

Abstract
Signaling mediated by the cellular kinase mammalian target of rapamycin (mTOR) activates cap-dependent translation under normal (nonstressed) conditions. However, translation is inhibited by cellular stress responses or rapamycin treatment, which inhibit mTOR kinase activity. We show that during human cytomegalovirus (HCMV) infection, viral protein synthesis and virus production proceed relatively normally when mTOR kinase activity is inhibited due to hypoxic stress or rapamycin treatment. Using rapamycin inhibition of mTOR, we show that HCMV infection induces phosphorylation of two mTOR effectors, eucaryotic initiation factor 4E (eIF4E) binding protein (4E-BP) and eIF4G. The virally induced phosphorylation of eIF4G is both mTOR and phosphatidylinositol 3-kinase (PI3K) independent, whereas the phosphorylation of 4E-BP is mTOR independent, but PI3K dependent. HCMV infection does not induce mTOR-independent phosphorylation of a third mTOR effector, p70S6 kinase (p70S6K). We show that the HCMV-induced phosphorylation of eIF4G and 4E-BP correlates with the association of eIF4E, the cap binding protein, with eIF4G in the eIF4F translation initiation complex. Thus, HCMV induces mechanisms to maintain the integrity of the eIF4F complex even when mTOR signaling is inhibited.
The cellular kinase mammalian target of rapamycin (mTOR) (6, 19) is a major regulator of cap-dependent translation initiation (12, 18). Under metabolically favorable conditions, mTOR kinase is activated by phosphorylation, leading to active translation. Activated mTOR kinase phosphorylates three major effectors: eucaryotic initiation factor 4E (eIF4E) binding protein (4E-BP), eIF4G, and p70 S6 kinase (p70S6K) (Fig. 1) (11, 12). Phosphorylation of 4E-BP prevents it from binding eIF4E, the cap binding protein. This permits eIF4E to join the eIF4F complex (Fig. 1), which also contains eIF4A, Mnk1, and the scaffolding protein eIF4G. In the eIF4F complex, eIF4E binds capped mRNAs productively, promoting cap-dependent translation. The significance of eIF4G phosphorylation by active mTOR remains to be established; it is possible that eIF4G functions more effectively as a scaffold to maintain an active eIF4F complex when it is phosphorylated. Activated mTOR kinase also phosphorylates and activates p70S6K, resulting in phosphorylation of ribosomal protein S6; this correlates with increased ribosome biogenesis, which supports increased protein synthesis (Fig. 1) (11, 12).
FIG. 1.

The mTOR signaling pathway. The phosphorylation characteristics of mTOR downstream effectors and the status of the eIF4F complex when mTOR is either active or inactive are shown. The points at which hypoxia and rapamycin inhibit mTOR are indicated. S6K, p70S6 kinase; Mnk1, mitogen-activated protein kinase signal-integrating kinase 1.
As intracellular parasites, viruses like human cytomegalovirus (HCMV) are highly dependent on the maintenance of translation for synthesis of their proteins. Given the central role of mTOR in controlling cap-dependent translation, we hypothesized that HCMV may actively preserve the phosphorylation of mTOR and/or its downstream effectors during the course of viral infection to ensure the integrity of the eIF4F complex. Activation of mTOR occurs via a number of signaling pathways, including phosphatidylinositol 3-kinase (PI3K)/Akt (8, 11, 12, 14). It has previously been shown that HCMV infection and the HCMV major immediate-early proteins (MIEPs) can activate PI3K/Akt signaling (16, 25, 26), potentially resulting in mTOR phosphorylation and activation under normal infection conditions. However, mTOR activation, and the resulting phosphorylation of its effectors, can be inhibited by cellular stress responses that may occur during a viral infection, such as nutrient deprivation or hypoxia (1).
To determine whether HCMV induces a mechanism(s) to maintain mTOR signaling, even when mTOR activation is prevented, we utilized the drug rapamycin, which prevents activated mTOR from phosphorylating its downstream effectors (9-12, 22). We also utilized hypoxia to determine whether HCMV has a mechanism for counteracting the mTOR inhibition resulting from virus-induced stress responses.
In the following studies we show that HCMV infection activates mTOR signaling under normal conditions. However, by inhibiting mTOR activity with rapamycin or hypoxia, we find that HCMV induces mTOR-independent mechanisms for the phosphorylation of 4E-BP and eIF4G. The virally induced phosphorylation of eIF4G is also PI3K independent. HCMV infection does not induce mTOR-independent activation of a third mTOR effector, p70S6K. Our data suggest that a major consequence of HCMV infection is the phosphorylation of mTOR effectors to promote the formation and integrity of the eIF4F complex (Fig. 1), thus ensuring the maintenance of cap-dependent translation even when mTOR activity is inhibited.
MATERIALS AND METHODS
Cell culture.
Life-extended human foreskin fibroblasts (LEHFFs) (5) were cultured at 37°C in 5% CO2 and atmospheric oxygen in D10 medium (Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum, 100 U of penicillin per ml, 100 μg of streptomycin per ml, and 2 mM Gluta-Max [all from GIBCO-BRL, Gaithersburg, Md.]). All cells used in experiments were between passages 9 and 14. For hypoxia experiments, cells were incubated in an InVivo2 hypoxic workstation (Biotrace, Inc.) with 1.5% O2 and 5%CO2 at 37°C or in a hypoxic incubator under similar conditions. All serum starvation experiments were done in D0 medium, which is D10 medium without the serum. Under serum starvation conditions, confluent monolayers were incubated in D0 medium for 48 h prior to infection and maintained in D0 medium throughout the infection.
Antibodies and reagents.
Rapamycin was purchased from Sigma (St. Louis, Mo.), and LY294002 (LY) was purchased from Cell Signaling Technologies (Beverly, Mass.). Both were dissolved in dimethyl sulfoxide (DMSO) according to the manufacturers' protocols; thus, all controls were done with DMSO without drug. 7Me-GTP-Sepharose 4B was purchased from Amersham Biosciences (Piscataway, N.J.). Anti-eIF4G (sc-11373) was purchased from Santa Cruz Biotechnology (Santa Cruz, Calif.); antiactin antibody was purchased from Chemicon International, Inc. (Temecula, Calif.); antibody to ICP36 was purchased from Advanced Biotechnologies, Inc. (Columbia, Md.); and anti-rabbit immunoglobulin G secondary antibodies were purchased from Pierce Biotechnologies (Rockford, Ill.). All other antibodies used were obtained from Cell Signaling Technologies, except for anti-exon2/3, a polyclonal antiserum prepared for this laboratory by Cocalico Biologicals, Inc. (Reamstown, Pa.); it recognizes the common 85 amino acids in all HCMV MIEPs (13).
Virus stock preparation and virus purification.
All experiments were done with a derivative of the Towne strain of HCMV that has been modified by replacement of a nonessential gene with the green fluorescent protein (GFP) gene expressed from the simian virus 40 early promoter (15). Virus stocks were prepared from LEHFFs infected at a multiplicity of infection (MOI) of 0.02. Infected plates were monitored for cytopathic effects (CPE), and fresh D10 medium was added to each plate when CPE was observed in 100% of the cells (approximately 10 to 12 days). After an additional 3 to 4 days, the cells were scraped into the medium, and media from all infected plates were collected and pooled in sterile centrifuge bottles. The bottles were spun for 10 min at 10,000 rpm at room temperature in a Sorvall RC-5B centrifuge. All but 3 ml of the supernatant was removed to a sterile glass bottle. The cell pellets were resuspended in the remaining 3 ml, sonicated 10 times with 1-s pulses, and then centrifuged at 13,000 rpm for 5 min in a Biofuge Pico Microcentrifuge. The supernatant from the sonicated cells were pooled with the rest of the collected supernatent. Viruses were purified by layering the supernatants over a d-sorbitol cushion (20% sorbitol, 50 mM Tris-HCl [pH 7.2], 1 mM MgCl2, 0.1 μg of Bacitracin per ml) and pelleting the viruses through the cushion by ultracentrifugation. Virus pellets were resuspended in D0 medium and again layered over a d-sorbitol cushion and pelleted. The final pellet was suspended in D0 medium, aliquoted, flash frozen in liquid nitrogen, and stored at −80°C.
Viral growth in the presence or absence of rapamycin.
LEHFFs (2 × 105) were grown in two six-well plates and infected at an MOI of 2 in the presence or absence of 50 nM rapamycin either with or without serum starvation as described above. After a 2-h incubation, the cells were washed three times with medium, and then 2 ml of medium with or without 50 nM rapamycin was added to each well. At designated time points, 1 ml of medium was removed from one well of each plate and placed in a 15-ml conical tube. The cells in the well were scraped into the remaining 1 ml of medium, sonicated as described above, and centrifuged for 10 min in a microcentrifuge at 4°C. The supernatant were combined with previously collected medium, aliquoted, and stored at −80°C.
Viral growth in hypoxia.
Cells (2 × 105) were plated in 10 35-mm-diameter dishes in D10 medium and infected at an MOI of 2 with purified HCMV in D10 medium. Five dishes were incubated at 37°C in 5% CO2 and atmospheric oxygen (normoxia), and five dishes were incubated in an InVivo2 hypoxic workstation at 37°C in 5% CO2 and 1.5% O2 (hypoxia). After a 4-h incubation, all dishes were washed three times with D0 medium and replaced with 3 ml of D10 medium. Conditions of normoxia or hypoxia were maintained. At designated time points after infection, one dish from the normoxic and hypoxic conditions was harvested for viruses as described above.
Viral titration.
Viral titration was carried out by the 50% tissue culture infective dose method (15).
Microscopy.
LEHFFs (2.5 × 105) were plated onto 35-mm-diameter dishes containing glass coverslips. After reaching confluency, the cells were serum starved for 48 h for the experiments done with rapamycin or maintained in D10 medium for the experiments done with hypoxia. Cells were then mock infected or infected with purified HCMV at an MOI of 3 in the presence or absence of 50 nM rapamycin or under hypoxic conditions, as described above. At designated time points, dishes were removed and washed three times with ice-cold standard phosphate-buffered saline and then fixed for 30 min in 4% paraformaldehyde. Coverslips were stained with 4′,6′-diamidino-2-phenylindole (DAPI), mounted on slides, and examined with a Nikon Eclipse E600 microscope.
Time course viral infections.
LEHFFs (106) were seeded onto 100-mm-diameter dishes and allowed to grow to 90% confluency. Cells were then serum starved for 48 h. One hour prior to infection, medium was removed from the plates and replaced with D0 medium containing either 50 nM rapamycin or an equivalent volume of DMSO (vehicle control). Cells were then either mock infected with D0 medium containing either 50 nM rapamycin or DMSO or infected with purified HCMV at an MOI of 2 in D0 medium containing either 50 nM rapamycin or DMSO. The time courses commenced with the addition of the viruses. Mock-infected plates were pretreated with serum starvation and rapamycin similarly to cells to be infected; D0 medium was added to the mock-infected cells at 0 h, and the cells were immediately harvested as described below. For several experiments we harvested mock-infected plates after 24 h, but we never noted significant differences in cellular proteins between the 0- and 24-h mock-infected samples. At designated time points after infection, the plates were harvested by washing three times with ice-cold phosphate-buffered saline, followed by a 10-min incubation at 4°C in cell lysis buffer (200 mM Tris-HCl [pH 7.5], 140 mM NaCl, 1% NP-40, 10 mM NaF, 1 mM EDTA, 200 μM NaVO4, 2 mM phenylmethylsulfonyl fluoride, 1.5 μg of aprotinin per ml, and 1 μg of leupeptin per ml). Cells were scraped and lysates were centrifuged for 10 min at 13,000 rpm in a Eppendorf microcentrifuge at 4°C. The supernatants were transferred to microcentrifuge tubes and stored at −80°C. Experiments with LY were performed as described above but with 50 μM LY replacing rapamycin and infection with purified HCMV at an MOI of 10. All hypoxia experiments were done in D10 medium with no serum starvation. When rapamycin was used with hypoxia, there was a 1-h preincubation with either 50 nM rapamycin or DMSO in D10 medium as described above.
Western Analyses.
Twenty micrograms of lysate protein was separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (8 or 12% polyacrylamide) and transferred to nitrocellulose. The membranes were blocked for 1 h in 5% nonfat dry milk in standard Tris-buffered saline (TBS) with 0.1% Tween 20. Specific proteins were detected by incubating the membranes overnight at 4°C with appropriate antibody diluted in 5% bovine serum albumin in TBS with 0.1% Tween 20. Transfers were then incubated for 1 h with horseradish peroxidase-conjugated secondary antibody diluted in 5% nonfat dry milk in TBS with 0.1% Tween 20. Specific proteins were visualized with ECL reagents (Amersham Biosciences) and autoradiography.
7Me-GTP Affinity Purification.
LEHFFs (8 × 105)were serum starved and infected in the presence or absence of rapamycin as described above. Cells were lysed and harvested in complete affinity purification buffer (0.5% Triton X-100, 50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1.5 mM EDTA, 20 mM β-glycerophosphate, 10 mM NaF, 200 μM NaVO4, 2 mM phenylmethylsulfonyl fluoride, 1.5 μg of aprotinin per ml, and 1 μg of leupeptin per ml). Two hundred micrograms of extract protein was incubated in a 25% slurry of 7Me-GTP Sepharose beads in affinity purification buffer for 4 h at 4°C with rocking. After three washes in ice-cold complete affinity purification buffer, beads were boiled in 20 μl of 3× SDS loading buffer (187.5 mM Tris-HCl [pH 6.8], 6% SDS, 30% glycerol, 0.025% bromphenol blue, 473 μM 2-mercaptoethanol). The eluted proteins were separated by SDS-12% polyacrylamide gel electrophoresis and analyzed by Western analysis as described above.
RESULTS
HCMV infection proceeds both in hypoxia and in the presence of rapamycin.
To determine whether HCMV can productively infect cells under hypoxic conditions or in the presence of rapamycin, LEHFFs (5) were grown on coverslips and then infected with purified HCMV at an MOI of 3. After 4 h, the viruses were removed and the cells were washed and fed. One set of cultures was incubated under normal growth conditions of atmospheric O2 (normoxia) and 5% CO2 at 37°C, another set was placed in a hypoxia incubator (1.5% O2 and 5% CO2) (1, 2) at 37°C, and a third set was incubated under normoxic conditions with added rapamycin (50 nM). Mock-infected cultures were incubated under the same conditions. Uninfected LEHFFs maintained viability under hypoxic conditions and in rapamycin for at least 144 h, the longest time point we have tested (data not shown). Coverslips were removed at various times after infection or mock infection and fixed for microscopic examination and nuclear staining with DAPI. The derivative of the HCMV Towne strain used contains a GFP gene transcribed from the simian virus 40 early promoter, inserted in place of a nonessential gene (15). Thus, fluorescence microscopy was used to detect GFP, which served as an indicator of viral infection and spread through the culture.
The micrographs in Fig. 2A show cells infected and maintained under hypoxic conditions for 72 h. The DAPI-stained nuclei are shown in red, and the infected cells are green due to expression of GFP. These images were superimposed on differential interference contrast micrographs; overlap of the red and green shows the infected cell nuclei as yellow. Note that in the normal 72-h infection, the viruses have spread throughout most of the culture, since adjacent cells are infected. In addition, the infected cells are enlarged, which is indicative of CPE. At 48 h after infection (not shown), there were many fewer green cells and less CPE. Under hypoxic conditions, the infected cultures appeared to be very similar to those grown under normoxic conditions, in terms of both extent of infection (green cells) and CPE. This result suggests that HCMV manages to conduct a relatively normal infection despite hypoxia.
FIG. 2.

HCMV infects and spreads in LEHFF cells under hypoxic conditions and in the presence of rapamycin. (A) LEHFFs were mock infected (Uninfected) or infected with HCMV (Infected) for 72 h in the presence of serum under normoxic or hypoxic conditions. Cells were fixed and stained with DAPI (red) for nuclear visualization. The Towne strain of HCMV contains an inserted GFP gene which indicated the infected cells (green). The images were superimposed on differential interference contrast micrographs; overlap of the red and green shows the infected cell nuclei as yellow. (B) Serum-starved LEHFFs were mock or HCMV infected and maintained in serum-free medium with or without 50 nM rapamycin for 48 or 72 h. Microscopy was the same as for panel A.
Figure 2B shows the effects of 48 or 72 h of rapamycin treatment on HCMV infection. The presence of rapamycin clearly lowered the rate of infection after 48 h, since there were fewer infected (green) cells in the rapamycin-treated samples than in the untreated (normal) samples. However, after 72 h of rapamycin treatment this difference was not as apparent (compare the number of infected cells in the panels for the 72-h normal and rapamycin-treated samples), suggesting that by this time the virus has overcome the inhibitory effects of rapamycin.
This conclusion is supported by the viral growth curves shown in Fig. 3. LEHFFs were infected as described above and then maintained under normal conditions, under hypoxic conditions, or in the presence of rapamycin. At various times after infection, the cells and medium were harvested and the viral titers were determined. In the presence of rapamycin there was at least a 12-h delay before the first detection of progeny viruses compared to normal growth conditions (36 h for normal conditions compared to at least 48 h with rapamycin). However, once viral production initiated in the rapamycin-treated cells it increased rapidly, with a final yield only 0.5 log unit below that under the normal conditions. The onset of growth in the rapamycin-treated cultures is not due to decay of the drug, since we have shown that 50 nM rapamycin maintains the ability to inhibit mTOR over the time course used in these experiments (not shown). Under hypoxic conditions, there was no delay in the appearance of progeny viruses and the yield was only 0.6 to 0.8 log unit less than under normoxic conditions.
FIG. 3.

HCMV viral growth curves. LEHFFs were infected under normal conditions (−Rap, +serum), under hypoxic conditions (−O2, +serum), and in the presence of rapamycin (+Rap, +serum) and harvested at various times after infection up to 144 h. Virus titers at each time point were determined by the 50% tissue culture infective dose method. In addition, viral growth curves were generated under conditions where the cells were serum starved for 48 h and then infected and maintained in the absence of serum (−Rap, −serum) or in the absence of serum with the addition of rapamycin (+Rap, −serum).
The data in Fig. 2 and 3 suggest that HCMV induces mechanisms which can counteract the inhibitory effects of hypoxia and rapamycin, allowing it to conduct a relatively normal infection with only modest reduction in the final yield.
In some of the experiments described below we examined the phosphorylation of mTOR effectors during the course of HCMV infection in the presence and absence of rapamycin. In order to avoid effects of exogenous growth factors, we serum starved the cells for 48 h prior to infection, infected the cells with highly purified viruses in serum-free medium (see Materials and Methods), and maintained the infected cells in serum-free medium. The growth curves in Fig. 3 show that virus production in the absence of rapamycin and serum was nearly identical to that in the presence of serum and rapamycin; in both cases there was at least a 12-h delay in the first appearance of progeny virions compared to that under normal conditions, followed by a rapid rise in virus production, with a final yield within 0.5 log unit of that under normal growth conditions. However, the combination of serum starvation and rapamycin did inhibit viral production. Besides the delay in the first appearance of progeny virions, the growth rate was lower throughout the time course, with a final virus yield 1 to 1.5 log units less than that under normal growth conditions. However, it is important to recognize that even under these very stringent conditions (serum starvation and rapamycin treatment prior to infection), a relatively robust infection was mounted.
The HCMV immediate-early and delayed-early proteins are synthesized during hypoxia and rapamycin treatment.
Western analysis was used to show the accumulation of HCMV MIEPs under normoxic and hypoxic conditions in HCMV-infected LEHFF cells (Fig. 4A). In addition, we determined the accumulation of the delayed-early protein ICP36 (UL44), which requires the functions of the MIEPs for full expression (20). There is little difference in the time of appearance and accumulation of these viral proteins under hypoxic conditions compared to normoxic conditions. MIEP (IEP72 and IEP86) and ICP36 levels were determined at 48 h after mock or HCMV infection in the presence of rapamycin or in the presence of hypoxia and rapamycin together (Fig. 4B). Again, there were only modest decreases in the accumulation of the MIEPs or ICP36 with rapamycin compared to normoxic conditions. There appears to be less IE86 and IE72 in the hypoxia-plus-rapamycin sample; however, we have noted that under these conditions the MIEPs often denature and remain in the well. We also note that IEP72 (IE1) from the hypoxia-plus-rapamycin samples migrates slower than IEP72 in the other samples. This suggests altered modification of the protein, e.g., increased phosphorylation, under these conditions; we have not examined this further. Under the more severe conditions of hypoxia plus rapamycin, we note a decrease in the levels of ICP36. In sum, the data suggest that levels of synthesis of HCMV MIEPs and ICP36 are not dramatically affected by hypoxia, rapamycin, or the combination of hypoxia and rapamycin; this conclusion is reiterated below with the rapamycin inhibition studies done under serum starvation conditions.
FIG. 4.

The appearance and accumulation of HCMV immediate-early and early proteins occur with both hypoxia and rapamycin. (A) Cultures of LEHFFs were grown under normal serum conditions and mock infected (uninfected) or HCMV infected under normoxic (Norm) and hypoxic (Hyp) conditions for various times (hours) after infection (hpi). Extracts were analyzed by Western analysis for HCMV MIEP and delayed-early ICP36 (UL44) protein synthesis. (B) A similar experiment showing only the 48-h time point examines MIEP and ICP36 production and mTOR phosphorylation in the presence of rapamycin (Rap.) and hypoxia (Hypox.). The asterisk represents denatured viral MIEPs which result from harvesting after hypoxia treatment.
Inhibition of mTOR phosphorylation by hypoxia is not reversed by HCMV infection.
In the mock-infected cells (Fig. 4B), hypoxia abolished the appearance of phosphorylated mTOR, in agreement with previous data showing that hypoxia inhibits mTOR phosphorylation and activation (1). Treatment with rapamycin under normoxic conditions did not inhibit mTOR phosphorylation, since rapamycin's effect is to prevent activated mTOR from phosphorylating its downstream effectors. The status of mTOR phosphorylation in hypoxia is not altered in HCMV-infected cells. Significantly, under conditions of hypoxia or hypoxia plus rapamycin, there was no detectable phosphorylation of mTOR in the HCMV-infected cells. These results suggest that HCMV has no mechanism to overcome the hypoxia-induced inhibition of mTOR phosphorylation (activation).
HCMV infection induces rapamycin-insensitive phosphorylation of eIF4G.
The above data suggest that HCMV infection can proceed when mTOR kinase is inactivated by hypoxia or inhibited by rapamycin. Thus, we predicted that HCMV may bypass mTOR by targeting its downstream effectors for phosphorylation, in order to ensure the high rates of translation needed to sustain viral protein synthesis. Therefore we examined the effect of HCMV on the levels and phosphorylation status of eIF4G, 4E-BP, p70S6K, and ribosomal protein S6.
When mTOR kinase is inhibited by rapamycin, most 4E-BP, p70S6K, and eIF4G should be hypophosphorylated at their mTOR kinase sites; thus, increased phosphorylation of these proteins during a viral infection in rapamycin would indicate a virally induced, rapamycin-insensitive mechanism. To test this, time course infections were done with serum-starved LEHFFs as described above. The time course studies were done under normal conditions (serum-free medium with no rapamycin) or in cells which had been pretreated with serum-free medium plus 50 nM rapamycin for 1 h before infection and maintained in serum-free medium plus 50 nM rapamycin throughout the time course of infection. Under these conditions, the virus must face the inhibitory effects of serum starvation and rapamycin pretreatment from the beginning of the infection; thus, the experiment tests the ability of the infection to become established under adverse conditions, for example, the conditions which cause the lag in the growth curve with rapamycin (Fig. 3).
Cell extracts were examined by Western analysis and probed for HCMV proteins and both the total and phosphorylated forms of mTOR effectors (Fig. 5A). Figure 5A shows that rapamycin had little effect on synthesis of the MIEPs (IEP72 and IEP86) and the delayed-early protein ICP36, again suggesting that HCMV infection can counteract the inhibitory effects of rapamycin.
FIG. 5.

Effect of HCMV infection on total levels and phosphorylation status of mTOR effectors in the presence and absence of rapamycin. (A) Analysis of the MIEPs (IEP86 and IEP72), ICP36, eIF4G, 4E-BP, mTOR, and actin. LEHFFs were serum starved for 48 h and then mock or HCMV infected in serum-free medium with or without 50 nM rapamycin. Cells were extracted at various times after infection for up to 24 h, and the phosphorylation status (p) and/or total (Tot.) levels of various proteins were determined by Western analyses as described in Materials and Methods. Arrows mark the positions of hyperphosphorylated forms of 4E-BP generated in the presence of rapamycin. (B) Rapamycin inhibits serum-induced phosphorylation of 4E-BP over a 24-h time course. LEHFFs were serum starved for 48 h and then changed to medium containing 10% fetal calf serum with or without 50 nM rapamycin. At various times after the addition of serum, the phosphorylation status of 4E-BP was determined by Western analysis. Arrows indicate where hyperphosphorylated forms of 4E-BP should appear if they formed in the rapamycin-treated samples (compare with arrows in panel A).
The level of total eIF4G showed little variation throughout the time course (Fig. 5A). Probing for eIF4G phosphorylated on S1108 (a rapamycin-sensitive phosphorylation site), we found little of the phosphorylated form in serum-starved, mock-infected LEHFFs and even less in mock-infected cells treated with rapamycin. The most intriguing aspect of these data is that within 30 min after the addition of HCMV, a large amount of phosphorylated eIF4G appeared. This elevated level of the phosphorylated form was maintained throughout the remainder of the infection time course. The rapid appearance of phosphorylated eIF4G suggests that its phosphorylation is induced by viral attachment to cellular receptors. This induced phosphorylation was only partially inhibited by rapamycin, suggesting that the mechanism induced by viral attachment is mTOR independent. Since activation due to viral attachment is transient (4, 25), we suggest that other viral proteins (tegument, IE, and early proteins) may maintain the levels of phosphorylated eIF4G throughout the infection, even in the presence of rapamycin.
We have also shown that high levels of phosphorylated eIF4G were present in infected cells that had been pretreated and maintained in hypoxia for 72 h, whereas there was no phosphorylated eIF4G in uninfected hypoxic cell controls (not shown). These data suggest that HCMV infection significantly induces eIF4G phosphorylation by mechanisms that are independent of mTOR kinase activity; this appears to be a very early event in the viral infection which is maintained throughout the infection.
It has been reported that eIF4G is targeted for proteolytic cleavage by caspase 3 in apoptotic cells (17). In Fig. 5A smaller bands, indicative of cleavage, are seen in both the total and phosphorylated eIF4G panels. In the total eIF4G analysis, these bands do not change in amount during the time course. In the analysis of phosphorylated eIF4G, the amounts of the smaller band are proportional to the amounts of the full-size phosphorylated forms throughout the time course. Thus, there are no data to suggest that the level of caspase cleavage of eIF4G changes during the time course of infection.
HCMV induces rapamycin-insensitive phosphorylation of 4E-BP.
Phosphorylation of 4E-BP occurs at a number of sites: T37, T46, S65, T70, S83, S101, and S112 (reviewed in reference 14). The effects of phosphorylation at T37, T46, S65, and T70 are best understood. Phosphorylation of these sites occurs in an ordered fashion; T37 and T46 are first and must be phosphorylated before T70 phosphorylation, which is followed by S65 phosphorylation. Although mTOR is believed to phosphorylate T37 and T46, phosphorylation of T70 and S65 is most reduced by rapamycin (14). T46 and S65 flank the eIF4E binding domain, and phosphorylation of S65 greatly inhibits eIF4E binding. Thus, it appears that the required order of phosphorylation (T37, T46, T70, and S65) leads to the combination of phosphorylation events which inhibits 4E-BP from binding to eIF4E. Thus, in the analysis of 4E-BP binding we expect to detect multiple phosphorylated forms, where the appearance of slower-migrating forms would indicate the hyperphosphorylation required to inhibit 4E-BP binding to eIF4E.
The antibody used for Western analysis recognizes 4E-BP phosphorylation at T37 and/or T46 and will also recognize the forms that are additionally phosphorylated at T70 and S65. A quantitative examination of the total phosphorylated forms (all forms) shows a significant increase over the time course (Fig. 5A, p4E-BP). In the mock-infected samples there are two phosphorylated forms which we suspect are phosphorylated T37 and/or T46. Rapamycin inhibits 4E-BP phosphorylation, as indicated by a decrease in the slower-migrating form. This inhibition of 4E-BP phosphorylation by rapamycin continues through the first 6 h of infection. Interestingly, there is a marked accumulation of the faster-migrating, least phosphorylated form in the rapamycin-treated samples; we have no explanation for this at this time. However, by 8 h postinfection in the untreated samples there is a dramatic increase in the slower-migrating form, indicating virally induced phosphorylation, and the first appearance of an even slower-migrating, hyperphosphorylated form which increases in intensity by 24 h after infection. Thus, by 8 h in a normal viral infection, mechanisms which result in multiply phosphorylated forms of 4E-BP are induced. These same multiply phosphorylated forms begin to appear in the rapamycin-treated samples at 12 h and increase by 24 h postinfection (Fig. 5A). These data suggest that the mechanisms induced by the virus to multiply phosphorylate 4E-BP can function in the presence of rapamycin, although the process is delayed and less efficient than in the infection in the absence of rapamycin. We suspect that the multiply phosphorylated forms are phosphorylated at T37, T46, T70, and S65; thus, 4E-BP binding to eIF4E should be decreased in these samples (see below).
The total amount of 4E-BP (Fig. 5A) increased no more that two- to threefold over the course of the 24-h infection; thus, the major effect of the viral infection on 4E-BP appears to be phosphorylation and not an alteration of its total amount.
It is possible that the rapamycin-insensitive phosphorylation of 4E-BP seen at 12 and 24 h after infection is due to a declining effect of the drug or incomplete inhibition of mTOR. To rule out this possibility, LEHFFs were serum starved for 48 h in the same manner used for the infections. The medium was then changed to medium containing 10% fetal calf serum to stimulate 4E-BP phosphorylation. This was done in the absence and presence of rapamycin. Cells were extracted at various times between 20 min and 24 h after serum addition, and the phosphorylation status of 4E-BP was determined. As shown in Fig. 5B, only the two least phosphorylated forms of 4E-BP were present in the serum starved samples (0 h); however, within 20 min (0.3 h) of serum addition the slower-migrating hyperphosphorylated forms of 4E-BP were detected. These hyperphosphorylated forms remained throughout the time course, although they declined somewhat by 24 h. At all time points the addition of rapamycin dramatically reduced the hyperphosphorylated forms. Thus, the rapamycin-insensitive phosphorylation of 4E-BP seen in infected cells at 12 and 24 h postinfection (Fig. 5A) cannot be attributed to failure of the drug or incomplete inhibition of mTOR.
HCMV infection maintains eIF4E in the eIF4F complex under conditions of mTOR kinase inhibition.
The above data suggest that HCMV infection induces mechanisms to maintain the integrity of the eIF4F complex (Fig. 1) even if mTOR kinase is inhibited. Specifically, 4E-BP is phosphorylated to prevent it from binding eIF4E, and a major component of eIF4F, eIF4G, is phosphorylated. The integrity of the eIF4F complex can be determined by allowing eIF4E to bind to 7Me-GTP eucaryotic cap analog coupled to Sepharose beads and isolating it along with its bound proteins. As shown in Fig. 6, under translationally active (mTOR-active) conditions, the eIF4E should be bound primarily to eIF4G, in the translationally active eIF4F complex. Since 4E-BP is phosphorylated under these conditions, little of it should be bound to eIF4E. Under translationally inactive conditions (mTOR inactive, e.g., with rapamycin), the opposite is expected; 4E-BP should be primarily bound to eIF4E, since 4E-BP is not phosphorylated. Binding of 4E-BP to eIF4E removes eIF4E from eIF4G but does not inhibit its ability to bind to the cap; thus, by using cap binding to select eIF4E and its associated proteins, we can determine by Western analysis whether eIF4E was bound to 4E-BP or eIF4G.
FIG. 6.

HCMV infection reduces eIF4E binding to 4E-BP and increases its binding to eIF4G. LEHFFs were serum starved for 48 h and mock or HCMV infected in serum-free medium with or without 50 nM rapamycin (Rap) for 8 or 24 h. Extracts were incubated with 7Me-GTP-Sepharose beads to capture eIF4E and associated proteins as described in Materials and Methods. Western analysis was used to detect total eIF4E, 4E-BP, and eIF4G.
LEHFFs were mock infected or HCMV infected for 8 or 24 h in the presence or absence of rapamycin. The extracts were incubated with Sepharose beads coupled to cap analog; the beads were then washed, and the bound proteins were subjected to Western analysis probing for eIF4E, 4E-BP, and eIF4G (Fig. 6). The top panel of the gel in Fig. 6 shows that equivalent amounts of eIF4E were selected from each sample. In the mock-infected samples, in which 4E-BP is hypophosphorylated (Fig. 5A), eIF4E is bound primarily to 4E-BP, suggesting translation inhibition, as expected in serum-starved cells. By 8 h after infection in the absence of rapamycin, increased amounts of eIF4G are detected, suggesting that HCMV infection has induced the phosphorylation of 4E-BP, thus releasing eIF4E and allowing eIF4E to bind with eIF4G. eIF4E binding to eIF4G is significantly increased by 24 h in the absence of rapamycin, coincident with a dramatic decrease in eIF4E binding to 4E-BP. This is consistent with the 4E-BP phosphorylation data in Fig. 5A, in which significant hyperphosphorylation of 4E-BP was observed by 24 h in the samples without rapamycin. As also noted in Fig. 5A, hyperphosphorylation of 4E-BP was delayed in the infected cells treated with rapamycin; however, by 24 h there was a significant amount of multiply phosphorylated 4E-BP. This is consistent with the cap binding data for the rapamycin-treated samples (Fig. 6); by 24 h a significant amount of eIF4G is bound to eIF4E, and, concomitantly, the amount of 4E-BP bound to eIF4E is significantly decreased.
These results suggest that the virus induces a mechanism that drives the interaction between eIF4E and eIF4G in order to maintain the translationally active eIF4F complex. This mechanism appears to be mTOR kinase independent and correlates with the virally induced, rapamycin-insensitive phosphorylation of eIF4G and 4E-BP.
HCMV does not induce an mTOR kinase-independent mechanism to phosphorylate p70S6K but does induce a low level of rapamycin-insensitive phosphorylation of ribosomal protein S6.
Activation of p70S6K requires phosphorylation on T229, T389, and S371; T389 and S371 are targets for mTOR. The anti-phospho-p70S6K antibody that we used for Western analyses (Fig. 7) recognizes phosphorylation on T389. In the untreated samples there was a modest increase in phosphorylated p70S6K by 1 h after infection compared with the mock-infected sample. However, this increase was transient, and phosphorylated p70S6K decreased to nearly undetectable levels by 4 h; this was very reproducible. By 8 h, phosphorylated p70S6K returned and was notably increased compared with that in the mock-infected sample. However, the most significant observation is that no phosphorylation of T389 was detected in samples treated with rapamycin, suggesting that HCMV induces no mechanism to phosphorylate this critical site when mTOR is inhibited. The observation that p70S6K phosphorylation is inhibited by rapamycin at all time points stands as additional proof that the drug remains affective throughout the infection time course.
FIG. 7.

Analysis of p70S6K and its substrate ribosomal protein S6. The same extracts and conditions used for Fig. 5A were used to analyze the total levels and phosphorylation status of p70S6K and S6 protein.
Given the apparent loss of p70S6K activity in the presence of rapamycin, we examined the phosphorylation status of its substrate, ribosomal protein S6 (Fig. 7). In the untreated samples, phosphorylated S6 (pS6) was easily detected (short exposure); in the presence of rapamycin, phosphorylation was severely inhibited, in agreement with the loss of phosphorylated p70S6K. The short exposure shows that there are modest increases in the levels of phosphorylated S6 through the time course, but this correlates with increased levels of total S6 protein. More interesting is the appearance of a low level of phosphorylated S6 beginning at 6 h after infection in the presence of rapamycin (long exposure). It is unclear at this point whether this is due to a low level of residual p70S6K activity or to a virally induced kinase activity that is both mTOR kinase and p70S6K independent. In addition, the functional significance of this low level of phosphorylated S6 protein remains to be determined.
PI3K signaling is required for HCMV-induced mTOR-independent phosphorylation of 4E-BP but not eIF4G.
We and others have previously shown that HCMV infection and the HCMV MIEPs can activate PI3K signaling (16, 26). This is expected to result in activation of Akt. As shown in Fig. 8, infection of serum-starved LEHFFs resulted in phosphorylation and activation of Akt within 30 min of infection, and, compared with mock-infected sample, the amount of phosphorylated Akt remained elevated throughout the 24-h infection time course.
FIG. 8.
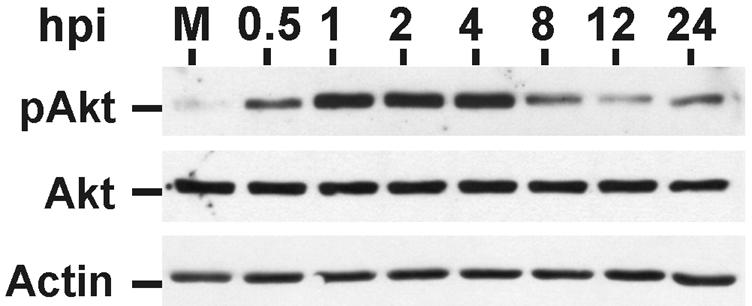
Phosphorylation (activation) of Akt during a 24-h HCMV infection of LEHFFs. LEHFFs were serum starved for 48 h and then mock (M) or HCMV infected in serum-free medium with or without 50 nM rapamycin for up to 24 h. Extracts were analyzed for phosphorylated Akt (pAkt), total Akt, and actin. hpi, hours postinfection.
The activation of PI3K/Akt signaling is one means by which HCMV may activate mTOR signaling during a normal infection. To determine whether the virus can maintain phosphorylation of mTOR effectors when PI3K is inhibited, we used the PI3K-specific inhibitor LY (23) in time course experiments similar to those done with rapamycin. Figure 9 shows the results.
FIG. 9.

Effect of HCMV infection on total levels and phosphorylation status of mTOR effectors in the presence and absence of the PI3K inhibitor LY. LEHFFs were serum starved for 48 h and then mock or HCMV infected in serum-free medium with or without 50 μM LY. Cells were extracted at various times (hours) after infection (hpi) for up to 24 h, and the phosphorylation status (p) and/or total (Tot.) levels of various proteins were determined by Western analyses as described in Materials and Methods. (A) Analysis of the MIEPs (IEP86 and IEP72), eIF4G, 4E BP, and actin. (B) Analysis of p70S6K and its substrate ribosomal protein S6.
Examination of HCMV MIEP production shows that the MIEPs first appeared at the same time between 2 and 4 h regardless of the presence of LY; however, their accumulation was less in the LY-treated cells than in untreated cells throughout the time course. Nonetheless, significant levels of the MIEPs were present in the LY-treated extracts. This result is in contrast to studies done with HEL cells, which reported that inhibition of PI3K with LY resulted in severely reduced production of the MIEPs (16). During the very early phase of the infection (0.5 to 1 h), LY inhibited eIF4G phosphorylation; however, by 2 h, LY-insensitive phosphorylation of eIF4G was detected, and this increased throughout the remainder of the time course. In the rapamycin inhibition studies (Fig. 5A) we noted rapamycin-insensitive phosphorylation beginning at 30 min after infection. The combination of the rapamycin and LY data suggests that at very early times after infection, phosphorylation of eIF4G is mediated by a PI3K-dependent, mTOR kinase-independent mechanism, but beginning at 2 h and continuing through the time course, a PI3K-independent, mTOR kinase-independent mechanism for phosphorylating eIF4G was steadily induced by the viral infection.
As previously seen, hyperphosphorylated forms of 4E-BP appeared in the untreated, infected cells. This was particularly evident by 8 h, when the least phosphorylated (fastest-migrating) form was no longer detected, being replaced by the slower-migrating multiply phosphorylated forms. Strikingly, hyperphosphorylation of 4E-BP mediated by the HCMV infection was dramatically inhibited by LY throughout the time course, suggesting that the hyperphosphorylation of 4E-BP seen in the presence of rapamycin (Fig. 5A) results from a PI3K-dependent, mTOR kinase-independent mechanism.
The phosphorylation of p70S6K seen during HCMV infection was inhibited by LY (Fig. 9B), similar to the results seen with rapamycin (Fig. 7). These data suggest that during a viral infection the only means to phosphorylated p70S6K is through the PI3K/Akt/mTOR pathway. In contrast, between 4 and 8 h after infection, phosphorylated S6 can be detected in the presence of LY (Fig. 9B). This observation supports the data in Fig. 7 and suggests that HCMV may induce a mechanism to phosphorylate S6 which is independent of PI3K, and possibly independent of mTOR/p70S6K; however, this remains to be verified. In addition, the functional significance of the relatively low levels of phosphorylated S6 produced under these conditions remains to be determined.
DISCUSSION
A successful viral infection requires that normal cellular functions undergo major adaptations. For example, the cell's nutrient supply, metabolism, and oxygen supply and utilization must increase, whereas stress responses and apoptosis must be inhibited. It is to the virus's advantage to target master cellular regulators which may adapt several of these processes simultaneously. An example of such adaptation is the activation of the cellular kinase Akt by HCMV infection and the HCMV MIEPs (7, 16, 21, 26). Activation of this central regulator inhibits apoptosis while activating cellular proliferation, growth, and metabolism (8), all of which benefit viral infection.
Another major cellular regulator is mTOR, which regulates cap-dependent translation (12). Our data suggest that under normal infection conditions HCMV can activate mTOR signaling, most likely by activating PI3K/Akt signaling. However, inhibition of mTOR kinase activity by hypoxia or rapamycin does not significantly affect the viral infection. Although rapamycin delayed the onset of viral production by at least 12 h, the final yield of virus from either rapamycin- or hypoxia-treated cells was only 0.5 to 0.8 log unit less than the yield of a normal infection. Previous data have also suggested that HCMV infection proceeds in the presence of rapamycin (16).
In several of the experiments presented we utilized serum starvation and preincubation with rapamycin prior to infection. From the perspective of the virus, this may represent a worst-case scenario, since the virus enters a cell in which mTOR kinase as well as serum-dependent cellular activities are already inhibited. Under normal in vivo conditions the viral infection would become established, adapting or counteracting cellular pathways, prior to the induction of stress responses resulting from increased metabolism and viral protein synthesis. Under the serum starvation-rapamycin preincubation conditions, the virus must face the inhibitory conditions from the beginning; thus, the experiments test the ability of the infection to become established under preexisting adverse conditions.
Our data suggest that stress- or rapamycin-induced inhibition of mTOR kinase activity can be bypassed in an HCMV infection through mTOR kinase-independent mechanisms which phosphorylate 4E-BP and eIF4G. These virally induced mechanisms may well function under normal growth conditions, but they become readily detectable during rapamycin or hypoxia treatment, which inhibits the mTOR pathway. Our data suggest that two different mechanisms are involved in mTOR kinase-independent phosphorylation of these proteins, since phosphorylation of 4E-BP is PI3K dependent, whereas phosphorylation of eIF4G appears to be biphasic, being PI3K dependent at very early times (30 to 90 min after infection) and PI3K independent as viral proteins are synthesized. The PI3K-sensitive phosphorylation of eIF4G within 30 min of infection is likely mediated by viral attachment to receptors (4, 25), which can activate PI3K and Akt. Since the effects of this type of signaling are transient (4, 16), we suggest that the continued PI3K-insensitive phosphorylation of eIF4G may be mediated by tegument proteins and/or by newly synthesized viral proteins as they appear during the course of the infection. In this scenario a temporal cascade of viral proteins would maintain the proper levels of phosphorylation throughout the course of the infection regardless of the inhibition of mTOR or PI3K.
In contrast to 4E-BP and eIF4G, HCMV appears to be unable to induce an mTOR kinase-independent mechanism to phosphorylate p70S6K. However, it is possible that this deficiency is bypassed by the induction of a mechanism which can promote a low level of ribosomal protein S6 phosphorylation which is independent of PI3K and possibly independent of mTOR/p70S6K. However, the functional relevance of low levels of phosphorylated S6 remains to be determined.
The targeting of 4E-BP and eIF4G for phosphorylation suggests that HCMV infection induces mechanisms which actively maintain the integrity of the eIF4F complex to ensure cap-dependent translation regardless of the cellular conditions. Our cap binding experiments (Fig. 6) support this hypothesis, showing that the mTOR kinase-independent phosphorylation of 4E-BP and eIF4G correlates with inhibition of eIF4E binding to 4E-BP and the promotion of its association with the eIF4F complex even when mTOR is inhibited. It is reasonable to speculate that other components of the eIF4F complex (eIF4E, Mnk1, and eIF4A) may also be modified by the HCMV infection to further ensure the integrity of the eIF4F complex. In agreement, a recent study (24) suggests that herpes simplex virus 1 infection actively maintains the eIF4F complex by promoting the hyperphosphorylation of 4E-BP. These studies also suggest that phosphorylated 4E-BP is degraded during the herpes simplex virus infection; however we have no evidence of 4E-BP degradation during HCMV infections.
Although the roles of mTOR and its effectors in cap-dependent translational control are well established, inhibition of mTOR kinase by rapamycin does not have a global inhibitory effect on translation in mammalian cells (11, 14). For example, rapamycin reduces cap-dependent translation in NIH 3T3 cells by no more than 30% (3). We have seen a 30 to 35% decrease in total [35S]methionine incorporation in pulse-labeling experiments with LEHFFs in rapamycin (data not shown). Within 24 h of HCMV infection this rapamycin-induced decrease is less than 20%, consistent with the translational activating effects of HCMV infection on mTOR effectors and the integrity of the eIF4F complex. However, this may not be indicative of the entire effect of the viral infection on translation of specific mRNAs. Among mRNAs dependent on the cap for translation there is a wide range in the degree of inhibition elicited by rapamycin treatment (11). Thus, it is possible that the mechanisms utilized by HCMV to increase the integrity of the eIF4F complex may be more beneficial to viral mRNAs, and some cellular mRNAs, than to the bulk of cellular mRNAs.
Acknowledgments
We thank Celeste Simon and Brian Keith for assistance with hypoxia experiments, Sherri Adams for critical reading of the manuscript, and all of the members of the Alwine laboratory for support and critical evaluation of the experiments and data.
This work was supported by Public Health Service grant CA28379 awarded to J.C.A. by the National Cancer Institute.
REFERENCES
- 1.Arsham, A. M., J. J. Howell, and M. C. Simon. 2003. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 278:29655-29660. [DOI] [PubMed] [Google Scholar]
- 2.Arsham, A. M., D. R. Plas, C. B. Thompson, and M. C. Simon. 2002. PI3-K/Akt signaling is neither required for hypoxic stabilization of HIF-1 nor sufficient for HIF-1-dependent target gene transcription. J. Biol. Chem. 277:15162-15170. [DOI] [PubMed] [Google Scholar]
- 3.Beretta, L., A. C. Gingras, Y. V. Svitkin, M. N. Hall, and N. Sonenberg. 1996. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 15:658-664. [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle, K. A., R. L. Pietropaolo, and T. Compton. 1999. Engagement of the cellular receptor for glycoprotein B of human cytomegalovirus activates the interferon-responsive pathway. Mol. Cell. Biol. 19:3607-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bresnahan, W. A., G. E. Hultman, and T. Shenk. 2000. Replication of wild type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816-10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown, E. J., M. W. Albers, T. B. Shin, K. Ichikawa, C. T. Keith, W. S. Lane, and S. L. Schreiber. 1994. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369:756-758. [DOI] [PubMed] [Google Scholar]
- 7.Dahl, J., A. Jurczak, L. A. Cheng, D. C. Baker, and T. L. Benjamin. 1998. Evidence of a role for phosphatidylinositol 3-kinase activation in the blocking of apoptosis by polyomavirus middle T antigen. J. Virol. 72:3221-3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905-2927. [DOI] [PubMed] [Google Scholar]
- 9.Gingras, A. C., S. P. Gygi, B. Raught, R. D. Polakiewicz, R. T. Abraham, M. G. Hoekstra, R. Aebersold, and N. Sonernberg. 1999. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13:1422-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gingras, A. C., B. Raught, S. P. Gygi, A. Niedzwiecka, M. Miron, S. K. Burley, R. D. Polakiewicz, A. Wyslouch-Cieszynska, R. Aebersold, and N. Sonenberg. 2001. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 15:2852-2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gingras, A. C., B. Raught, and N. Sonenberg. 2004. mTOR signaling to translation. Curr. Top. Microbiol. Immunol. 279:169-197. [DOI] [PubMed] [Google Scholar]
- 12.Gingras, A. C., B. Raught, and N. Sonernberg. 2001. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 15:807-826. [DOI] [PubMed] [Google Scholar]
- 13.Harel, N. Y., and J. C. Alwine. 1998. Phosphorylation of the human cytomegalovirus 86-kilodalton immediate early protein IE2. J. Virol. 72:5481-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris, T. H., and J. C. Lawrence, Jr. 2003. TOR signaling. Science STKE 212:1-17. [DOI] [PubMed] [Google Scholar]
- 15.Heider, J. A., Y. Yu, T. Shenk, and J. C. Alwine. 2002. Characterization of a human cytomegalovirus with phosphorylation site mutations in the immediate-early 2 protein. J. Virol. 76:928-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson, R. A., X. Wang, X. L. Ma, S. M. Huong, and E. S. Huang. 2001. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol. 75:6022-6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marissen, W. E., and R. E. Lloyd. 1998. Eukaryotic translation initiation factor 4G is targeted for proteolytic cleavage by caspase 3 during inhibition of translation in apoptotic cells. Mol. Cell Biol. 18:7565-7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raught, B., A. C. Gingras, and N. Sonenberg. 2001. The target of rapamycin (TOR) proteins. Proc. Natl. Acad. Sci. USA 98:7037-7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabatini, D. M., H. Erdjument-Bromage, M. Lui, P. Tempst, and S. H. Snyder. 1994. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78:35-43. [DOI] [PubMed] [Google Scholar]
- 20.Stasiak, P. C., and E. S. Mocarski. 1992. Transactivation of the cytomegalovirus ICP36 gene promoter requires the alpha gene product TRS1 in addition to IE1 and IE2. J. Virol. 66:1050-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Summers, S. A., L. Lipfert, and M. J. Birnbaum. 1998. Polyoma middle T antigen activates the Ser/Thr kinase Akt in a PI3-kinase-dependent manner. Biochem. Biophys. Res. Commun. 246:76-81. [DOI] [PubMed] [Google Scholar]
- 22.Vezina, C., A. Kudelski, and S. N. Sehgal. 1975. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 28:721-726. [DOI] [PubMed] [Google Scholar]
- 23.Vlahos, C. J., W. F. Matter, K. Y. Hui, and R. F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241-5248. [PubMed] [Google Scholar]
- 24.Walsh, D., and I. Mohr. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 18:660-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang, X., S.-M. Huong, N. Chiu, N. Raab-Traub, and E.-S. Huang. 2003. EGF receptor is a cellular receptor for human cytomegalovirus. Nature 424:456-461. [DOI] [PubMed] [Google Scholar]
- 26.Yu, Y., and J. C. Alwine. 2002. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and cellular kinase Akt. J. Virol. 76:3731-3738. [DOI] [PMC free article] [PubMed] [Google Scholar]