

Abstract
Human cytomegalovirus (HCMV) infection leads to dysregulation of multiple cell cycle-regulatory proteins. In this study, we examined the effects of inhibition of cyclin-dependent kinase (cdk) activity on viral replication. With the drug Roscovitine, a specific inhibitor of cyclin-dependent kinases 1, 2, 5, 7, and 9, we have shown that during the first 6 h of infection, cyclin-dependent kinase-dependent events occurred that included the regulated processing and accumulation of the immediate-early (IE) UL122-123 transcripts and UL36-37 transcripts. Altered processing of UL122-123 led to a loss of IE1-72 and an increase in IE2-86. The ratio of spliced to unspliced UL37 transcripts also changed. These effects did not require de novo protein synthesis or degradation of proteins by the proteasome. Addition of Roscovitine at the beginning of the infection was also associated with inhibition of expression of selected viral early gene products, viral DNA replication, and late viral gene expression. When Roscovitine was added after the first 6 h of infection, the effects on IE gene expression were no longer observed and viral replication proceeded through the late phase, but viral titers were reduced. The reduction in viral titer was observed even when Roscovitine was first added at 48 h postinfection, indicating that cyclin-dependent kinase activity is required at both IE and late times. Flavopiridol, another specific inhibitor of cyclin-dependent kinases, had similar effects on IE and early gene expression. These results underscore the importance of accurate RNA processing and reiterate the significant role of cell cycle-regulatory factors in HCMV infection.
Human cytomegalovirus (HCMV), a member of the family Herpesviridae, is the major viral cause of birth defects and is associated with significant morbidity in immunocompromised individuals (52). Its genome is 230 kbp and has the capacity to encode approximately 150 open reading frames (19, 44). HCMV cell entry, gene expression, replication, and virion maturation are multilayered processes that require many viral as well as cellular factors during a productive infection. There are three major phases of viral gene expression. Viral immediate-early (IE) genes are the first to be expressed in an infected cell, and their transcription requires no de novo protein synthesis. A major site of IE transcription includes two genetic units, IE1 and IE2 (for review, see references 27 and 44) (Fig. 1B). The predominant IE RNA (IE1) consists of four exons; a single open reading frame (UL123) initiates in exon 2 and specifies a 72-kDa nuclear protein designated IE1-72. The major IE2 gene product, IE2-86 (open reading frame UL122), is an 86-kDa protein that is encoded by an alternatively spliced RNA that contains the first three exons of IE1 and a different terminal exon.
FIG. 1.

HCMV IE transcripts. A schematic diagram representing (A) the HCMV genome, (B) the major IE gene transcripts (UL122-123), and (C) transcripts originating from the UL36-38 open reading frame and their respective gene products. In panels B and C, the thick lines represent transcripts, the broken lines represent splicing events, and thin lines are noncoding regions of the transcript. Poly(A) tails and promoter sequences are depicted as arrowheads and boxes with bent arrows, respectively. Solid boxes indicate gene products translated from the respective transcripts. The splice acceptor (SA) sites for exon 5 of IE2-86 and exon 2 of UL37 and UL37M are shown.
IE1-72 and IE2-86 are essential transactivators of both early and late viral gene expression. Included in the early class are genes that are important for viral DNA replication. Some examples are the UL112-113 nuclear phosphoproteins, the viral polymerase (UL54), a DNA polymerase processivity factor (UL44), and the single-stranded-DNA-binding protein (UL57) (2, 3, 29, 35, 37). Late gene expression occurs after the initiation of viral DNA synthesis at approximately 24 h postinfection (p.i.), and these late genes encode primarily the structural components of the virion. Tegument proteins such as pp28 (UL99) and pp65 (UL83) as well as components of the capsid are products of the late genes (for review, see reference 44).
A second locus of IE transcription is UL36-38. This region includes three promoters and gives rise to at least five transcripts (Fig. 1C). One of the IE promoters directs the synthesis of several spliced 3.2- to 3.4-kb RNAs (UL37 and UL37M) that are present in small amounts only at IE times. It also is the promoter for an abundant 1.7-kb unspliced RNA that encodes the UL37 exon 1 (UL37X1) gene product, which is present throughout the infection. The cleavage-polyadenylation site for the UL37X1 RNA is located 8 nucleotides upstream of the splice acceptor site for exon 2 of the UL37 and UL37M RNAs. The second IE promoter is responsible for the synthesis of a 1.65-kb spliced RNA (UL36) that increases in abundance at early times and is 3′ coterminal with the UL37 spliced RNAs (36, 66, 67). In addition, another promoter within UL37X1 directs the synthesis of an abundant early transcript of 1.35 kb (UL38) that is 3′ coterminal with the UL37X1 RNA. The start site of the UL38 transcript is 70 nucleotides downstream of the splice donor site for the UL37 and UL37M RNAs (66, 67).
The UL37X1, UL37, and UL36 proteins have regulatory activity, and the locus encoding these proteins is required for HCMV DNA replication in a transient complementation assay (17, 30, 50, 51, 62, 74). In addition, the UL37X1 protein and the UL37 and UL37M glycoproteins have a common amino terminus of 162 amino acids. Within this common domain are a hydrophobic signal sequence (amino acids 1 to 22) and an acidic domain (amino acids 81 to 108). The UL37 glycoprotein also has 17 N-glycosylation sites, a basic domain, a transmembrane region, and a cytosolic tail. UL37X1 encodes an antiapoptotic protein (viral mitochondrion-localized inhibitor of apoptosis) that prevents release of cytochrome c from the mitochondria (16, 28, 41).
In addition to the viral proteins, many cellular proteins play an important role in the replication of HCMV. Some of these proteins also play pivotal roles in the regulation of the cell cycle. The cell cycle includes four phases (G1, S, G2, and M), and both entry into and exit from these phases are largely controlled by regulating the activity of the cyclin-dependent kinases (cdks) and expression of their cyclin partners (for review, see references 23 and 54). Multiple studies have addressed the effect of HCMV infection on the expression of these cyclins and the progression of the cell cycle (6-12, 14, 15, 21, 25, 26, 31, 33, 34, 38, 40, 42, 45-47, 49, 59, 69-71). The general picture that emerges from these studies is that HCMV activates or induces the expression of many host cell proteins to create a cellular environment that is optimal for gene expression and DNA replication; however, the virus inhibits selective host cell functions to ensure that its own replication is favored over that of the host. The net effect is that the cell cycle is blocked prior to the initiation of cellular DNA synthesis.
In this study, we used the drugs Roscovitine and Flavopiridol to assess the role of cyclin-dependent kinases in HCMV infection. Roscovitine is a purine-derived compound that specifically inhibits the activity of cdk1/cyclin B, cdk2/cyclin E, cdk2/cyclin A, cdk5/p25 (expressed in neural cells), cdk7/cyclin H, and cdk9/cyclin T1 (P-TEFb) with a 50% inhibitory concentration (IC50) of approximately 0.7 μM in in vitro kinase assays; cdk4/cyclin D and cdk6/cyclin D are not inhibited (20, 22, 43, 61, 68). Flavopiridol, a flavanoid, is another specific inhibitor of cyclin-dependent kinases that acts as a competitor with ATP to inhibit cdk1/cyclin B (IC50 = 30 to 40 nM in in vitro kinase assays), cdk2/cyclin A and cdk2/cyclin E (IC50 = 100 nM), cdk4/cyclin D (IC50 = 20 to 40 nM), cdk6/cyclin D (IC50 = 60 nM), and cdk7/cyclin H (IC50 = 110 to 300 nM). It is also a potent inhibitor of P-TEFb that binds to P-TEFb with 1:1 stoichiometry and is not competitive with ATP (for review, see reference 18).
Both of these drugs are being used in clinical trials as potential anticancer agents and appear to have low toxicity. The inhibition of the replication of the herpesviruses herpes simplex virus type 1, herpes simplex virus type 2, Epstein-Barr virus, varicella-zoster virus, and HCMV as well as human immunodeficiency virus type 1 by Roscovitine has led to the proposal that productive infection by these viruses requires the activity of one or more cyclin-dependent kinases (for review, see reference 60). Flavopiridol also inhibits human immunodeficiency virus type 1 replication primarily through its effect on P-TEFb (IC50 < 10 nM).
In a prior study on HCMV, Roscovitine was found to inhibit viral DNA synthesis in density-arrested human lung fibroblasts and in a transformed astrocytoma/glioblastoma cell line (U373) (9). The authors concluded from the results of transient expression assays with a dominant negative cdk2 expression vector in infected U373 cells that this cyclin-dependent kinase was required for late gene expression; however, the low percentage of cells transfected with the vector (<5%) coupled with the inability to infect more than 30% of the cells complicated the interpretation of these experiments.
In this work, we sought to determine whether the requirement for cyclin-dependent kinase activity was restricted to the early period of infection and to decipher what molecular processes were affected by the absence of these kinases. We have shown that the addition of Roscovitine at the beginning of the infection resulted in altered processing and accumulation of UL122-123 and UL37 transcripts and inhibition of the expression of selected viral early genes. These effects did not require de novo protein synthesis or degradation of proteins via the proteasome. Delaying the addition of Roscovitine until 6 h p.i. abolished these effects and viral replication proceeded through late gene expression, but viral titers were still significantly reduced. We also demonstrated that inhibiting cyclin-dependent kinase activity with the drug Flavopiridol had similar effects on IE and early gene expression.
MATERIALS AND METHODS
Cell culture and virus.
Human foreskin fibroblasts (HFF) were obtained from the University of California, San Diego, Medical Center and cultured in Earle's minimal essential medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 50 μg of gentamicin sulfate (Invitrogen) per ml, 1.5 μg of amphotericin B (Invitrogen) per ml, 2 mM l-glutamine (Invitrogen), 100 U of penicillin (Invitrogen) per ml, and 100 μg of streptomycin (Invitrogen) per ml. Cells were kept in incubators maintained at 37°C and 7% CO2. The Towne strain of HCMV was obtained from the American Type Culture Collection (VR 977) and propagated as previously described (65).
Cell synchronization and infections.
Cells were synchronized in the G0 phase by allowing them to grow to confluence as previously described (59). Three days after confluence, the cells were trypsinized, replated at a lower density to allow progression into the cell cycle, and infected at a multiplicity of infection (MOI) of 3 to 5 with HCMV Towne or mock infected with tissue culture supernatants. At designated times p.i., Roscovitine (Calbiochem) or Flavopiridol (gift from J. Brady, National Institutes of Health) was added to the medium. The Roscovitine stock solution was 10 mM in dimethyl sulfoxide, and the Flavopiridol stock solution was 0.1 mM in dimethyl sulfoxide. Control samples were treated with appropriate volumes of dimethyl sulfoxide. At various times p.i., cells were washed with phosphate-buffered saline, scraped or trypsinized, and processed as described.
Cytotoxicity assays.
The toxic effects of Roscovitine on HFF cells were evaluated with the LIVE/DEAD viability/cytotoxicity assay kit (Molecular Probes) according to the manufacturer's protocol. This assay uses two-color fluorescence to simultaneously determine the presence of live and dead cells in a culture. In live cells, the nonfluorescent cell-permeable calcein acetoxymethyl (AM) dye is converted by intracellular esterase activity to calcein, which fluoresces green. Ethidium homodimer-1 (EthD-1) enters cells with damaged membranes and undergoes a 40-fold enhancement of red fluorescence upon binding to nucleic acids. Cells were plated on coverslips at a density of 50% confluency in the absence or presence of 5, 15, 25, or 50 μM Roscovitine. In cultures treated with Roscovitine for more than 24 h, fresh medium with the appropriate concentration of the drug was added every 24 h. Coverslips were washed in Dulbecco's phosphate-buffered saline prior to incubation with reagents for the viability assay. As a positive control for dead cells, cells were treated with ice-cold methanol for 10 min and washed with Dulbecco's phosphate-buffered saline. Coverslips were incubated in 150 μl of combined assay reagents (0.5 μM EthD-1 and 0.25 μM calcein AM) for 30 min at room temperature. To ensure minimal damage to the cells, coverslips were mounted on 10 μl of Dulbecco's phosphate-buffered saline on a microscope slide and sealed to prevent evaporation. Green and red fluorescent cells were counted, and the percentage of live and dead cells was determined for mock- and virus-infected cells treated with increasing concentrations of Roscovitine at 24, 48, 72, 96, 120, and 144 h p.i.
Determining the effects of Roscovitine on virus titer.
G0-synchronized cells were released from confluence and infected with the Towne strain of HCMV at an MOI of 3 or 5. At the designated times p.i., the indicated concentration of Roscovitine or dimethyl sulfoxide was added to the medium. The medium was changed every 24 h, and fresh drug or control dimethyl sulfoxide was added. Viral supernatants were collected at days 4, 5, and 6, and the titers were determined by plaque assay (65).
Cycloheximide and actinomycin D experiments.
Cells were synchronized in G0, trypsinized, and reseeded at a lower density. After allowing the cells to recover for 1 h, the cells were treated for an hour with either 100 μg of cycloheximide (stock solution, 10 mg/ml in distilled H2O) per ml in medium or medium alone prior to infection. Cycloheximide was kept on the treated cells until 6 h p.i. All cells were infected with the Towne virus at an MOI of 3, and 15 μM Roscovitine was added at the time of infection to designated cell cultures. At 6 h p.i., all cells were washed twice with phosphate-buffered saline and fresh medium was added. Roscovitine and actinomycin D (stock solution, 10 mg/ml in methanol) were added to the designated cell cultures to final concentrations of 15 μM and 20 μg/ml, respectively, and the cells were harvested at 18 h p.i.
Experiments with the proteasome inhibitor MG132.
G0-synchronized cells were released from confluence and infected with Towne at an MOI of 3 at the time of replating; 15 μM Roscovitine and 2.5 μM MG132 (stock solution, 10 mM in dimethyl sulfoxide) were added to designated plates at the time of infection. At 8 h p.i., all cells were washed twice with phosphate-buffered saline, and fresh medium with the appropriate drugs was added back to the designated cell cultures. Cells were harvested at various times p.i.
Slot blot analysis.
DNA was isolated from infected cells at 24, 48, and 72 h p.i. with the Blood and Cell Culture minikit (Qiagen). DNA samples (5, 25, and 125 ng of each sample) were spotted onto 45-μm Nytran (Osmonics) with a slot blot apparatus (Schleicher & Schuell) according to the manufacturer's instructions. The DNA was then cross-linked to the blot with a Stratalinker (Stratagene) as recommended by the manufacturer. The blot was probed with a radiolabeled ApaLI-NcoI fragment of the EcoRI B fragment of HCMV strain AD169 (63) per standard protocols and exposed to autoradiograph film. Band intensity was quantified by measuring the integrated pixel densities as determined with NIH Image and Photoshop 7.0 software. Differences were determined for samples within the linear range.
Western blot analysis.
Cells were lysed in Laemmli reducing sample buffer (2% sodium dodecyl sulfate, 10% glycerol, 100 mM dithiothreitol, 60 mM Tris, pH 6.8, 2 μg each of aprotinin and leupeptin per ml, 1 mM phenylmethylsulfonyl fluoride, 50 mM NaF, 0.5 mM Na3VO4, 4 mM EDTA, 10 mM Na4P2O7, 1 mM benzamidine, and 1 mM NaS2O5). The lysates were then sonicated, boiled for 5 min, and centrifuged for 1 min at 16,000 × g. Samples were run on 10% polyacrylamide gels. Proteins were transferred to Immobilon P (Millipore) or Protran (Schleicher & Schuell), and the blots were stained with amido black to ensure that each lane had an equivalent amount of protein. Lysates were also assayed by Western blot for actin. Western blot analysis was performed with the appropriate mouse or rabbit antibody followed by the appropriate horseradish peroxidase-linked secondary antibody (Calbiochem). Proteins were visualized with the West Femto or West Pico (Pierce) detection method (per the manufacturer's instructions). Band intensity was quantified by measuring integrated pixel densities as determined with NIH Image and Photoshop 7.0 software.
Antisera.
The antibodies used in Western analysis were anti-IE1 and IE2 (1203; Goodwin Institute), anti-UL112-113 (73), anti-UL44 (1202; Goodwin Institute), anti-UL57 (1209, Goodwin Institute), anti-pp28 (UL99), and anti-major capsid protein (UL86) (gifts from William Britt, University of Alabama), and anti-pp65 (UL83) (1205S; Goodwin Institute).
Northern blot analysis.
Total RNA was isolated at 24 h p.i. with the RNAqueous midikit (Ambion). RNA samples (10 μg of each sample) were separated by electrophoresis on a 1% agarose-formaldehyde gel and transferred to 0.45-μm Nytran (Osmonics) as previously described (4). Following cross-linking in a Stratalinker (Stratagene), the blot was probed for the UL44 viral transcripts according to standard protocols with the 183-bp EcoRI fragment of the UL44 gene (a gift from Greg Pari, University of Nevada) that was radiolabeled with 32P by random priming. The blot was then exposed to autoradiograph film. Band intensity was quantified by measuring integrated pixel densities as determined with NIH Image and Photoshop 7.0 software.
Quantitative real-time PCR.
Cells (107 per sample) were infected with the Towne virus at an MOI of 5, and 15 μM Roscovitine or dimethyl sulfoxide alone was added to the medium at designated times p.i. The cells were harvested at various times p.i., and total RNA was isolated from the harvested cells with a NucleoSpin RNA II purification kit (BD Biosciences Clontech, Palo Alto, Calif.). The concentration of each sample was determined by UV spectrophotometry. Quantitative real-time reverse transcription-PCR was performed in an Applied Biosystems ABI Prism 7700 sequence detection system or in an Applied Biosystems ABI Prism 7000 sequence detection system with the TaqMan One-Step reverse transcription-PCR master mix reagents kit (Applied Biosystems) and oligonucleotide primers and TaqMan dual-labeled (5′ fluorescein (FAM) 3′ Black hole quencher) probes (Integrated DNA Technologies, Coralville, Iowa) (see Table 1 for sequences).
TABLE 1.
Quantitative real-time PCR primers and TaqMan probes
| RNA | Sequence
|
||
|---|---|---|---|
| Forward Primer | Reverse Primer | TaqMan Probe | |
| UL37X1 | 5′ CGGATGCTGCAGCACAAC 3′ | 5′ AGCAATAGCGGTAAAGTCCCTTCT 3′ | 5′ AGTCTCACCAGTAAGCAG 3′ |
| UL37 | 5′ CGGATGCTGCAGCACAAC 3′ | 5′ CGTGTCCCGTGCTCCAA 3′ | 5′ TCTCACCATGCCGCGGT 3′ |
| IE1-72 | 5′ CAAGTGACCGAGGATTGCAA 3′ | 5′ CACCATGTCCACTCGAACCTT 3′ | 5′ TCCTGGCAGAACTCGTCAAACAGA 3′ |
| IE2-86 | 5′ TGACCGAGGATTGCAACGA 3′ | 5′ CGGCATGATTGACAGCCTG 3′ | 5′ TGGCAGAACTCGGTGACATCCTCGCC 3′ |
| Glucose-6-phosphate dehydrogenase | 5′ TCTACCGCATCGACCACTACC 3′ | 5′ GCGATGTTGTCCCGGTTC 3′ | 5′ ATGGTGCTGAGATTTGCCAACAGGA 3′ |
Primers and probes for RNAs expressing UL37, IE1-72, IE2-86, and glucose-6-phosphate dehydrogenase have been previously described (64, 72) and spanned the splice junction when applicable. The primers and probes were added to reagents from the TaqMan One-Step reverse transcription-PCR master mix reagents kit (Applied Biosystems) and then mixed with 50 ng of each total RNA sample. The RNA isolated at 8 h p.i. from the untreated infected cells was used to generate a standard curve for each gene examined. The standard curve was then used to calculate the relative amount of specific RNA present in a sample, from which the induction of transcription of the gene was calculated by comparison to the value obtained for the specific RNA from untreated infected cells that were harvested at 8 h p.i. As an additional control for the amount of RNA in each reaction, samples were analyzed with primers and a TaqMan probe specific to the cellular housekeeping gene glucose-6-phosphate dehydrogenase.
RESULTS
Inhibition of cyclin-dependent kinase activity results in a decrease in virus titers.
Previous studies (9) showed that HCMV replication is inhibited during the early phase when the drug Roscovitine is added at the beginning of the infection. In these experiments, human embryonic lung cells and the astrocytoma-glioblastoma U373 cell line were used, and the cells were density arrested at the time of infection. No cell toxicity was observed in uninfected human embryonic lung cells that were maintained in the presence of 15 μM Roscovitine for 96 h. In another study (75), confluent human embryonic lung cells that were treated with 33 μM Roscovitine for 5 days showed a 50% reduction in viability as assayed by neutral red uptake. In our experiments on the dysregulation of the cell cycle by HCMV, we used primary human foreskin fibroblasts (HFF) that are synchronized in G0 phase by allowing them to grow for 3 days past confluence. Just prior to infection, the cells are trypsinized and replated at lower density to allow them to enter G1 phase. Since toxicity depends on the cell type and the conditions of growth, we tested the effects of various concentrations of Roscovitine on the viability of the uninfected cells and on viral titer under our experimental conditions.
To assess the cytotoxicity of Roscovitine on mock- and virus-infected HFF cells, we observed the cells for changes in morphology and tested them for viability with the LIVE/DEAD viability/cytotoxicity assay kit (Molecular Probes). Cells were plated on coverslips at a density of 50% confluency and infected with HCMV Towne or mock infected with tissue culture supernatant. At the time of infection, HCMV- and mock-infected cultures were treated with 5, 15, 25, or 50 μM Roscovitine or dimethyl sulfoxide, and cell viability was assayed at 24, 48, 72, 96, 120, and 144 h p.i. In the LIVE/DEAD viability/cytotoxicity assay, calcein dye retained by live cells was observed as green fluorescence, while EthD-1 staining in dead cells was observed as red fluorescence.
During the entire time course, there was no detectable cytotoxicity in mock-infected cultures treated with 5, 15, or 25 μM Roscovitine (<0.1% red cells), although the cultures treated with 15 or 25 μM Roscovitine took longer to reach confluence (data not shown). When treated with 50 μM Roscovitine, the mock-infected cells began to show morphological changes at 72 h postplating, and an increased number of cells exhibited red fluorescence (≈1%). This was an underestimate, as the cell density decreased by 50% during the 72 h of incubation, and most of the dead cells were likely removed during the wash steps of the assay. As expected, in the mock-infected culture treated with ice-cold methanol for 10 min prior to the assay, all of the cells were dead and showed bright red fluorescence (data not shown).
We then determined the effects of these increasing concentrations of Roscovitine on the infected cells and viral titers when the drug was added at the time of infection. As shown in Fig. 2, we observed a dose-dependent response in viral titers. The peak titer at day 6 for untreated cultures was 3.8 × 106 PFU/ml. At this time point, cells treated with 5 μM Roscovitine produced approximately the same amount of virus (5.2 × 106 PFU/ml). A greater effect on the infection and viral titer was observed when Roscovitine concentrations of 15 μM and above were tested. We first observed a difference in cytopathic effect at 24 h p.i. in cells treated with these higher concentrations of the drug. At this time, both untreated cells and those treated with 5 μM Roscovitine were completely rounded, an effect of early gene expression, while the cells treated with 15, 25, and 50 μM Roscovitine showed progressively less cytopathic effect.
FIG. 2.

Increasing amounts of Roscovitine have differential effects on viral titers. HFF cells were infected at an MOI of 5 with HCMV Towne in the presence of 5, 15, 25, and 50 μM Roscovitine or treated with dimethyl sulfoxide. At days (d) 4, 5, and 6, supernatants were collected and titers were determined by plaque assay. At day 6, cells were harvested and counted. The titer is plotted on a logarithmic scale.
Starting at approximately 72 h p.i., the infected cells treated with 25 and 50 μM Roscovitine began to die, and this process continued through the rest of the time course. This cytotoxic effect on infected cells was not observed at Roscovitine concentrations of 5 and 15 μM; however, at a concentration of 15 μM Roscovitine, infected cells remained rounded and did not flatten as normally happens late in infection (data not shown). Cultures treated with 15 μM Roscovitine contained approximately the same number of cells as the dimethyl sulfoxide-treated control cultures at day 6, and there was an approximately 75-fold reduction in virus titer (5.1 × 104 PFU/ml) compared to the untreated culture. Only a very low level of virus was detected in the cultures treated with 25 and 50 μM Roscovitine (140 and 20 PFU/ml, respectively).
In the experiments presented here, we used Roscovitine at a concentration of 15 μM, which did not completely inhibit infection. To determine whether the requirement for the cyclin-dependent kinases in a productive infection was time dependent, we investigated the production of infectious virus when Roscovitine was added at various times during the infection. In the experiment shown in Fig. 3, the titer of wild-type HCMV (Towne) in the absence of Roscovitine at day 6 p.i. was 1.1 × 106 PFU/ml. The addition of 15 μM Roscovitine at the time of infection resulted in a 20-fold reduction in virus production. A similar decrease in viral titer occurred when the drug was added at 12 h p.i. Even when the drug was added at 24 h p.i. (after the onset of viral DNA replication) or at 48 h p.i. (when viral assembly is occurring), virus titers were still reduced by approximately fivefold. In other experiments, a higher level of inhibition was seen at all time points, but the relative difference in inhibition when the drug was added at early and late times was similar. These results suggest that the requirement for cyclin-dependent kinase activity is not restricted to the early phase of the infection.
FIG. 3.

Virus titers are reduced in the presence of the drug Roscovitine. HFF cells were infected at a MOI of 3 with HCMV Towne. Roscovitine (15 μM) was added at various times p.i., and cell culture supernatants were collected at day 6 for determination of viral titer by plaque assay. The data presented are the mean results of two experiments in duplicate, and the range is shown.
Inhibition of cyclin-dependent kinase activity abolishes viral DNA replication.
To further define the temporal kinetics of the requirement for cyclin-dependent kinase activity during infection and to determine if the inhibition of virus production when Roscovitine was added at early times was due to a lack of viral DNA replication, the following experiment was performed. Cells were infected with HCMV, and at defined times p.i., 15 μM Roscovitine was added to the culture medium. DNA was isolated and subjected to slot blot analysis as described in Materials and Methods. In the absence of Roscovitine, viral DNA replication proceeded normally (Fig. 4). From 24 through 72 h p.i., there was a 22-fold increase in the amount of viral DNA present in the untreated cells. When 15 μM Roscovitine was added at the time of infection, however, there was no increase in the level of viral DNA during the same 48-h interval. Surprisingly, this defect was not observed when the drug was added at 6 h p.i., which marks the transition from IE to early gene expression. Although there was a lag in viral DNA replication, at 72 h p.i. the level of viral DNA was only threefold lower than that in the untreated cells at the same time point. These results suggested that the viral DNA polymerase was not directly inhibited by the drug and that viral DNA replication requires cyclin-dependent kinase-dependent events that occur at very early times in the infection, prior to the initiation of DNA synthesis.
FIG. 4.

Delayed addition of Roscovitine abolishes its effect on HCMV DNA replication. HFF cells were infected at an MOI of 5 with HCMV Towne in the absence or presence of 15 μM Roscovitine, added at the time of infection or at 6 h p.i. DNA was isolated at 24, 48, and 72 h p.i. The samples were then subjected to slot blot analysis and probed with an HCMV-specific radiolabeled DNA fragment as discussed in Materials and Methods. The plots show the distribution of pixel densities within each band. Determination of the relative accumulation of DNA was based on scans where the intensity of the bands was within the linear range.
Steady-state levels of several viral proteins are altered when infected cells are treated with Roscovitine.
The life cycle of HCMV is temporally regulated, and each step in the life cycle requires the products of the previous step before it can proceed. For example, early gene expression cannot occur without IE gene expression, and viral DNA replication cannot occur without viral early gene expression. The effects of Roscovitine on viral DNA replication may be direct or may occur through an indirect mechanism, such as alteration of viral IE or early gene expression. To investigate these possibilities, we examined the steady-state levels of a number of viral gene products in the presence of Roscovitine.
Figures 5A and B show that when 15 μM Roscovitine was added at the time of infection, there was a decrease in the amount of IE1-72, while IE2-86 levels increased. At 24 h p.i., the level of IE1-72 protein in untreated cells was approximately two- to threefold higher than in cells treated with the drug. At this time point, there was an increase of approximately 1.5-fold in IE2-86 levels in cells treated with Roscovitine. Coincident with the alterations in viral IE gene expression, viral early gene expression was also affected by the addition of 15 μM Roscovitine. The differential effects on the expression of three genes that play a role in viral DNA replication are shown. The levels of the two major early proteins (43 and 50 kDa) encoded by the UL112-113 locus were only modestly affected by the presence of Roscovitine (Fig. 5A). A greater inhibitory effect was seen on the expression of UL44, the processivity factor for the viral polymerase (Fig. 5A and B). At 24 and 48 h p.i., the levels of UL44 were at least twofold higher in lysates from untreated samples than from Roscovitine-treated cells.
FIG. 5.

Expression of viral IE, early, and late proteins is altered in the presence of the drug Roscovitine. HFF were infected at an MOI of 5 with HCMV Towne in the absence or presence of 15 μM Roscovitine. (A) Cells were harvested at the indicated times postinfection and subjected to Western blotting for IE gene products of UL122-123, early gene products of UL112-113, UL44, and UL57, and late gene products pp28 and major capsid protein. (B) Quantification of Western blots for IE1-72, IE2-86, UL44, and UL57. In all cases, the value of the untreated sample at 14 h p.i. was set to 1.
The most striking effect of Roscovitine, however, was the almost complete absence of UL57, the major single-stranded-DNA-binding protein (Fig. 5A and B). Since viral early gene products are required for viral DNA replication and viral DNA replication is required for viral late gene expression, it was not surprising that the drug also inhibited the synthesis of late proteins. Representative examples of the effect of Roscovitine on the viral tegument protein pp28 (UL99) and on the major capsid protein (UL86) are shown in Figs. 5A. Figure 5A also shows the inhibitory effect on expression of the late proteins (84 and 34 kDa) encoded by the UL112-113 gene.
Delaying addition of Roscovitine until 6 h p.i. abolishes its effects on the steady-state levels of viral proteins.
Because the DNA slot blot experiment suggested that there was a window of time within which cyclin-dependent kinase activity was required for viral DNA replication, we asked if this was also true for the effects of Roscovitine on the levels of viral gene products. In the experiment shown in Fig. 6, cells were infected in the absence of Roscovitine or the drug was added at the beginning of the infection or at 6 or 12 h p.i. The infected cells were then harvested at 24 h p.i. In contrast to what was observed when the drug was added at the time of infection, addition of Roscovitine at 6 or 12 h p.i. did not affect the accumulation of IE1-72. The levels of both UL44 and UL57 were partially restored to the levels in the untreated cells by delaying addition of the drug to 6 h p.i. and completely restored by delaying addition of the drug to 12 h p.i.
FIG. 6.
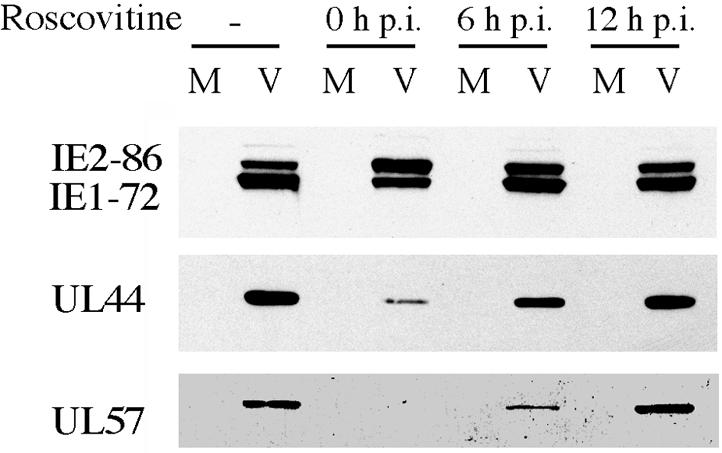
Delayed addition of Roscovitine abolishes its ability to alter IE and early viral gene expression. HFF were infected at an MOI of 5 with HCMV Towne in the absence or presence of 15 μM Roscovitine (added at the time of infection or 6 or 12 h p.i.). Cells were harvested at 24 h p.i. and subjected to Western blot analysis for the IE gene products of UL122-123 and the early gene products of UL44 and UL57. M, mock infected; V, HCMV infected.
To more precisely determine the time interval at the beginning of the infection during which addition of Roscovitine affects IE1-72-IE2-86 protein expression, we repeated the experiment by adding the drug at 30-min intervals p.i. Roscovitine was added to the infected cells at 30-min intervals up to 4 h p.i., at 5 h p.i., and at 6 h p.i. All cells were harvested at 24 h p.i. and analyzed by Western blot for IE protein expression. Figure 7 shows that delaying the addition of Roscovitine up to 3.5 h p.i. had a negligible effect IE protein levels. After 3.5 h p.i., the drug had a smaller effect on the accumulation of IE1-72, and the level of IE1-72 was comparable to that in the untreated infected cells. Although the amount of IE2-86 decreased when Roscovitine was added after 4 h p.i., the level still remained slightly higher than that in the untreated cells.
FIG. 7.

Effect of Roscovitine on the levels of the IE1-72 and IE2-86 proteins occurs during the first 4 h of infection. (A) HFF were infected at an MOI of 5 with HCMV Towne in the absence or presence of 15 μM Roscovitine, added at the indicated times postinfection. Samples were harvested at 24 h p.i. and subjected to Western blot analysis for the IE gene products of UL122-123. (B) Quantification of the Western blot. The levels of IE1-72 and IE2-86 in the untreated sample were set to 1.
Transcription of viral genes is altered by Roscovitine treatment.
In order to determine if Roscovitine treatment was affecting viral gene expression at the level of RNA transcription, we measured the amounts of selected viral transcripts by quantitative real-time PCR and by Northern blot analysis. For quantitative real-time PCR, cells were infected with virus either in the presence of Roscovitine or in medium alone, and total RNA was isolated at 8, 16, and 24 h p.i. and quantified with primers and probes specific for IE1-72, IE2-86, and glucose-6-phosphate dehydrogenase (Table 1). Differential splicing of the UL122-123 transcript gives rise to two major transcripts, a 1.9-kb transcript that encodes IE1-72 and a 2.25-kb transcript that encodes IE2-86 (Fig. 1B) (for review, see reference 27). Since IE1-72 and IE2-86 share the first three exons and differ in their terminal exon, the probes for IE1-72 and IE2-86 spanned the splice junction of exons 3 and 4 and exons 3 and 5, respectively. Glucose-6-phosphate dehydrogenase, whose RNA levels do not change during the infection, was included as a control.
The reverse transcription-PCR results for IE1-72 and IE2-86 (Fig. 8A and B) are consistent with the previous Western analysis of IE protein levels. Addition of Roscovitine at the time of infection led to a reduction in IE1-72 mRNA levels and an increase in the levels of IE2-86 mRNA. This was particularly evident at the 8-h time point.
FIG. 8.

Analysis of viral RNA synthesized in the presence of Roscovitine. (A and B) HFF were infected at an MOI of 5 with HCMV Towne in the absence (dimethyl sulfoxide, DMSO) or presence of 15 μM Roscovitine. Cells were then harvested at 8, 16, and 24 h p.i. Total RNA was isolated from these samples and analyzed by quantitative real-time PCR for the mRNA transcripts for IE1-72 (A) and IE2-86 (B). Glucose-6-phosphate dehydrogenase (G6PD) was used as an internal control, and the values for IE1-72 and IE2-86 were normalized to that of glucose-6-phosphate dehydrogenase for each sample. The sequences for the primers and probes used are in Table 1. The RNA isolated at 8 h p.i. from the infected cells in the absence of drug was used to generate a standard curve for each gene examined. The standard curve was then used to calculate the relative amount of specific RNA present in a sample, from which the induction of transcription of the gene was calculated by comparison to the value obtained for the specific RNA at 8 h p.i. in the absence of the drug (which was assigned a value of 1). The lighter bars represent samples without Roscovitine treatment, and the darker bars indicate samples with Roscovitine added at the time of infection. The values are based on RNA isolatedfrom at least two different experiments, and the range is shown. (C) HFF were infected at an MOI of 5 with HCMV Towne in the absence or presence of 15 μM Roscovitine and harvested at 24 h p.i. Total RNA was isolated and subjected to Northern blot analysis for UL 44 RNA. The rRNAs are shown in the ethidium bromide-stained gel. M, mock infected; V, HCMV infected.
We also analyzed the amount of unspliced early UL44 mRNA with Northern blot analysis. In this case, cells were treated with 15 μM Roscovitine at the time of infection, and total RNA was harvested at 24 h p.i. Following electrophoresis and transfer to Nytran, the blot was probed with a radiolabeled DNA fragment specific for UL44 (Fig. 8C). As a control for the amount of RNA in each lane, the gel was stained with ethidium bromide to visualize the rRNA. In accord with the Western blot analysis, Roscovitine treatment reduced the expression of the early 4.4-kb RNA encoding UL44 by approximately sixfold.
Roscovitine affects the differential processing and accumulation of the IE UL37 RNAs.
The major UL37 IE promoter directs the synthesis of several spliced 3.2- to 3.4-kb RNAs (UL37 and UL37M) that are present in small amounts only at IE times. It is also the promoter for an abundant 1.7-kb unspliced RNA (UL37X1) that is present throughout the infection (see Fig. 1C). Examination of the relative position of the sequences used for processing the HCMV unspliced and spliced UL37 IE RNAs (64) revealed a striking similarity to the region between UL123 exon 4 and UL122 exon 5 that is used for the alternative cleavage-polyadenylation and splicing that generates the IE1-72 and IE2-86 RNAs (Fig. 9A). The cleavage-polyadenylation site for the IE1-72 transcript (a binding site for the cleavage factors CFI and CFII) lies just upstream (33 nucleotides) of the splice acceptor site for the terminal exon of IE2. It also lies within a consensus mammalian splice branch point (UACUAAG).
FIG. 9.

Quantitative real-time PCR analysis of the effect of Roscovitine on the levels of the UL37 IE RNAs. (A) Sequences used for alternative cleavage-polyadenylation and splicing to generate the UL37 RNAs and IE1/IE2 RNAs. The cleavage-polyadenylation site of the UL37X1 and IE1-72 RNAs and binding site for the cleavage factors CFI and CFII are indicated by a vertical arrow. Poly A, AAUAAA signal that binds the cleavage and polyadenylation specificity factor (CPSF) complex; U/GU, U/GU-rich sequence that serves as the binding site for the 64-kDa subunit of the polyadenylation cleavage stimulatory factor (CstF) complex; BP, splice branch point; PPY, polypyrimidine tract that lies upstream of the splice acceptor (SA) site for UL37 exon 2 and IE2-86 exon 5. PPY serves as the binding site for the essential U2AF-65 splicing factor. (B and C) The same RNA samples that were used in the analysis shown in Fig. 8 were also analyzed by quantitative real-time PCR for the mRNA transcripts for the IE gene UL37X1 (B) and the spliced UL37 RNA (C). Glucose-6-phosphate dehydrogenase (G6PD) was used as an internal control, and the values for UL37X1 and UL37 were normalized to that of glucose-6-phosphate dehydrogenase for each sample. The lighter bars represent samples without the addition of Roscovitine, and the darker bars represent those samples with Roscovitine added at the time of infection. The values are based on RNA isolated from two different experiments, and the range is shown.
Preceding the splice acceptor site is a U-rich sequence within a polypyrimidine tract that could be used for the binding of the polyadenylation factor CstF, the polypyrimidine tract binding protein (a suppressor of splicing), or the essential U2AF-65 splicing factor. Likewise, the cleavage-polyadenylation site for the UL37X1 RNA is just 8 nucleotides upstream of the splice acceptor site for exon 2 of the UL37 and UL37M RNAs, and several U-rich sequences within a polypyrimi-dine tract overlap both sites. The polyadenylation signal (AAUAAA) for UL37X1 also overlaps the putative splice branch point for the spliced UL37 RNAs. For both IE regions, the cleavage-polyadenylation site is preferentially used at IE times to generate either IE1-72 or UL37X1 RNA. Since treatment with Roscovitine affected the processing and accumulation of IE1-72 RNA, we were interested in determining whether a similar effect would be observed for the UL37X1 RNA.
Samples of RNA used for the quantitative real-time PCR analysis shown in Fig. 8 were assayed with primers and probes specific for the unspliced UL37X1 and the spliced UL37 mRNAs (Table 1). Figure 9 shows that addition of Roscovitine decreased the mRNA levels of the unspliced UL37X1 transcript (Fig. 9B) and at the same time increased the amount of spliced UL37 mRNA (Fig. 9C). Thus, Roscovitine had the same effect on the processing and accumulation of the UL37 RNA as it did on the UL122-123 RNA.
Effects of Roscovitine on IE gene expression occur in the absence of de novo protein synthesis.
In order to determine if the effects of Roscovitine on IE1-72 and IE2-86 expression required new protein synthesis, we infected cells in the presence of cycloheximide, Roscovitine, cycloheximide plus Roscovitine, or medium alone (Fig. 10). In the presence of cycloheximide, no host or viral proteins are synthesized, and therefore only the IE RNAs are transcribed. At 6 h p.i., the cycloheximide was removed so that protein synthesis could resume. Cells that were infected in the presence of cycloheximide alone (Fig. 10, lanes 3 and 6) were refed with medium alone (Fig. 10, lane 3) or medium plus actinomycin D to inhibit further transcription of IE and early RNAs (Fig. 10, lane 6). Cells that had been infected in the presence of Roscovitine alone (Fig. 10, lanes 2 and 7) or Roscovitine plus cycloheximide (Fig. 10, lanes 5 and 8) were refed with medium containing Roscovitine (Fig. 10, lanes 2 and 5) or with Roscovitine plus actinomycin D (Fig. 10, lanes 7 and 8). As a control, cells that were infected in the absence of both cycloheximide and Roscovitine (Fig. 10, lanes 1 and 4) were refed with medium alone (Fig. 10, lane 1) or with medium plus actinomycin D (Fig. 10, lane 4). All cells were harvested at 18 h p.i. and analyzed by Western blot for the expression of the IE1-72 and IE2-86 proteins.
FIG. 10.

Effect of Roscovitine on IE gene expression does not require de novo protein synthesis. Cells were infected at an MOI of 3 with HCMV Towne in the presence of Roscovitine, cycloheximide, and actinomycin D, added at the times indicated (top). Samples were harvested at 18 h p.i. and subjected to Western blot analysis for IE proteins IE1-72 and IE2-86.
In all of the samples where Roscovitine was added, its effect on the relative amounts of IE1-72 and IE2-86 was observed regardless of the presence of cycloheximide (compare Fig. 10, lanes 2 and 5 and lanes 7 and 8). As expected, the overall level of the IE proteins was higher in cells treated with cycloheximide from 0 to 6 h p.i. and lower in cells that were maintained in actinomycin D from 6 to 18 h p.i. These results suggest that the effect of Roscovitine on the transcriptional processing of the IE transcripts does not require de novo protein synthesis and that Roscovitine is most likely affecting existing cellular factors or input viral proteins.
Effects of Roscovitine on IE gene expression are proteasome independent.
An alternative explanation for the altered transcriptional processing was that Roscovitine affected the ubiquitin proteasome degradation pathway such that a positive factor for the processing of the IE1-72 RNA was rapidly degraded. If this were the case, then inhibiting the proteasome to prevent protein degradation should allow IE1-72 protein levels to be restored. To test this hypothesis, we infected cells in the presence of Roscovitine alone, MG132 alone (a proteasome inhibitor), Roscovitine plus MG132, or medium. The cells were harvested at 8, 16, and 24 h p.i. and analyzed by Western blot for IE1-72 and IE2-86 gene expression. Figure 11 shows that inhibition of the proteasome did not have an effect on the levels of the IE proteins in the absence of Roscovitine and did not alter the Roscovitine-mediated change in the relative amounts of IE1-72 and IE2-86. Based on these results, we conclude that Roscovitine is not causing the rapid proteasome-mediated turnover of a factor needed for IE1-72 gene expression.
FIG. 11.

Effect of Roscovitine is not proteasome dependent. HFF were infected at an MOI of 3 with HCMV Towne in the presence of Roscovitine alone, MG132 alone, or both Roscovitine and MG132. Cells were harvested at the times indicated and then analyzed by Western blot for IE proteins IE1-72 and IE2-86.
Flavopiridol and Roscovitine have similar effects on HCMV IE and early gene expression.
Although Roscovitine is a highly specific inhibitor of cyclin-dependent kinase activity, we sought additional evidence that cyclin-dependent kinases were required at early times in the infection. Flavopiridol is another cyclin-dependent kinase inhibitor that differs significantly from the purine-based drug Roscovitine. It is a potent inhibitor of cdk9/cyclin T (P-TEFb) and also inhibits the activity of cdk1, cdk2, cdk4, cdk6, and cdk7. We tested the effect of two different concentrations of Flavopiridol (25 and 50 nM) that show no cytotoxicity in uninfected cells (data not shown). Cells were infected in the presence of 25 or 50 nM Flavopiridol or medium alone. At 8 h p.i., 25 or 50 nM Flavopiridol was added to some of the cultures that were infected in the presence of medium alone (Fig. 12). The cells were harvested at 12 and 24 h p.i. and analyzed by Western blot for IE1-72 and IE2-86 gene expression. The cells harvested at 24 h p.i. were also analyzed for the expression of UL57 and UL44. Figure 12 shows that 50 nM Flavopiridol and Roscovitine had similar effects on the relative amounts of IE1-72 and IE2-86. The inhibitory effects on the expression of UL57 and UL44 were also comparable. At 25 nM Flavopiridol, only partial effects were seen. Analogous to what was observed with Roscovitine, delaying the addition of Flavopiridol to 8 h p.i. eliminated the effects of the drug on the expression of these proteins. Thus, two different inhibitors of cyclin-dependent kinases had similar effects on viral IE and early gene expression.
FIG. 12.

Flavopiridol and Roscovitine have similar effects on HCMV IE and early gene expression. HFF were infected at an MOI of 5 with HCMV Towne strain in the absence or presence of 20 or 50 nM Flavopiridol (added at the time of infection or at 8 h p.i.). Cells were harvested at the time points indicated and analyzed by Western blot for the IE proteins IE1-72 and IE2-86 and early gene products of UL44 and UL57. The asterisk indicates that Flavopiridol was added at 8 h p.i. M, mock infected; V, HCMV infected.
DISCUSSION
The drug Roscovitine is a purine analogue that inhibits cyclin-dependent kinase activity by binding to the active site of the enzyme. It is an effective and specific inhibitor of cyclin-dependent kinases 1, 2, 5, 7, and 9 at very low concentrations (20, 22, 43, 61, 68). In our experiments, uninfected cells treated with up to 25 μM Roscovitine for 6 days showed no cytotoxicity. For this study, data are presented from experiments in which the cells were treated with 15 μM Roscovitine. Previously, Evers et al. (24) reported that in an HCMV plaque reduction assay, the inhibition of plaque formation in human foreskin fibroblasts showed a steep dose-response curve to Roscovitine. In their experiments, they saw no reduction in the number of plaques at concentrations of up to 20 μM and complete inhibition at a concentration of 50 μM or higher. This likely explains the variability that we observed at a concentration of 15 μM (20- to 100-fold reduction in viral titer), as we also noted that when the concentration was increased from 15 to 25 μM, there was a large decrease in viral titer.
In accord with those of others (9, 24), our experiments showed that addition of Roscovitine to the medium at the beginning of the infection significantly reduced HCMV titers. Viral DNA replication was also inhibited; however, both of these phenomena could have been due to the lack of viral gene expression. Given that HCMV infection proceeds in a temporal fashion, where one event is required before the next can occur, the loss of viral gene expression would lead to the loss of replication and in turn the loss of virus production.
When Roscovitine was added at the beginning of the infection, the amount of the IE1-72 protein decreased, while the levels of IE2-86 increased. Because IE1-72 is a known transactivator of early gene expression, it was not surprising that the levels of some early gene products (UL44 and UL57) were also reduced. Along similar lines, because UL44 and UL57 are required for HCMV replication (51), it followed that viral DNA replication would also be affected. With immunofluorescence analysis, we noted that replication centers formed but never grew and coalesced in the presence of Roscovitine (data not shown). Morphologically, the replication centers in Roscovitine-treated cells at late times of infection were similar in appearance to the replication centers that are seen very early in untreated cells.
Importantly, it is unlikely that the effects of Roscovitine are due entirely to decreased levels of IE1-72, as it has been shown that an IE1-72 deletion mutant can replicate efficiently at a high MOI. Since all of the experiments reported in this paper were conducted at a high MOI, it is almost certain that other events are occurring early during the infection that are cyclin-dependent kinase dependent. Most of these events likely occur prior to 6 h p.i., because a 6-h delay in the addition of Roscovitine to the infected cells allowed viral gene expression to progress normally to the late phase, although viral titers were still affected. These results suggest that although cyclin-dependent kinase activity is not directly required for viral DNA replication, it is required at later times, possibly for virion assembly.
The early effect on IE1-72 and IE2-86 gene expression appears to be at the level of processing of the RNA. During the first 24 h of the normal infection, IE1-72 RNA accumulates to a higher level than IE2-86 RNA. As time progresses, more IE2-86 transcripts accumulate and there is a slight increase in IE1-72 transcripts. However, when Roscovitine was present during the first 24 h of the infection, the ratio of IE1-72 RNA to IE2-86 RNA was reversed; there was a decrease in IE1-72 RNA and an increase in the level of the IE2-86 transcript. The most likely explanation for these data is that the differential splicing of the UL122-123 transcript was altered by Roscovitine treatment, but we cannot exclude the possibility that there was a change in the relative stability of the RNAs. Treatment with the drug appeared to affect the differential splicing and polyadenylation of the IE UL37 RNAs in a similar way.
We hypothesize that for both the UL122-123 and UL37 transcripts, Roscovitine and Flavopiridol treatment leads to suppression of the first cleavage-polyadenylation site and enhanced utilization of the adjacent downstream 3′ splice acceptor site. Relevant to this are the recent studies of Su et al. (64). Although their experiments used mutant target minigenes and transient transfection assays in uninfected cells, the results indicated that the cis polyadenylation and 3′ splice acceptor sites for the UL37 RNAs caused steric hindrance on the precursor mRNA for the alternative processes of polyadenylation and splicing, with polyadenylation of UL37X1 being dominant. In the case of the UL122-123 transcripts, the polyadenylation site generating the IE1-72 RNA is also juxtaposed to the 3′ splice acceptor site that is used to produce the IE2-86 RNA. In this case, the U-rich polypyrimidine sequence upstream of the 3′ splice acceptor could provide a binding site for the cleavage-polyadenylation complex CstF, the splicing suppressor polypyrimidine tract binding protein, or the essential U2AF-65 splicing factor (Fig. 9).
Likewise, the sequence where the cleavage occurs at the 3′ end of the IE1-72 RNA could be used for the binding of the cleavage factors CFI/II. However, this sequence also lies within the sequence UACUAAG, which matches the mammalian splicing branch point consensus sequence (YNYURAY) except for the last position, which can be changed without a significant decrease in activity (53), and thus it could serve as the site for the binding of SF1 and U2 splicing factors. Su et al. additionally reported that during HCMV infection, there was a transient increase in CstF64, the splicing suppressor polypyrimidine tract binding protein, and the hypophosphorylated form of the splicing factor SF2 at 4 h p.i. We also examined the expression of these factors but did not detect these changes during the first 8 h p.i. even when the infected cells were analyzed every hour (data not shown). Their expression was also not affected by treatment of the cells with Roscovitine. The reason for the discrepancy is not clear but may be due to the use of different strains of HCMV (AD169 in the studies of Su et al. versus Towne in our studies) or the state of the fibroblasts (unsynchronized versus synchronized).
There is a large body of evidence showing that the processes of mRNA capping, splicing, and cleavage-polyadenylation occur cotranscriptionally and are highly interdependent. Given the proximity of the polyadenylation site and the splice acceptor site, it seems likely that there is competition for sites on the RNA that may be required for both processes. Since numerous factors are involved in both splicing and cleavage-polyadenylation, a change in the abundance, activity, or localization of any of the factors could alter the balance. The phosphorylation state of many of the factors involved in both processes determines their activity, and therefore the cyclin-dependent kinases may play a direct or indirect role in their phosphorylation. Because multiple negative and positive splicing factors are differentially phosphorylated, it is difficult to predict whether inhibition of the cyclin-dependent kinases results in suppression of the cleavage-polyadenylation of IE1-72 and UL37X1 by the loss of activity of a positive factor or gain of activity of a negative factor. It remains to be seen which cyclin-dependent kinase might be the important one for the differential processing of the UL122-123 and UL37 transcripts and which specific proteins are important for this level of regulation of viral gene expression. Based on the results of the experiments with cycloheximide and MG132, it appears that the effects of Roscovitine on the IE RNAs do not require de novo protein synthesis or proteasome-mediated degradation of proteins.
It is also possible that the effects of Roscovitine and Flavopiridol are related to the phosphorylation of the C-terminal domain of the large subunit of RNA polymerase II. There is increasing evidence that the C-terminal domain, which in human cells consists of 52 repeats of the consensus heptapeptide sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser, plays a central regulatory role in all steps of transcription by serving as the binding domain and transporter of factors involved in RNA initiation, elongation, 5′ capping, splicing, and cleavage-polyadenylation (for review, see references 5, 48, and 57). The C-terminal domain is differentially phosphorylated primarily at the serine 2 and serine 5 positions, and the level of phosphorylation varies considerably during the transcription cycle (for review, see reference 39 and 55).
Briefly, hypophosphorylated-RNA polymerase II is recruited to the initiation complex. The C-terminal domain is then phosphorylated, with the phosphorylation of the serines in position 5 by cdk7/cyclin H (which is a component of the basal transcription factor complex TFIIH) mostly occurring prior to the phosphorylation of the serines in position 2 by cdk9/cyclin T (P-TEFb). The commitment of the RNA polymerase II complex to the elongation step is associated with this phosphorylation, particularly with the modification at position 2 by P-TEFb. Multiple proteins involved in 5′ capping, elongation, and processing of the RNA are recruited to the C-terminal domain, and their pattern of binding appears to be influenced by the differential phosphorylation of serine 2 and serine 5 (and possibly by ubiquitylation, glycosylation, and phosphorylation of other residues) within the 52 repeats. The rules governing this association in uninfected cells have yet to be elucidated, and it is likely that viral infections will introduce new layers to the complexity.
In this regard, several studies have already shown that the recruitment of P-TEFb to RNA polymerase II by human immunodeficiency virus type 1 Tat and the resulting phosphorylation of the C-terminal domain are necessary for the activation of human immunodeficiency virus long terminal repeat transcription (for review, see reference 56). Human immunodeficiency virus type 1 transcription is much more sensitive than cellular transcription to low concentrations of Flavopiridol; the IC50 for human immunodeficiency virus transcription is less than 10 nM, while significant inhibition of cellular transcription is only seen at concentrations above 100 nM (13). In herpes simplex virus type 1-infected cells, a new intermediate form of phosphorylated RNA polymerase II has been described (32, 58). Although preliminary, we also have evidence that there is a change in the phosphorylation of the C-terminal domain during HCMV infection, but it appears to differ from that reported for herpes simplex virus type 1.
The question of why a delay in the addition of Roscovitine and Flavopiridol until 6 to 8 h p.i. abolished the effects of the drug on IE and early RNAs is an interesting one. It is possible that by this time, transcription of the HCMV genome is localized to a nuclear domain (possibly next to a residual ND10 domain) that is inaccessible to the drug. Alternatively, once the RNA polymerase II is committed to the transcription of the viral genes, it might be able to engage in multiple rounds of transcription without having to completely disengage from the DNA and repeat all of the initiation steps. Relevant to this are recent reports showing that there are many protein-protein interactions between the factors involved in initiation at the promoter and termination of transcription, suggesting that these two processes are interconnected (for review, see reference 48).
These are only a few of the possible ways that the cyclin-dependent kinases might affect infection. Unfortunately, the use of inhibitors such as Roscovitine and Flavopiridol, which inhibit more than one cyclin-dependent kinase, makes it difficult to distinguish between these possibilities. There are likely many more functions for cyclin-dependent kinase activity than transcription during the course of an HCMV infection, and this is evidenced by the fact that addition of Roscovitine at later times still resulted in a decrease in viral titers. Another potential role for cyclin-dependent kinase activity during an HCMV infection is the phosphorylation of viral proteins. In a related virus, herpes simplex virus type 1, it was demonstrated that UL42 (the herpes simplex virus equivalent of UL44) bound to and was phosphorylated by cdk1 (1). The effects that this may have on viral replication and the potential downstream targets of this kinase complex are unknown; however, the implications of these data as they pertain to HCMV infection are promising. Experiments with dominant negative cyclin-dependent kinases and the technology of small interfering RNAs coupled with analysis of the splicing machinery and of viral targets of cyclin-dependent kinase activity are in progress and aim to elucidate the role of the individual cyclin-dependent kinases during infection.
Acknowledgments
We thank William Britt for providing antibodies for pp28 and the major capsid protein, Greg Pari for the gift of the 183-bp EcoRI fragment of the UL44 gene, J. Brady for the gift of Flavopiridol, and Randall Johnson for help with the real-time reverse transcription-PCR assays. We are grateful to Elizabeth White for helpful suggestions and comments on the manuscript.
This work was supported by NIH grants CA73490 and CA34729 to D.H.S. A.K.M. was supported by NIH training grant CA09345. J.Y.Y. was supported by NIH training grant GM07240.
REFERENCES
- 1.Advani, S. J., R. R. Weichselbaum, and B. Roizman. 2001. cdc2 cyclin-dependent kinase binds and phosphorylates herpes simplex virus 1 UL42 DNA synthesis processivity factor. J. Virol. 75:10326-10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anders, D. G., and W. Gibson. 1988. Location, transcript analysis, and partial nucleotide sequence of the cytomegalovirus gene encoding an early DNA-binding protein with similarities to ICP8 of herpes simplex virus type 1. J. Virol. 62:1364-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anders, D. G., A. Irmiere, and W. Gibson. 1986. Identification and characterization of a major early cytomegalovirus DNA-binding protein. J. Virol. 58:253-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1989. Current protocols in molecular biology. Greene Publishing Associates Wiley-Interscience, New York, N.Y.
- 5.Bentley, D. 2002. The mRNA assembly line:transcription and processing machines in the same factory. Curr. Opin. Cell Biol. 14:336-342. [DOI] [PubMed] [Google Scholar]
- 6.Biswas, N., V. Sanchez, and D. H. Spector. 2003. Human cytomegalovirus infection leads to accumulation of geminin and inhibition of the licensing of cellular DNA replication. J. Virol. 77:2369-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonin, L. R., and J. K. McDougall. 1997. Human cytomegalovirus IE2 86-kilodalton protein binds p53 but does not abrogate G1 checkpoint function. J. Virol. 71:5831-5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bresnahan, W. A., T. Albrecht, and E. A. Thompson. 1998. The cyclin E promoter is activated by human cytomegalovirus 86-kDa immediate early protein. J. Biol. Chem. 273:22075-22082. [DOI] [PubMed] [Google Scholar]
- 9.Bresnahan, W. A., I. Boldogh, P. Chi, E. A. Thompson, and T. Albrecht. 1997. Inhibition of cellular CDK2 activity blocks human cytomegalovirus replication. Virology 231:239-247. [DOI] [PubMed] [Google Scholar]
- 10.Bresnahan, W. A., I. Boldogh, E. A. Thompson, and T. Albrecht. 1996. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 224:156-160. [DOI] [PubMed] [Google Scholar]
- 11.Bresnahan, W. A., E. A. Thompson, and T. Albrecht. 1997. Human cytomegalovirus infection results in altered Cdk2 subcellular localization. J. Gen. Virol. 78:1993-1997. [DOI] [PubMed] [Google Scholar]
- 12.Castillo, J. P., A. Yurochko, and T. F. Kowalik. 2000. Role of human cytomegalovirus immediate-early proteins in cell growth control. J. Virol. 74:8028-8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao, S.-H., and D. H. Price. 2001. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 276:31793-31799. [DOI] [PubMed] [Google Scholar]
- 14.Chen, Z., E. Knutson, A. Kurosky, and T. Albrecht. 2001. Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J. Virol. 75:3613-3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi, K. S., S.-J. Kim, and S. Kim. 1995. The retinoblastoma gene product negatively regulates transcriptional activation mediated by the human cytomegalovirus IE2 protein. Virology 208:450-456. [DOI] [PubMed] [Google Scholar]
- 16.Colberg-Poley, A. M., M. B. Patel, D. P. Erezo, and J. E. Slater. 2000. Human cytomegalovirus UL37 immediate-early regulatory proteins traffic through the secretory apparatus and to mitochondria. J. Gen. Virol. 81:1779-1789. [DOI] [PubMed] [Google Scholar]
- 17.Colberg-Poley, A. M., L. D. Santomenna, P. P. Harlow, P. A. Benfield, and D. J. Tenney. 1992. Human cytomegalovirus US3 and UL36-38 immediate-early proteins regulate gene expression. J. Virol. 66:95-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai, Y., and S. Grant. 2003. Cyclin-dependent kinase inhibitors. Curr. Opin. Pharmacol. 3:362-370. [DOI] [PubMed] [Google Scholar]
- 19.Davison, A. J., A. Dolan, P. Akter, C. Addison, D. J. Dargan, D. J. Alcendor, D. J. McGeoch, and G. S. Hayward. 2003. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J. Gen. Virol. 84:17-28. [DOI] [PubMed] [Google Scholar]
- 20.De Azevedo, W. F., S. Leclerc, L. Meijer, L. Havlicek, M. Strnad, and S. H. Kim. 1997. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 243:518-526. [DOI] [PubMed] [Google Scholar]
- 21.Dittmer, D., and E. S. Mocarski. 1997. Human cytomegalovirus infection inhibits G1/S transition. J. Virol. 71:1629-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edamatsu, H., C. L. Gau, T. Nemoto, L. Guo, and F. Tamanoi. 2000. Cdk inhibitors, roscovitine and olomoucine, synergize with farnesyltransferase inhibitor (FTI) to induce efficient apoptosis of human cancer cell lines. Oncogene 19:3059-3068. [DOI] [PubMed] [Google Scholar]
- 23.Ekholm, S. V., and S. I. Reed. 2000. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 12:676-684. [DOI] [PubMed] [Google Scholar]
- 24.Evers, D. L., J. M. Breitenbrach, K. Z. Borysko, L. B. Townsend, and J. C. Drach. 2002. Inhibition of cyclin-dependent kinase 1 by purines and pyrrolo[2,3-d]pyrimidines does not correlate with antiviral activity. Antimicrob. Agents Chemother. 46:2470-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fortunato, E. A., V. Sanchez, J. Y. Yen, and D. H. Spector. 2002. Infection of cells with human cytomegalovirus during S phase results in a blockade to immediate-early gene expression that can be overcome by inhibition of the proteasome. J. Virol. 76:5369-5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fortunato, E. A., and D. H. Spector. 1998. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J. Virol. 72:2033-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fortunato, E. A., and D. H. Spector. 1999. Regulation of human cytomegalovirus gene expression. Adv. Virus Res. 54:61-128. [DOI] [PubMed] [Google Scholar]
- 28.Goldmacher, V. S., L. M. Bartle, A. Skaletskaya, C. A. Dionne, N. L. Kedersha, C. A. Vater, J. W. Han, R. J. Lutz, S. Watanabe, E. D. C. McFarland, E. D. Kieff, E. S. Mocarski, and T. Chittenden. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 96:12536-12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heilbronn, R., G. Jahn, A. Burkle, U.-K. Fresse, B. Fleckenstein, and H. ZurHausen. 1987. Genomic localization, sequence analysis, and transcription of the putative human cytomegalovirus DNA polymerase gene. J. Virol. 61:119-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iskenderian, A. C., L. Huang, A. Reilly, R. M. Stenberg, and D. G. Anders. 1996. Four of eleven loci required for transient complementation of human cytomegalovirus DNA replication cooperate to activate expression of replication genes. J. Virol. 70:383-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jault, F. M., J.-M. Jault, F. Ruchti, E. A. Fortunato, C. Clark, J. Corbeil, D. D. Richman, and D. H. Spector. 1995. Cytomegalovirus infection induces high levels of cyclins, phosphorylated RB, and p53, leading to cell cycle arrest. J. Virol. 69:6697-6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins, H. L., and C. A. Spencer. 2001. RNA polymerase II holoenzyme modifications accompany transcription reprogramming in herpes simplex virus type 1-infected cells. J. Virol. 75:9872-9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalejta, R. F., J. T. Bechtel, and T. Shenk. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalejta, R. F., and T. Shenk. 2003. The human cytomegalovirus UL82 gene product (pp71) accelerate progression through the G1 phase of the cell cycle. J. Virol. 77:3451-3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kemble, G. W., A. L. McCormick, L. Pereira, and E. S. Mocarski. 1987. A cytomegalovirus protein with properties of herpes simplex virus ICP8; partial purification of the polypeptide and map position of the gene. J. Virol. 61:3143-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kouzarides, T., A. T. Bankier, A. C. Satchwell, E. Preddy, and B. G. Barrell. 1988. An immediate early gene of human cytomegalovirus encodes a potential membrane glycoprotein. Virology 165:151-164. [DOI] [PubMed] [Google Scholar]
- 37.Kouzarides, T., A. T. Bankier, A. C. Satchwell, K. Weston, P. Tomlinson, and B. G. Barrell. 1987. Sequence and transcription analysis of the human cytomegalovirus DNA polymerase gene. J. Virol. 61:125-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu, M., and T. Shenk. 1996. Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J. Virol. 70:8850-8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majello, B., and G. Napolitano. 2001. Control of RNA polymerase II activity by dedicated CTD kinases and phosphatases. Front. Biosci. 6:D1358-D1368. [DOI] [PubMed] [Google Scholar]
- 40.Margolis, M. J., S. Panjovic, E. L. Wong, M. Wade, R. Jupp, J. A. Nelson, and J. C. Azizkhan. 1995. Interaction of the 72-kilodalton human cytomegalovirus IE1 gene product with E2F1 coincides with E2F-dependent activation of dihydrofolate reductase transcription. J. Virol. 69:7759-7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCormick, A. L., V. L. Smith, D. Chow, and E. S. Mocarski. 2003. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 77:631-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McElroy, A. K., R. S. Dwarakanath, and D. H. Spector. 2000. Dysregulation of cyclin E gene expression in human cytomegalovirus-infected cells requires viral early gene expression and is associated with changes in the Rb-related protein p130. J. Virol. 74:4192-4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meijer, L., A. Borgne, O. Mulner, J. P. J. Chong, J. J. Blow, N. Inagaki, M. Inagaki, J. G. Delcros, and J. P. Molinoux. 1997. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243:527-536. [DOI] [PubMed] [Google Scholar]
- 44.Mocarski, E. S., and C. T. Courcelle. 2001. Cytomegaloviruses and their replication, p. 2629-2673. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 45.Muganda, P., O. Mendoza, J. Hernandez, and Q. Qian. 1994. Human cytomegalovirus elevates levels of the cellular protein p53 in infected fibroblasts. J. Virol. 68:8028-8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murphy, E. A., D. N. Streblow, J. A. Nelson, and M. F. Stinski. 2000. The human cytomegalovirus IE86 protein can block cell cycle progression after inducing transition into the S phase of the cell cycle. J. Virol. 74:7108-7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noris, E., C. Zannetti, A. Demurtas, J. Sinclair, M. DeAndrea, M. Gariglio, and S. Landolfo. 2002. Cell cycle arrest by human cytomegalovirus 86-kilodalton IE2 protein resembles premature senescence. J. Virol. 76:12135-12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orphanides, G., and D. Reinberg. 2002. A unified theory of gene expression. Cell 108:439-451. [DOI] [PubMed] [Google Scholar]
- 49.Pajovic, S., E. L. Wong, A. R. Black, and J. C. Azizkhan. 1997. Identification of a viral kinase that phosphorylates specify E2Fs and pocket proteins. Mol. Cell. Biol. 17:6459-6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979-6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pari, G. S., M. A. Kacica, and D. G. Anders. 1993. Open reading frames UL44, IRS1/TRS1, and UL36-38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J. Virol. 67:2575-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pass, R. F. 2001. Cytomegalovirus, p. 2675-2705. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 53.Peled-Zehavi, H., J. A. Berglund, M. Rosbash, and A. D. Frankel. 2001. Recognition of RNA branch point sequences by the KH domain of splicing factor 1 (mammalian branch point binding protein) in a splicing factor complex. Mol. Cell. Biol. 21:5232-5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pines, J. 1993. Cyclins and cyclin-dependent kinases: take your partners. Trends Biochem. Sci. 18:195-197. [DOI] [PubMed] [Google Scholar]
- 55.Prelich, G. 2002. RNA polymerase II carboxy-terminal domain kinases: emerging clues to their function. Eukaryot. Cell 1:153-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price, D. H. 2000. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 20:2629-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Proudfoot, N. J., A. Furger, and M. J. Dye. 2002. Integrating mRNA processing with transcription. Cell 108:501-512. [DOI] [PubMed] [Google Scholar]
- 58.Rice, S. A., M. C. Long, V. Lam, and C. A. Spencer. 1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following Herpes Simplex virus infection. J. Virol. 68:988-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salvant, B. S., E. A. Fortunato, and D. H. Spector. 1998. Cell cycle dysregulation by human cytomegalovirus: Influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol. 72:3729-3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schang, L. M. 2004. Effects of pharmacological cyclin-dependent kinase inhibitors on viral transcription and replication. Biochim. Biophys. Acta 1697:197-209. [DOI] [PubMed] [Google Scholar]
- 61.Schang, L. M., A. Bantly, M. Knockaert, F. Shaheen, L. Meijer, M. H. Malim, N. S. Gray, and P. A. Schaffer. 2002. Pharmacological cyclin-dependent kinase inhibitors inhibit replication of wild-type and drug-resistant strains of herpes simplex virus and human immunodeficiency virus type 1 by targeting cellular, not viral, proteins. J. Virol. 76:7874-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith, J. A., and G. S. Pari. 1995. Expression of human cytomegalovirus UL36 and UL37 genes is required for viral DNA replication. J. Virol. 69:1925-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spector, D. H., L. Hock, and J. C. Tamashiro. 1982. Cleavage maps for human cytomegalovirus DNA strain AD169 for restriction endonucleases EcoRI, BglII, and HindIII. J. Virol. 42:558-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su, Y., R. Adair, C. D. Davis, N. L. DiFronzo, and A. M. Colberg-Poley. 2003. Convergence of RNA cis elements and cellular polyadenylation factors in the regulation of human cytomegalovirus UL37 exon 1 unspliced RNA production. J. Virol. 77:12729-12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tamashiro, J. C., L. J. Hock, and D. H. Spector. 1982. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169). J. Virol. 42:547-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tenney, D. J., and A. M. Colberg-Poley. 1991. Expression of the human cytomegalovirus UL36-38 immediate early region during permissive infection. Virology 182:199-210. [DOI] [PubMed] [Google Scholar]
- 67.Tenney, D. J., and A. M. Colberg-Poley. 1991. Human cytomegalovirus UL36-38 and US3 immediate-early genes: temporally regulated expression of nuclear, cytoplasmic, and polysome-associated transcripts during infection. J. Virol. 65:6724-6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang, D., C. de la Fuente, L. Deng, L. Wang, I. Zilberman, C. Eadie, M. Healey, D. Stein, T. Denny, L. E. Harrison, L. Meijer, and F. Kashanchi. 2001. Inhibition of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent kinase inhibitors. J. Virol. 75:7266-7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weibusch, L., J. Asmar, R. Uecker, and C. Hagemeier. 2003. Human cytomegalovirus immediate-early over protein 2 (IE2)-mediated activation of cyclin E is cell-cycle-independent and forces S-phase entry in IE2-arrested cells. J. Gen. Virol. 84:51-60. [DOI] [PubMed] [Google Scholar]
- 70.Weibusch, L., and C. Hagemeier. 1999. Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G1. J. Virol. 73:9274-9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weibusch, L., and C. Hagemeier. 2001. The human cytomegalovirus immediate early 2 protein dissociates cellular DNA synthesis from cyclin dependent kinase activation. EMBO J. 20:1086-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.White, E. A., C. L. Clark, V. Sanchez, and D. H. Spector. 2004. Small internal deletions in the human cytomegalovirus IE2 gene result in nonviable recombinant viruses with differential defects in viral gene expression. J. Virol. 78:1817-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wright, D. A., S. I. Staprans, and D. H. Spector. 1988. Four phosphoproteins with common amino termini are encoded by human cytomegalovirus AD169. J. Virol. 62:331-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang, H., H. O. al-Barazi, and A. M. Colberg-Poley. 1996. The acidic domain of the human cytomegalovirus UL37 immediate early glycoprotein is dispensable for its transactivating activity and localization but is not for its synergism. Virology 223:292-302. [DOI] [PubMed] [Google Scholar]
- 75.Zimmermann, A., H. Wilts, M. Lenhardt, M. Hahn, and T. Mertens. 2000. Indolocarbazoles exhibit strong antiviral activity against human cytomegalovirus and are potent inhibitors of the pUL97 protein kinase. Antivir. Res. 48:49-60. [DOI] [PubMed] [Google Scholar]