type 1 diabetes patients.
β‐Cell function is decreased by approximately 80% in patients with impaired glucose tolerance, and is even less in patients with type 2 diabetes39. Short‐term intensive insulin therapy in patients with newly diagnosed type 2 diabetes can induce a glycemic remission and has favorable outcomes on the maintenance of β‐cell function40. Besides impaired β‐cell function, several studies also reported a significant (30–60%) decrease in β‐cell mass in patients with type 2 diabetes41, 42. According to the present results, maintaining a moderate elevated BG level for a short‐term instead of short‐term intensive insulin therapy might stimulate β‐cell proliferation, markedly increase the β‐cell mass and therefore show better results. However, type 2 diabetes patients are often associated with elevated free fatty acid levels, which have been shown to restrain glucose‐stimulated β‐cell proliferation in vivo and in vitro 43, and to further promote apoptotic cell death25. Increased pro‐inflammatory cytokines and islet amyloid deposition are evident in type 2 diabetes44, which also link to β‐cell failure and might have negative effects on mild hyperglycemia‐induced β‐cell regeneration. Therefore, the therapeutic effects of our approach in type 2 diabetes patients should be carefully evaluated in the future.
The present results showed a slight increase of α‐cell proliferation rate and α‐cell number after mild hyperglycemia incubation. However, inconsistent with α‐cell mass expansion, glucagon secretion in the mHG group was far below that in the HG group, and showed no significant difference with the normal controls. For isolated α‐cells, glucose (16 mmol/L) was mildly stimulatory for glucagon secretion in the absence of other islet cell types, whereas the stimulatory effect was prevented on intact islets as intra‐islet β‐cell secretory products were proved to directly inhibit glucagon secretion45, 46. In addition, with an α‐cell‐specific insulin receptor knockout mouse, Dan Kawamori et al.47 provided genetic evidence that insulin signaling is critical for the regulation of glucagon secretion in vivo. We postulated that the enhanced secretion of β‐cell secretory product from the rapidly expanded β‐cell mass in the mHG group inhibited glucagon secretion of α‐cells, therefore avoiding α‐cell dysfunction, as in the HG group.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1 | Recovery from diabetes after mild hyperglycemia incubation.
Figure S2 | Rapid β‐cell regeneration after mild hyperglycemia treatment.
Figure S3 | Little evidence for the presence of embryonic endocrine progenitor cells in the regenerating pancreas.
Figure S4 | Detection of a few cells co‐expressing insulin and glucagon in regenerating islets.
Table S1 | List of sequences of forward and reverse primers.
Acknowledgments
This research was supported in part by the National Basic Science and Development Program (2012CB518103), the 863 Projects of Ministry of Science and Technology of China (2013AA020105 and 2012AA020502), and the National Nature Science Foundation of China (no. 81200615 and 81121004). The funders were not involved in study design, data collection and analysis, or the decision to publish.
J Diabetes Investig 2017; 8: 44–55
Contributor Information
Yiming Mu, Email: muyiming@301hospital.com.cn.
Weidong Han, Email: hanwdrsw69@yahoo.com.
References
- 1. Atkinson MA, Bluestone JA, Eisenbarth GS, et al How does type 1 diabetes develop?: the notion of homicide or beta‐cell suicide revisited. Diabetes 2011; 60: 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell 2013; 153: 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3. Porat S, Weinberg‐Corem N, Tornovsky‐Babaey S, et al Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 2011; 13: 440–449. [DOI] [PubMed] [Google Scholar]
- 4. Hugl SR, White MF, Rhodes CJ. Insulin‐like growth factor I (IGF‐I)‐stimulated pancreatic β‐cell growth is glucose‐dependent: synergistic activation of insulin receptor substrate‐mediated signal transduction pathways by glucose and IGF‐I in INS‐1 cells. J Biol Chem 1998; 273: 17771–17779. [DOI] [PubMed] [Google Scholar]
- 5. Assmann A, Ueki K, Winnay JN, et al Glucose effects on beta‐cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol 2009; 29: 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chick WL. Beta cell replication in rat pancreatic monolayer cultures. Effects of glucose, tolbutamide, glucocorticoid, growth hormone and glucagon. Diabetes 1973; 22: 687–693. [DOI] [PubMed] [Google Scholar]
- 7. Metukuri MR, Zhang P, Basantani MK, et al ChREBP mediates glucose‐stimulated pancreatic beta‐cell proliferation. Diabetes 2012; 61: 2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang H, Li W, Wang Q, et al Glucose‐mediated repression of menin promotes pancreatic beta‐cell proliferation. Endocrinology 2012; 153: 602–611. [DOI] [PubMed] [Google Scholar]
- 9. Bonner‐Weir S, Deery D, Leahy JL, et al Compensatory growth of pancreatic beta‐cells in adult rats after short‐term glucose infusion. Diabetes 1989; 38: 49–53. [DOI] [PubMed] [Google Scholar]
- 10. Bernard C, Thibault C, Berthault MF, et al Pancreatic beta‐cell regeneration after 48‐h glucose infusion in mildly diabetic rats is not correlated with functional improvement. Diabetes 1998; 47: 1058–1065. [DOI] [PubMed] [Google Scholar]
- 11. Paris M, Bernard‐Kargar C, Berthault MF, et al Specific and combined effects of insulin and glucose on functional pancreatic beta‐cell mass in vivo in adult rats. Endocrinology 2003; 144: 2717–2727. [DOI] [PubMed] [Google Scholar]
- 12. Alonso LC, Yokoe T, Zhang P, et al Glucose infusion in mice: a new model to induce beta‐cell replication. Diabetes 2007; 56: 1792–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res 2001; 50: 537–546. [PubMed] [Google Scholar]
- 14. Lenzen S. The mechanisms of alloxan‐ and streptozotocin‐induced diabetes. Diabetologia 2008; 51: 216–226. [DOI] [PubMed] [Google Scholar]
- 15. Nie J, Liu X, Lilley BN, et al SAD‐A kinase controls islet beta‐cell size and function as a mediator of mTORC1 signaling. Proc Natl Acad Sci USA 2013; 110: 13857–13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Das F, Ghosh‐Choudhury N, Dey N, et al High glucose forces a positive feedback loop connecting Akt kinase and FoxO1 transcription factor to activate mTORC1 kinase for mesangial cell hypertrophy and matrix protein expression. J Biol Chem 2014; 289: 32703–32716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu X, D'Hoker J, Stange G, et al Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008; 132: 197–207. [DOI] [PubMed] [Google Scholar]
- 18. Solar M, Cardalda C, Houbracken I, et al Pancreatic exocrine duct cells give rise to insulin‐producing beta cells during embryogenesis but not after birth. Dev Cell 2009; 17: 849–860. [DOI] [PubMed] [Google Scholar]
- 19. Thorel F, Nepote V, Avril I, et al Conversion of adult pancreatic alpha‐cells to beta‐cells after extreme beta‐cell loss. Nature 2010; 464: 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chung CH, Hao E, Piran R, et al Pancreatic beta‐cell neogenesis by direct conversion from mature alpha‐cells. Stem Cells 2010; 28: 1630–1638. [DOI] [PubMed] [Google Scholar]
- 21. Poungvarin N, Lee JK, Yechoor VK, et al Carbohydrate response element‐binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012; 55: 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanchang W, Semprasert N, Limjindaporn T, et al Testosterone protects against glucotoxicity‐induced apoptosis of pancreatic beta‐cells (INS‐1) and male mouse pancreatic islets. Endocrinology 2013; 154: 4058–4067. [DOI] [PubMed] [Google Scholar]
- 23. Maedler K, Sergeev P, Ris F, et al Glucose‐induced beta cell production of IL‐1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002; 110: 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bensellam M, Laybutt DR, Jonas JC. The molecular mechanisms of pancreatic beta‐cell glucotoxicity: recent findings and future research directions. Mol Cell Endocrinol 2012; 364: 1–27. [DOI] [PubMed] [Google Scholar]
- 25. Kim JW, Yoon KH. Glucolipotoxicity in pancreatic beta‐cells. Diabetes Metab J 2011; 35: 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vuguin PM, Charron MJ. Novel insight into glucagon receptor action: lessons from knockout and transgenic mouse models. Diabetes Obes Metab 2011; 13(Suppl 1): 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luyckx A. The role of glucagon in hyperglycemia. A review (author's transl). Diabete Metab 1975; 1: 201–208. [PubMed] [Google Scholar]
- 28. Unger RH, Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1975; 1: 14–16. [DOI] [PubMed] [Google Scholar]
- 29. Tsuchiyama N, Takamura T, Ando H, et al Possible role of alpha‐cell insulin resistance in exaggerated glucagon responses to arginine in type 2 diabetes. Diabetes Care 2007; 30: 2583–2587. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y, Zhang Y, Bone RN, et al Regeneration of pancreatic non‐beta endocrine cells in adult mice following a single diabetes‐inducing dose of streptozotocin. PLoS One 2012; 7: e36675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bonner‐Weir S, O'Brien TD. Islets in type 2 diabetes: in honor of Dr. Robert C. Turner. Diabetes 2008; 57: 2899–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Z, Karlsson FA, Sandler S. Islet loss and alpha cell expansion in type 1 diabetes induced by multiple low‐dose streptozotocin administration in mice. J Endocrinol 2000; 165: 93–99. [DOI] [PubMed] [Google Scholar]
- 33. Yoon KH, Ko SH, Cho JH, et al Selective beta‐cell loss and alpha‐cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 2003; 88: 2300–2308. [DOI] [PubMed] [Google Scholar]
- 34. Bernal‐Mizrachi E, Kulkarni RN, Scott DK, et al Human beta‐cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes 2014; 63: 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kulkarni RN, Mizrachi EB, Ocana AG, et al Human beta‐cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 2012; 61: 2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levitt HE, Cyphert TJ, Pascoe JL, et al Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD‐severe combined immunodeficiency (SCID) mice. Diabetologia 2011; 54: 572–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Borowiak M, Melton DA. How to make beta cells? Curr Opin Cell Biol 2009; 21: 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao Y, Jiang Z, Zhao T, et al Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood‐derived multipotent stem cells. BMC Med 2012; 10: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeFronzo RA, Eldor R, Abdul‐Ghani M. Pathophysiologic approach to therapy in patients with newly diagnosed type 2 diabetes. Diabetes Care 2013; 36(Suppl 2): S127–S138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weng J, Li Y, Xu W, et al Effect of intensive insulin therapy on beta‐cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel‐group trial. Lancet 2008; 371: 1753–1760. [DOI] [PubMed] [Google Scholar]
- 41. Butler AE, Janson J, Bonner‐Weir S, et al Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110. [DOI] [PubMed] [Google Scholar]
- 42. Rahier J, Guiot Y, Goebbels RM, et al Pancreatic beta‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008; 10(Suppl 4): 32–42. [DOI] [PubMed] [Google Scholar]
- 43. Pascoe J, Hollern D, Stamateris R, et al Free fatty acids block glucose‐induced beta‐cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes 2012; 61: 632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011; 11: 98–107. [DOI] [PubMed] [Google Scholar]
- 45. Franklin I, Gromada J, Gjinovci A, et al Beta‐cell secretory products activate alpha‐cell ATP‐dependent potassium channels to inhibit glucagon release. Diabetes 2005; 54: 1808–1815. [DOI] [PubMed] [Google Scholar]
- 46. Salehi A, Vieira E, Gylfe E. Paradoxical stimulation of glucagon secretion by high glucose concentrations. Diabetes 2006; 55: 2318–2323. [DOI] [PubMed] [Google Scholar]
- 47. Kawamori D, Kurpad AJ, Hu J, et al Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 2009; 9: 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 | Recovery from diabetes after mild hyperglycemia incubation.
Figure S2 | Rapid β‐cell regeneration after mild hyperglycemia treatment.
Figure S3 | Little evidence for the presence of embryonic endocrine progenitor cells in the regenerating pancreas.
Figure S4 | Detection of a few cells co‐expressing insulin and glucagon in regenerating islets.
Table S1 | List of sequences of forward and reverse primers.