

Abstract
Coxsackievirus B4 (CBV4), a member of the Picornavirus genus, has long been implicated in the development of insulin-dependent diabetes mellitus (IDDM) caused by virus-induced pancreatic cell damage. The progressive destruction of pancreatic β cells is responsible for the development of IDDM. It has recently been suggested that CBV4 infection can induce the production of proinflammatory cytokines, and these cytokines seem to be involved in the damage to the insulin-producing cells. In this study we investigated whether toll-like receptors (TLRs) are responsible for triggering the proinflammatory cytokine production in human pancreatic cells in response to CBV4. Here we demonstrate that CBV4 triggers cytokine production through a TLR4-dependent pathway. This interaction seems to be independent of virus attachment and cell entry.
Epidemiological studies have shown that enteroviruses are associated with chronic inflammatory and autoimmune diseases in humans (2). One such disease is insulin-dependent diabetes mellitus (IDDM), also known as type I diabetes, which is caused by progressive destruction of pancreatic β cells. Enterovirus and, in particular, coxsackievirus infections are associated with the development of type I diabetes (14, 33). The most commonly detected strain in diabetic and prediabetic patients is coxsackievirus B4 (CBV4) (14).
Although the pancreatropic nature of this virus is well documented, how it causes degenerate destruction of large regions of exocrine pancreas as well as β islets, leading to IDDM, is not very well understood. It has been shown that CBV4 infection can induce the production of proinflammatory cytokines such as interleukin-1 (IL-1) beta and tumor necrosis factor-alpha (TNF-α) (31). Even though the known role of these cytokines in host defense against infection leads us to believe that they may have a protective role, recent studies suggest that proinflammatory cytokines might have a negative effect on pancreatic function. It has been suggested that the cytokines produced in response to the virus might play an important role in the pathogenesis of IDDM (5). In particular, the involvement of TNF-α in the damage of the insulin producing cells has been reported previously (5).
Viruses have long been known to be able to activate the innate immune response and to induce the production of proinflammatory cytokines (24, 25). The mechanism by which viruses trigger this inflammatory process upon infection has long remained unknown. It is now emerging that toll-like receptors (TLRs) are involved in “sensing” microbial pathogens and triggering the induction and secretion of cytokines as part of the innate immune response. To date, a number of viruses have been shown to trigger innate responses via TLRs. Respiratory syncytial virus (18) and mouse mammary tumor virus (22) have been shown to trigger through TLR4, while measles virus (4) and human cytomegalovirus seem to trigger via TLR2 (7). It seems that at the very earliest stages of the virus-cell interactions, TLRs sense the virus and mount an inflammatory response against them.
In the case of the pancreatropic CBV4, the mechanism by which it triggers the production of proinflammatory cytokines upon infection of the pancreas is still unknown. In this study we investigated whether TLRs are also involved in the innate sensing of CBV4 and responsible for triggering the inflammatory cytokines that eventually lead to the destruction of insulin-producing cells. Using reporter cell lines, we demonstrate that CBV4 triggers the production of cytokines in human pancreatic cells through a TLR4-dependent pathway. TLR4-blocking monoclonal antibodies (MAbs) seem to be able to inhibit the production of proinflammatory cytokines in response to CBV4.
Interestingly, fluorescent imaging techniques demonstrated that TLR4 does not associate with the known virus receptor CAR (coxsackie adenovirus receptor) protein (3), thus demonstrating that the interaction of the virus with TLR4 is independent of virus attachment and cell entry.
MATERIALS AND METHODS
Cell lines.
A human pancreatic cell line (PANC-1) was maintained in Dulbecco's modified Eagle's medium with Glutamax and 4,500 mg of glucose medium (Gibco)/liter containing 10% heat-inactivated fetal bovine serum. Another human pancreatic cell line (ASPC-1) was maintained in RPMI medium with Glutamax (Gibco) containing 10% heat-inactivated fetal bovine serum. Chinese hamster ovary (CHO) cells transfected with human CD14 (hCD14) or with hCD14 and human (hTLR4) cDNA or with hCD14 and hTLR2 cDNA in a reporter background were constructed as previously described (9). CHO cells were maintained in Ham's F12 medium (Gibco BRL) supplemented with 2 mM l-glutamine, 7.5% fetal calf serum, and 500 μg of gentamicin sulfate (G418; Sigma)/ml. Cells were grown in 80-cm3 tissue culture flasks (Nunc). Trypsin-EDTA (0.05% trypsin-0.53 mM EDTA) was used for passaging the cells.
Human embryonic kidney 293 (HEK-239) and HEK-239 cells transfected with CD14 and TLR4 or with CD14 and TLR2 were maintained in Dulbecco's modified Eagle's medium containing 4.6 g of glucose/liter with 10% fetal calf serum, 5 μg of puromycin/ml, and 0.5 mg of G418 sulfate/ml.
Viruses.
The diabetogenic isolated strain CBV4-E2 was kindly provided by J.W. Yoon (33). UV inactivation of CBV4-E2 virions was performed in an UV transluminator for 30 min. UV inactivation was confirmed by performing plaque assays. The UV treatment described was sufficient to completely abolish virus infection.
Materials.
All fine chemicals were obtained from Sigma (Gillingham, United Kingdom). HTA-125, a TLR4-specific MAb which is known to be a function-blocking MAb, was obtained from Cambridge Biosciences (Cambridge, United Kingdom). TLR4-specific goat polyclonal serum was obtained from Santa Cruz (Santa Cruz, Calif.). The CAR-specific MAb C1079 was obtained from Europa Bioproducts. Horse polyclonal serum specific for CBV4 was purchased from the American Tissue Type Collection. Cholera toxin-tetramethyl rhodamine isothiocyanate was purchased from List Labs. The antibodies used for fluorescence resonance energy transfer (FRET) studies were conjugated to either Cy3 or Cy5 by use of labeling kits from Amersham Pharmacia (Bucks, United Kingdom).
Cytokine assays.
PBMCs were left unstimulated or were stimulated with CBV4 virions (103 PFU/ml). The cultures were incubated for the designated times. The supernatants were collected and frozen until the cytokine assays were performed. A Becton Dickinson cytometric bead array (CBA) system was used to determine the levels of multiple cytokines at the same time. The CBA system employs a series of beads with discrete fluorescence intensities that enable us to simultaneously detect multiple soluble cytokines. Each bead provides a capture surface for a cytokine and is analogous to a coated well in an enzyme-linked immunosorbent assay. In this study the human Th1/Th2 cytokine CBA kit-II was used to quantitatively measure IL-2, IL-4, IL-6, IL-10, TNF-α, and gamma interferon levels in a single sample.
Virus binding assayed by flow cytometry.
PANC-1 or ASPC-1 pancreatic cells (5 × 105) were harvested by scraping them off the flasks and fixed in 4% paraformaldehyde. The cells were washed with phosphate-buffered saline (PBS) and were subsequently incubated with 150 PFU of CBV4 in binding buffer (PBS supplemented with 2 mM CaCl2 and 1 mM MgCl2) at room temperature for an hour. Unbound viruses were removed by being washed three times in binding buffer followed by fixation using 4% paraformaldehyde (PFA). Cells were then washed and incubated with the appropriate dilution (1/500) of CBV4 polyclonal serum. Staining was visualized with fluorescein isothiocyanate (FITC)-conjugated goat anti-horse immunoglobulin (Stratech Immunochemicals) (1/500). Fluorescent staining was analyzed by flow cytometry using a FACSCalibur system (Becton Dickinson, Oxford, United Kingdom) and counting 10,000 cells per sample.
Virus binding inhibition.
PANC-1 or ASPC-1 pancreatic cells (5 × 105) were harvested by scraping them off the flasks. The cells were washed with PBS and incubated with different concentrations (1, 2, 4, 6, 8, and 10 μg/ml) of HTA-125, TLR4-specific MAb, TLR4-goat polyclonal serum, or irrelevant antibodies for 30 min on ice. After washing twice with binding buffer, virus binding was assayed as described above.
Plaque assays (infectivity assays).
For the production of virus plaques, 5 × 105 cells grown in six-well plates were infected with virus (multiplicity of infection of 0.01) and covered with a plaquing overlay. The plaquing overlay consisted of an appropriate medium to which 0.5% (wt/vol) carboxymethylcellulose was added. Plaques were visualized by staining with 0.2% (wt/vol) crystal violet in 1% (vol/vol) ethanol.
Virus entry inhibition.
For virus entry inhibition experiments, 5 × 105 cells in six-well plates were used as well as different concentrations (using a range of 1 to 10 μg/ml) of HTA-125, TLR4-specific antibody, TLR4-specific goat polyclonal serum, or irrelevant antibodies. The antibodies were added for 30 min in 1 ml of serum-free medium before the addition of the virus (multiplicity of infection of 0.01) and a further incubation for 50 min.
The cell monolayer was washed twice with culture medium and overlaid with 0.5% (wt/vol) carboxymethylcellulose in culture medium. The incubation was continued for another 72 h in a 7% CO2 incubator before plaque visualization. The efficiency of viral infection was analyzed by plaque assays. Plaques were visualized with 0.2% (wt/vol) crystal violet in 1% (vol/vol) ethanol.
Cell labeling for FRET.
PANC-1 or ASPC-1 cells on microchamber culture slides (Lab-tek; Gibco) were labeled with 100 μl of a mixture of donor-conjugated antibody Cy3 and acceptor-conjugated antibody Cy5. The cells were either stimulated with CBV4 or left unstimulated and were rinsed twice in PBS-0.02% bovine serum albumin prior to fixation with 4% formaldehyde for 15 min. The cells were fixed to prevent potential reorganization of the proteins during the course of the experiment.
Cells were imaged on a Carl Zeiss, Inc., LSM510 META confocal microscope (with an Axiovert 200 fluorescent microscope) using a 1.4-numerical aperture (NA) 63× Zeiss objective. The images were analyzed using LSM 2.5 image analysis software (Carl Zeiss, Inc.). Cy3 and Cy5 were detected using the appropriate filter sets. When typical exposure times (less than 5 s) for image acquisition were used, no fluorescence from a Cy3-labeled specimen was observed using the Cy5 filters and no Cy5 fluorescence was detected using the Cy3 filter sets.
FRET measurements.
FRET involves nonradiative transfer of energy from the excited state of a donor molecule to an appropriate acceptor (32). The rate of energy transfer (E) is inversely proportional to the 6th power of the distance r between the donor and the acceptor:
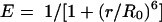 |
In the present study FRET was measured using a previously described method (17, 26, 27). Briefly, human pancreatic cells were cultured on microchamber culture slides (Lab-tek; Gibco) and labeled with 100 μl of a mixture of donor-conjugated antibody (Cy3) and acceptor-conjugated cholera toxin (Cy5). Energy transfer was detected as an increase in donor fluorescence (dequenching) after complete photobleaching of the acceptor molecule. Cells were imaged on a Carl Zeiss, Inc., LSM510 META confocal microscope (with an Axiovert 200 fluorescent microscope) using a 1.4-NA 63× Zeiss objective. Cy3 was excited with a helium-neon laser line emitting at 543 nm, whereas Cy5 was excited with a 633-nm laser line. Tracks were scanned sequentially, with only one laser and respective detector channel active per scan. The images were analyzed using LSM 2.5 image analysis software (Carl Zeiss, Inc.). FRET was calculated from the increase in donor fluorescence after acceptor photobleaching by the following equation:
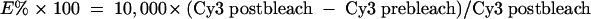 |
The scaling factor of 10,000 was used to expand E to the scale of the 12-bit images.
Isolation of lipid rafts.
Human pancreatic cells (100 × 106) were lysed in 500 μl of MEB buffer (150 mM NaCl, 20 mM morpholineethanesulfonic acid, pH 6.5) containing 1% Triton X-100 and protease inhibitors (500 μM phenylmethylsulfonyl fluoride and 5 mM iodoacetamide) for 1 h on ice. The cells were mixed with an equal volume of 90% sucrose in MEB buffer and placed at the bottom of a centrifuge tube. The sample was overlaid with 5.5 ml of 30% sucrose-4.5 ml of 5% sucrose in MEB buffer and centrifuged at 100,000 × g for 16 h. Fractions (1 ml) were gently removed from the top of the gradient, and n-octylglucoside was added to each fraction (60 μM final concentration) to solubilize rafts. For isolation of cellular membranes following CBV4 stimulation, PANC-1 cells were stimulated for 30 min at 37°C prior to solubilization in MEB buffer.
RESULTS
CBV4 activation of human pancreatic cells.
It has been previously shown that CBV4 can induce the production of proinflammatory cytokines in freshly isolated human leukocytes (31). Although CBV4 can infect many cell types, it displays a strong pancreatropic nature in vivo when a mouse model is used. To investigate its pancreatropic nature we infected human pancreatic cells (PANC-1) and performed plaque assays. It was shown that CBV4 was able to efficiently infect pancreatic cells (Fig. 1A). Similar results were obtained when we performed infectivity assays on ASPC-1 cells (data not shown). Thus, we proceeded to investigate whether CBV4 can induce inflammatory cytokines in human pancreatic cells.
FIG. 1.

CBV4 activation of human pancreatic cells investigated by infectivity assays. The results of plaque assays performed with non-UV-inactivated CBV4 (A) and UV-inactivated CBV4 (B) on PANC-1 cells are shown. Human pancreatic (PANC-1) cells were incubated with CBV4 virions (103 PFU/ml) (white bar charts) or with UV-inactivated CBV4 (black bar charts) for 1 h or were left untreated (grey bar charts) (C). The supernatants were harvested and assayed for cytokine secretion by use of a CBA system (Becton Dickinson). Fluorescence was detected using a FACSCalibur system (Becton Dickinson). The data represent the means of three independent experiments.
Human pancreatic cells (PANC-1) were incubated with CBV4 virions for different time periods. It was shown that CBV4 can induce TNF-α and, mainly, IL-6 cytokine production in these cells (Fig. 1C).
To further investigate whether CBV4 infection of pancreatic cells is required for the induction of cytokines, we incubated PANC-1 cells with UV-inactivated CBV4 particles, which were found not to be able to infect pancreatic cells (Fig. 1B). Although these virus particles are unable to infect cells, they were able to stimulate cytokine production (Fig. 1C), thus demonstrating that the initial contact between the viral particle and the cells is sufficient to stimulate the proinflammatory cytokine production.
CBV4-induced activation is TLR4 dependent.
To investigate the role that TLRs might play in CBV4-induced activation of human pancreatic cells we utilized transfected cell lines. HEK cells transfected with CD14 and TLR2 or with CD14 and TLR4 were utilized. HEK cells do not express TLR2 and TLR4 and thus were found not to be able to produce cytokines in response to the presence of CBV4 (Fig. 2). Similarly CBV4 did not trigger cytokine production in HEK cells transfected with CD14 and TLR2 (Fig. 2). In contrast, HEK cells transfected with CD14 and TLR4 produced cytokines after incubation with CBV4 virions (Fig. 2), thus suggesting that CBV4 induced activation in mediated via TLR4. Control cultures were stimulated with known TLR2 or TLR4 ligands.
FIG. 2.

Cytokine production is TLR4 dependent. HEK-293 (grey bar charts), HEK/CD14/TLR4 (black bar charts), and HEK/CD14/TLR2 (white bar charts) cells were incubated with no stimulus (medium alone) (Mock), 100 ng of LPS/ml, 10 μg of lipoteichoic acid/ml, CBV4 virions (103 PFU/ml), or UV-inactivated CBV4. The supernatants were harvested and assayed for cytokine secretion by use of a CBA system (Becton Dickinson). Fluorescence was detected using a FACSCalibur system (Becton Dickinson). The data represent the means of three independent experiments.
To determine whether the induction of cytokines was dependent on replication competence, we stimulated the transfected cell lines with UV-inactivated CBV4 virions. We found that UV-inactivated CBV4 virions were able to stimulate the production of proinflammatory cytokines only on HEK/CD14/TLR4 cells, thus demonstrating that infection of cells by CBV4 is not required to activate the innate immune response. In contrast, it seems that CBV4 triggers the innate immune response in its initial interaction with the cells and that the production of proinflammatory cytokines is independent of internalization and replication of CBV4 virions.
NF-κB transcriptional responses to CBV4 are mediated by TLR4.
Many of the inflammatory cytokines that were secreted in response to CBV4 stimulation are under the control of genes containing NF-κB elements in their promoters. TLRs seem to act upstream of NF-κB activation. TLR signaling pathways have been shown to ultimately result in the release of NF-κB from its endogenous inhibitor (1) and subsequent nuclear translocation that leads to the transcription of inflammatory cytokines.
To determine whether the CBV4-induced TLR4 activation that we observed leads to NF-κB-driven transcription response, we utilized reporter cell lines for our experiments. CHO cells expressing CD14 and human TLR2 or TLR4 which contains an NF-κB-CD25 reporter construct (12) were utilized. In these cell lines, when there is NF-κB-driven activation CD25 is expressed on the cell surface. Flow cytometry was used to test for CD25 expression on these cell lines in response to the presence of CBV4.
CD25 expression was found to be induced after stimulation with CBV4 virions in CHO/CD14/TLR4 cells but not in CHO/CD14/TLR2 cells (Fig. 3). These experiments demonstrate that CBV4 activates NF-κB in a TLR4-dependent manner. Control experiments with known TLR2 or TLR4 ligands were also performed.
FIG. 3.

TLR4-dependent NF-κB activation in response to CBV4. CHO (grey bar charts), CHO/CD14/TLR4 (black bar charts), and CHO/CD14/TLR2 (white bar charts) reporter cells were incubated with no stimulus (medium alone) (Mock), 1 μg of isotype control antibody/ml, 100 ng of LPS/ml, 10 μg of lipoteichoic acid/ml, CBV4 virions (103 PFU/ml), or UV-inactivated CBV4. At 1 h posttreatment, cell surface expression of CD25 was measured by flow cytometry. Fluorescence was detected using a FACSCalibur system (Becton Dickinson), counting 10,000 cells per sample. The percentages of CD25-positive cells are shown as the means and standard deviations from three independent experiments.
Inhibition of CBV4 activation of human pancreatic cells by TLR4-specific antibodies.
Since we had already demonstrated the importance of TLR4 in CBV4-mediated activation in model cell lines, we investigated whether TLR4 was also important for triggering the inflammatory response in human pancreatic cells.
Human pancreatic (PANC-1) cells were incubated with TLR4-specific MAb prior to incubation with CBV4 virions. Cytokine assays were performed after the designated incubation times. It was shown that preincubation with TLR4-specific MAbs inhibited CBV4-induced cellular activation (Fig. 4A), thus suggesting the importance of TLR4 in CBV4-mediated activation of human pancreatic cells. Similar results were obtained with ASPC-1 cells (data not shown).
FIG. 4.

TLR4-dependent IL-6 secretion in response to CBV4. Human pancreatic (PANC-1) cells were incubated with CBV4 virions (103 PFU/ml) (white bar charts) or UV-inactivated-CBV4 (black bar charts) for 1 h in the presence or absence of TLR4-specific MAb or TLR4-goat-specific serum (A). The supernatants were harvested and assayed for cytokine secretion using a CBA system (Becton Dickinson). Human pancreatic cells were incubated with CBV4 virions (103 PFU/ml) for 6 h. The supernatants were harvested every hour and assayed for cytokine secretion using a CBA system (Becton Dickinson) (B). Fluorescence was detected using a FACSCalibur system (Becton Dickinson). The data represent the means of three independent experiments.
TLR4-specific antibodies do not inhibit CBV4 infection of pancreatic cells.
Our experiments suggested that TLR4 was involved in triggering a proinflammatory cytokine production in response to CBV4 and that the TLR4-CBV4 interaction was independent of the internalization and replication competence of CBV4. We had concluded that CBV4 must interact with TLR4 in the initial stages of the virus-cell interaction. The results of the time course cytokine assays also supported this notion (Fig. 4B). During the 6-h infectious cycle of CBV4 it was shown that the maximum cytokine production occurred within the first hour of the virus-host interaction.
If TLR4 was associating with CBV4 in the initial stages of attachment, then TLR4 might play a role in the attachment step as a virus-receptor molecule. To investigate the role that TLR4 played in CBV4 binding, we preincubated human pancreatic (PANC-1) cells with TLR4-specific MAb and subsequently incubated them with CBV4 virions. Virus particles were detected using an antibody against the virus followed by the addition of a secondary antibody conjugated to FITC. The level of virus binding was determined by flow cytometry as previously described (28, 30). It was found that TLR4-specific MAbs could not inhibit CBV4 binding to human pancreatic cells (Fig. 5). The level of CBV4 binding was the same in pancreatic cells both in the presence and absence of TLR4-specific MAbs, thus leading us to believe that CBV4 does not directly bind to TLR4. Similar results were obtained with ASPC-1 cells (data not shown).
FIG. 5.

CBV4 binding is TLR4 independent. Human pancreatic (PANC-1) cells were incubated with CBV4 virions (102 PFU/ml) (white bar charts) or UV-inactivated-CBV4 (black bar charts) for 1 h. CBV4 binding was detected by using a CBV4 horse polyclonal serum followed by incubation with FITC-goat anti-horse immunoglobulin. Fluorescence was detected using a FACSCalibur system (Becton Dickinson), counting 10,000 cells per sample. The data represent the means of three independent experiments.
Since the attachment and internalization of certain viruses are often two different stages involving different receptors, we continued to investigate whether TLR4 was involved in the infectious cycle of CBV4. For these studies we performed plaque assays. Human pancreatic (PANC-1) cells were incubated with TLR4-specific MAbs prior to CBV4 infection. The level of infection was determined by plaque assays. TLR4-specific MAbs had no effect on the infectivity of CBV4. CBV4 appeared to have the same infectivity in the presence and absence of TLR4-specific MAbs (Fig. 6), thus suggesting that as we had previously speculated, TLR4 does not play any role in the attachment or internalization or replication of CBV4.
FIG. 6.

CBV4 infectivity is TLR4 independent. The results of plaque infectivity assays performed with CBV4 on PANC-1 cells in the presence (A) and absence (B) of TLR4-specific (HTA-125) MAb, CHO (C), CHO-CAR (D), and CHO-TLR4 (E) cells are shown.
In addition, to demonstrate that TLR4 is not involved in the CBV4 infectious cycle, we performed control experiments using CHO cells transfected with CAR, TLR4, or empty vector. Untransfected CHO cells are not permissive to virus infection. It was found that only CHO cells transfected with CAR were infected by the virus (Fig. 6), thus suggesting that TLR4 in not required for virus infection.
TLR4 is recruited in lipid rafts following CBV4 infection.
Accumulating evidence is suggesting the existence of specific partitioning of the plasma membrane in the form of cholesterol-enriched microdomains that are essential for cellular function (8, 11, 15, 16). These membrane microdomains are believed to perform diverse functions by providing a specialized microenvironment in which the relevant molecules for the specific biological functions accumulate (13).
The results of recent studies have indicated that these membrane microdomains seem to be involved in the recognition of microbial products and particularly in virus-cell interactions. Lipid rafts seem to play a role in the budding of human immunodeficiency virus (19, 21, 20) and influenza virus (23), comprising assembly sites of enveloped viruses, while involvement of rafts in virus entry has also been postulated (10).
In this study we wanted to investigate whether lipid rafts play an important role in CBV4-induced cellular activation. TLR4 has previously been found not to reside in lipid rafts but to be recruited there in response to bacterial stimuli such as lipopolysaccharide (LPS) (29). Here, using initially biochemical methods we isolated lipid raft fractions on the basis of their insolubility in Triton X-100 and low-level buoyant density in sucrose gradients (6).
GM-1 ganglioside, a raft-associated lipid, was detected using horseradish peroxidase-conjugated cholera toxin. We found that GM-1 ganglioside migrated near the top of the sucrose gradient (fractions 2 to 5), indicating that this procedure was effective in separating membrane rafts from the rest of the cellular membrane (Fig. 7). As expected, TLR4 was found not to reside in lipid rafts before incubation with CBV4 virions, but surprisingly it was recruited there following CBV4 stimulation (Fig. 7). Similarly, CAR was also recruited there upon CBV4 stimulation. Control experiments utilizing transferrin receptor MAb (since transferrin receptor is a well-known non-raft marker) as well as TLR2 (TL2.1) MAb revealed that neither transferrin receptor (Fig. 7) nor TLR2 (data not shown) was recruited in lipid rafts before or after CBV4 stimulation.
FIG. 7.

TLR4 recruitment within lipid rafts following CBV4 stimulation. Human pancreatic (PANC-1) cells were either treated with no stimulus (A, B, D, and F) or incubated with CBV4 virions (C, E, and G) prior to solubilization with 1% Triton X-100 buffer for 1 h on ice and then subjected to sucrose density gradient centrifugation. Fractions were collected from the top of the gradient, and equivalent portions of each fraction were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting. The lipid raft marker was detected using horseradish peroxidase-conjugated cholera toxin (A); the nitrocellulose membranes were also probed with TLR4-specific MAb (B and C), CAR-specific MAb (D and E), and transferrin-specific MAb (F and G). The relative positions of the raft and nonraft (soluble) fractions are indicated.
The concentration of CAR, the CBV4 receptor, as well as that of TLR4 within lipid rafts before and after stimulation by CBV4 virions was examined using FRET. We used cholera toxin conjugated to Cy5 to probe for lipid rafts, whereas we used Cy3-labeled antibodies against CAR and TLR4 as the donor.
FRET was measured in terms of dequenching of donor fluorescence after complete photobleaching of the acceptor fluorophore. Increased donor fluorescence after complete destruction of the acceptor indicated association between the two molecules of interest. Prior to our experiments, the energy transfer efficiency in our system was measured using a positive control as well as a negative control for the energy transfer (Table 1). As expected, maximum energy transfer efficiency (E) was 38% ± 0.5%.
TABLE 1.
Energy transfer efficiency values for donor-acceptor pairs
| Cell category | Donor (Cy3) | Acceptor (Cy5) | E ± ΔE (%)a |
|---|---|---|---|
| Unstimulated PANC-1 cells | MHC-Ib | MHC-I | 38 ± 0.5 |
| TLR4 | GM-1 ganglioside | 2 ± 0.5 | |
| TLR4 (treated with nystatin) | GM-1 ganglioside | 3 ± 0.8 | |
| CAR | GM-1 ganglioside | 9 ± 0.6 | |
| CAR (treated with nystatin) | GM-1 ganglioside | 10 ± 1.5 | |
| PANC cells stimulated with CBV4 | MHC-I | MHC-I | 38 ± 0.5 |
| TLR4 | GM-1 ganglioside | 34 ± 3.0 | |
| TLR4 (treated with nystatin) | GM-1 ganglioside | 5 ± 1.0 | |
| CAR | GM-1 ganglioside | 33 ± 2.0 | |
| CAR (treated with nystatin) | GM-1 ganglioside | 8 ± 0.4 |
Energy transfer for different pairs was detected from the increase in donor fluorescence after acceptor photobleaching. Data represent means ± standard deviations from a number of independent experiments.
MHC-I, major histocompatibility complex class I.
To examine whether TLR4 was localized in lipid rafts prior to stimulation with CBV4, we measured FRET on pancreatic (PANC-1) cells between TLR4 (by use of Cy3-conjugated TLR4-specific MAb) and GM-1 ganglioside, a raft-associated lipid (by use of Cy5-cholera toxin). The results showed that there was no energy transfer efficiency (2% ± 0.5%), thus verifying that TLR4 does not reside in lipid rafts.
We similarly examined whether CAR protein localizes in lipid rafts prior to CBV4 infection by using Cy3-C1079 specific for CAR protein and Cy5-cholera toxin for the GM-1 ganglioside. Our results showed there was a small percentage of CAR protein associated with lipid rafts, giving FRET values of 9% ± 0.6% (Table 1).
We also examined whether TLR4 or CAR molecules were recruited into the raft after infection with CBV4. We therefore performed FRET experiments with TLR4 and GM-1 ganglioside as well as with CAR protein and GM-1 ganglioside in the presence of CBV4. We found that there was an increased association between CAR and GM-1 ganglioside (E = 33% ± 2.0%) in the presence of CBV4 particles as well as an increased association of TLR4 with GM-1 ganglioside (E = 34% ± 3.0%) (Fig. 5). Our data show that TLR4 and CAR molecules are recruited in the rafts upon CBV4 infection.
To rule out the possibility that the FRET observed was due to random distribution, we varied the ratio of donors and acceptors used to label the proteins of interest. E was found to be independent of acceptor density, to be sensitive to the donor:acceptor ratio, and not to go to 0 at low surface density, thus suggesting that the FRET values observed were due to clustered molecules and not random associations.
The functional significance of TLR4 recruitment in lipid rafts was investigated by disrupting lipid raft formation by the use of lipid raft-disrupting drugs such as nystatin and by subsequently stimulating the cells with CBV4. The incubation with nystatin led to the disruption of TLR4 and CAR recruitment in lipid rafts (Table 1). We found that cytokine secretion was inhibited when pancreatic (PANC-1) cells were treated with nystatin prior to CBV4 stimulation, thus suggesting that raft integrity is crucial for CBV4-induced inflammation.
TLR4 does not associate with CBV4 receptor molecules.
Since both TLR4 and CAR were found to be recruited in lipid rafts following incubation with CBV4, we wanted to investigate whether TLR4 and CAR also associated. FRET was utilized to study CAR-TLR4 associations before and after CBV4 stimulation of PANC-1 cells.
Experiments to investigate FRET between TLR4 and CAR were performed prior to stimulation with CBV4 particles (Fig. 8A and B). It was found that after acceptor photobleaching, there was no significant energy transfer between TLR4 and CAR (E = 4% ± 1.3%); thus, TLR4 and CAR did not associate prior to CBV4 infection.
FIG. 8.

Energy transfer between TLR4 and CAR before and after stimulation by CBV4. Energy transfer between TLR4 (Cy3-TLR4) and CAR (Cy5-CAR) before (A and B) and after (C and D) stimulation by CBV4 can be detected by the increase in donor fluorescence after acceptor photobleaching. Donor (Cy3) images after acceptor photobleaching (A and C) and E images (images resulting from the subtraction of the Cy3 image before photobleaching from the Cy3 image after photobleaching) (B and D) are shown. Bar, 5 μm.
We also examined whether TLR4 and CAR molecules associated after stimulation with CBV4 particles (Fig. 8C and D). Although we had found both molecules to be recruited to lipid rafts after CBV4 stimulation, we found that, surprisingly, there was no energy transfer between TLR4 and CAR following CBV4 stimulation. Thus, CBV4 does not interact with TLR4 through a close proximity with CAR, its known receptor molecule.
DISCUSSION
The secretion of proinflammatory cytokines in response to microbial pathogens is an ancient protective measure that our innate immune system employs as part of its nonspecific host defense mechanism. It has long been known that this protective mechanism is mounted in response to pathogens such as conserved bacterial structures as well as viral products. Although the role of these proinflammatory cytokines is meant to be protective in the case of bacterial products, it can in some circumstances become harmful, resulting in often-fatal sepsis or septic shock. The possibility that a dysregulated cytokine secretion in response to some viruses might result in inflammatory and autoimmune diseases in humans has been suggested. One such disease is IDDM, for which CBV4 has been implicated as a cause.
The exact mechanism by which CBV4 destroys human pancreatic β islets and eventually leads to the development of IDDM has not yet been elucidated. It has been suggested that the virus might directly destroy pancreatic cells, but accumulating evidence is suggesting that CBV4 might trigger a dysregulated inflammatory response that could eventually be harmful for the host (5). By unraveling the mechanism by which CBV4 is able to trigger the production of inflammatory cytokines, we might be able to manipulate this cytokine response and find ways to prevent the development of IDDM.
In this study we investigated the possibility that CBV4 triggers the secretion of proinflammatory cytokines in human pancreatic cells. Our experiments suggest that CBV4 is capable of inducing the secretion of inflammatory cytokines, which include TNF-α and, to a greater extent, IL-6, within the first hour of its interaction with the host. This ability to induce an inflammatory response seems not to require the internalization of CBV4 or the successful infection of human pancreatic cells, since UV-inactivated CBV4 virions (which are incapable of infecting cells) were able to trigger exactly the same response. This leads us to believe that CBV4 must interact with some receptor of the innate immune system in the early attachment stage of the viral infectious cycle.
TLRs have been emerging as pattern recognition receptors that are part of the innate immune defense machinery that recognizes viral products and triggers an inflammatory response against them (4, 7, 18, 22). Thus, we investigated whether TLRs are also involved in the inflammatory response that is induced by CBV4 in human pancreatic cells. Our experiments suggest that the CBV4-induced inflammatory response is mediated through TLR4. The presence of TLR4 was required for CBV4 to trigger the production of cytokines, but when we investigated whether TLR4 was required for either viral attachment or internalization, we found that the presence of TLR4 was not sufficient for efficient viral binding and infection. Our findings suggest that TLR4 does not play an important role in the CBV4 infectious cycle but must be part of the innate immune system machinery and act as a trigger for a protective immune response to be mounted.
Interestingly, we also found that TLR4 is recruited within lipid rafts following stimulation by CBV4 particles. This recruitment seems to be crucial for the CBV4-induced inflammatory response, since lipid-raft-disrupting drugs were able to inhibit cytokine production. CAR protein, one of the known CBV4 receptors, was also found to be recruited to lipid rafts following stimulation by CBV4 particles. Surprisingly, TLR4 was found not to associate with CAR either before or after CBV4 stimulation, ruling out the possibility that a TLR4-CAR association is the mechanism by which CBV4 manages to trigger an inflammatory response via TLR4.
In this study we present evidence that CBV4 is capable of triggering the production of inflammatory cytokines in human pancreatic cells. Such a response has been suggested to play a role in the development in IDDM. Although our findings suggest that TLR4 is the main pattern recognition receptor that triggers this response, TLR4 seems not to be part of the CBV4 infectious cycle. It seems to simply act as sensing machinery for the innate immune system. Even though we have revealed a big piece of the puzzle of CBV4-induced inflammation, the question still remains: how does TLR4 sense CBV4 particles? Our findings suggest that TLR4 does not directly bind CBV4 particles, since its presence was not necessary for CBV4 binding. One mechanism could be that TLR4 must associate with a CBV4 receptor within lipid rafts and thus trigger an immune response against it. The definition of all host proteins that recognize CBV4 could prove to be very important, since unraveling the mechanism by which CBV4 induces this inflammatory response might help us find new therapeutic targets for IDDM.
Acknowledgments
This work was supported by Diabetes UK and the Hayling Island Diabetes UK Group.
We thank J. W. Yoon for providing us with CBV-E2, J. M. Bergelson for providing us with his CHO-CAR tranfectants, D. T. Golenbock for providing us with his HEK transfectants, and Jon Cohen for critical reading of the manuscript.
REFERENCES
- 1.Akira, S. 2001. Toll-like receptors and innate immunity. Adv. Immunol. 78:1-56. [DOI] [PubMed] [Google Scholar]
- 2.Barret-Connor, E. 1985. Is insulin-dependent diabetes mellitus caused by coxsackie B infection? A review of the epidemiologic evidence. Rev. Infect. Dis. 7:207-215. [DOI] [PubMed] [Google Scholar]
- 3.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 4.Bieback, K., E. Lien, I. M. Klagge, E. Avota, J. Schneider-Schaulies, W. P. Duprex, H. Wagner, and C. J. Kirschning. 2002. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 76:8729-8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bopegamage, S. A., and A. Petrovicova. 1998. Tumour necrosis factor alpha and glucose levels in sera of mice infected with coxsackie B4 and A7 viruses. Acta Virol. 42:409-411. [PubMed] [Google Scholar]
- 6.Brown, D. A., and J. K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68:533-544. [DOI] [PubMed] [Google Scholar]
- 7.Compton, T., E. A. Kurt-Jones, K. W. Boehme, J. Belko, E. Latz, D. T. Golenbock, and R. W. Finberg. 2003. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 77:4588-4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Damjanovitch, S., L. Bene, L. Matko, Z. Krasznai, G. Szabo, C. Pieri, R. Gaspar, and J. Szollosi. 1999. Two-dimensional receptor patterns in the plasma membrane of cells. A critical evaluation of their identification, origin and information content. Biophys. Chem. 82:99-108. [DOI] [PubMed] [Google Scholar]
- 9.Delude, R. L., R. Savedra, H. L. Zhao, R. Thieringer, S. Yamamoto, M. J. Fenton, and D. T. Golenbock. 1995. Cd14 enhances cellular-responses to endotoxin without imparting ligand-specific recognition. Proc. Natl. Acad. Sci. USA 92:9288-9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrov, D. S. 2000. Cell biology of virus entry. Cell 101:697-702. [DOI] [PubMed] [Google Scholar]
- 11.Edidin, M. 2001. Shrinking patches and slippery rafts: scales of domains in the plasma membrane. Trends Cell Biol. 11:492-496. [DOI] [PubMed] [Google Scholar]
- 12.Golenbock, D. T., Y. Liu, F. H. Millham, M. W. Freeman, and R. A. Zoeller. 1993. Surface expression of human CD14 in Chinese hamster ovary fibroblasts imparts macrophage-like responsiveness to bacterial endotoxin. J. Biol. Chem. 268:22055-22059. [PubMed] [Google Scholar]
- 13.Horejsi, V., K. Drbal, M. Cebecauer, J. Cerny, T. Brdicka, P. Angelisova, and H. Stockinger. 1999. GPI-microdomains: a role in signalling via immunoreceptors. Immunol. Today 20:356-361. [DOI] [PubMed] [Google Scholar]
- 14.Horwitz, M. 1990. The adenoviridae and their replication, p. 1679-1721. In B. N. Fields and D. M. Knipe (ed.), Virology. Raven Press, New York, N.Y.
- 15.Jacobson, K., and C. Dietrich. 1999. Looking at lipid rafts? Trends Cell Biol. 3:87-91. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson, K., E. D. Sheets, and R. Simson. 1995. Revisiting the fluid mosaic model of membranes. Science 268:1441-1442. [DOI] [PubMed] [Google Scholar]
- 17.Kenworthy, A. K., and M. Edidin. 1998. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 A using imaging fluorescence resonance energy transfer. J. Cell Biol. 142:69-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediate responses to respiratory syncytial virus. Nat. Immunol. 1:398-401. [DOI] [PubMed] [Google Scholar]
- 19.Lindwasser, O. W., and M. D. Resh. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75:7913-7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen, D. H., and J. E. K. Hildreth. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 74:3264-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono, A., and E. O. Freed. 2001. Plasma membrane rafts play a critical role in HIV1 assembly and release. Proc. Natl. Acad. Sci. USA 98:13925-13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rassa, J. C., J. L. Meyers, Y. Zhang, R. Kudaravalli, and S. R. Ross. 2002. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. USA 99:2281-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheiffele, P., A. Rietveld, T. Wilk, and K. Simons. 1999. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 274:2038-2044. [DOI] [PubMed] [Google Scholar]
- 24.Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255-281. [DOI] [PubMed] [Google Scholar]
- 25.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 26.Triantafilou, K., D. Fradelizi, K. M. Wilson, and M. Triantafilou. 2002. GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J. Virol. 76:633-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Triantafilou, K., M. Triantafilou, and R. L. Dedrick. 2001. A CD14-independent LPS receptor cluster. Nat. Immunol. 4:338-345. [DOI] [PubMed] [Google Scholar]
- 28.Triantafilou, M., K. M. Wilson, and K. Triantafilou. 2001. Identification of Echovirus 1 and Coxsackievirus A9 receptor molecules via a novel flow cytometric quantification method. Cytometry 43:279-289. [DOI] [PubMed] [Google Scholar]
- 29.Triantafilou, M., K. Miyake, D. Golenbock, and K. Triantafilou. 2002. Mediators of the innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 115:2603-2611. [DOI] [PubMed] [Google Scholar]
- 30.Triantafilou, M., K. Triantafilou, K. M. Wilson, Y. Takada, and N. Fernandez. 1999. Involvement of b2-microglobulin and integrin avb3 molecules in the Coxsackievirus A9 infectious cycle. J. Gen. Virol. 80:1-2600. [DOI] [PubMed] [Google Scholar]
- 31.Vreugdenhil, G. R., P. G. Wijnands, M. G. Netea, J. W. van der Meer, W. J. Melchers, and J. M. Galama. 2000. Enterovirus-induced production of pro-inflammatory and T-helper cytokines by human leukocytes. Cytokine 12:1793-1796. [DOI] [PubMed] [Google Scholar]
- 32.Wu, P., and L. Brand. 1994. Resonance energy transfer: methods and applications. Anal. Biochem. 218:1-13. [DOI] [PubMed] [Google Scholar]
- 33.Yoon, J.-W., M. Austin, and T. N. A. L. Onodera. 1979. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 300:1173-1179. [DOI] [PubMed] [Google Scholar]