

Primary murine HSCs show divergent fate decisions with biomaterial engagement and due to marrow-inspired biophysical cues.
Keywords: hematopoietic stem cell, stem cell niche, extracellular matrix, biophysical cues
Abstract
Hematopoiesis is the physiological process where hematopoietic stem cells (HSCs) continuously generate the body’s complement of blood and immune cells within unique regions of the bone marrow termed niches. Although previous investigations have revealed gradients in cellular and extracellular matrix (ECM) content across the marrow, and matrix elasticity and ligand type are believed to be strong regulators of stem cell fate, the impact of biophysical signals on HSC response is poorly understood. Using marrow-inspired ECM ligand–coated polyacrylamide substrates that present defined stiffness and matrix ligand cues, we demonstrate that the interplay between integrin engagement and myosin II activation processes affects the morphology, proliferation, and myeloid lineage specification of primary murine HSCs within 24 hours ex vivo. Notably, the impact of discrete biophysical signals on HSC fate decisions appears to be correlated to known microenvironmental transitions across the marrow. The combination of fibronectin and marrow matrix-associated stiffness was sufficient to maintain hematopoietic progenitor populations, whereas collagen and laminin enhanced proliferation and myeloid differentiation, respectively. Inhibiting myosin II–mediated contraction or adhesion to fibronectin via specific integrins (α5β1 and ανβ3) selectively abrogated the impact of the matrix environment on HSC fate decisions. Together, these findings indicate that adhesive interactions and matrix biophysical properties are critical design considerations in the development of biomaterials to direct HSC behavior in vitro.
INTRODUCTION
Hematopoietic stem cells (HSCs) are responsible for hematopoiesis, the production of the full complement of blood and immune cells (1). Processes associated with HSC fate decisions occur within distinct subregions of the marrow, termed niches (2). Matrix biophysical cues (for example, mechanical properties and matrix-associated ligands) (3–5), biomolecular signals (for example, cytokines, hypoxia, and CXCL12) (6), and local cell populations [for example, osteoblasts, mesenchymal stem cells (MSCs), and differentiated hematopoietic progeny] (7) vary considerably across the marrow. HSCs exhibit dynamic behavior within the marrow (1, 2, 7), with quiescent HSCs remaining engrafted in the marrow and subfractions of activated HSCs mobilizing from and homing to the marrow via the peripheral blood (2, 7–10). In addition, hematopoietic progenitor cells at discrete stages of differentiation have been observed to localize nonuniformly within the marrow (11, 12). The dynamic interplay between HSCs and discrete niches across the bone marrow suggests that microenvironmental signals may be key regulators of HSC fate decisions such as quiescence, self-renewal, and myeloid versus lymphoid lineage specification (13).
Although the identity and composition of discrete niches remain unclear, putative HSCs have been identified near the bone endosteum (endosteal niche), near the arterioles (arteriolar niche), in the central marrow cavity (central medullary niche), and near the sinusoidal endothelium (perivascular niche) (2). Recent data suggest that the endosteal niche, marked by a rich population of premature osteoblastic cells and arteriolar structures near the bone surface, houses quiescent, long-term repopulating HSCs (2). Found more deeply within the marrow, the sinusoidal endothelium (perivascular niche) has been implicated in housing more active, self-renewing, short-term, repopulating HSCs (11, 12). These subtle localization differences support the contention that quiescent and active niches exist in close spatial register across the marrow, but teasing apart the complex constellations of signals responsible for maintaining hematopoietic homeostasis remains challenging because of the rarity of HSCs within the marrow (<1:50,000 marrow cells) (1, 4) and limitations of in situ imaging approaches (14). As a result, although soluble biomolecules and putative niche cells have been correlated with HSC localization within the marrow (4, 7) and despite the known structural, mechanical, and biomolecular diversity of the marrow itself (3–5, 13), there exists no biophysical explanation for the niche-mediated regulation of HSC fate specification. This insight is critical for the design of biomaterial analogs of the marrow for ex vivo stem cell biomanufacturing applications. Recent efforts in the field using in vitro cultures of whole bone marrow (WBM) or subsets of hematopoietic stem and progenitor cells (HSPCs) suggest that changes in matrix properties [substrate elasticity (15, 16) and surface topography (17, 18)] and biomolecule availability [matrix ligand engagement (19, 20) and control of cytokine feedback (21)] may contribute to partial regulation of HSC fate decisions (22). However, these findings have largely precluded a more detailed study of the role matrix biophysical signals play in early HSC fate decisions.
Here, we provide evidence of a direct link between the in vitro biophysical niche microenvironment and early HSC fate decisions. We use marrow-inspired, extracellular matrix (ECM) protein–coated polyacrylamide (PA) substrates presenting combinations of matrix stiffness (3.7 and 44 kPa) and ligand type (type I collagen, fibronectin, and laminin). The design of this platform was motivated by the observation that the bone marrow is composed of discrete regions of variable matrix biophysical and compositional properties. Notably, near the bone surface is defined by stiffer microenvironments consisting of osteoid and collagenous bone (~40 kPa) with heightened fibronectin content, whereas the microenvironments near the marrow vasculature are noted with heightened laminin content and soft marrow rich in adipocytes (<3 kPa), compared to more general distribution of collagens I and IV within the marrow cavity (3, 5). The marrow is viscoelastic, containing gradations in mechanical properties reported to range from 0.25 to 24.7 kPa in porcine models (23). We also prepared ECM protein–coated glass substrates as control substrates that are several orders of magnitude stiffer (~70 GPa) than the ECM protein–coated PA gels (24). We cultured primary murine Lin−Sca-1+c-Kit+ (LSK) HSCs on the prepared substrates and, after 24 hours of culture, examined the impact of marrow-inspired matrix cues on HSC viability, morphology, and proliferation. We also mapped changes in HSC functional capacity via changes in myeloid lineage specification. Our findings show that substrates resembling the endosteal region (44 kPa; fibronectin-coated) promoted maintenance of primitive myeloid progenitors, whereas those resembling the vascular region (laminin-coated) favored differentiation particularly toward erythroid lineages. These results suggest that the biophysical elements of putative HSC niches modulate HSC fate decisions directly, particularly in line with the current HSC niche model.
RESULTS
Biophysical changes in HSCs occur within 24 hours of culture and are linked to matrix cues
After 24 hours of culture, phase-contrast imaging showed morphological changes in cultured HSCs (initially LSK cells) as a function of matrix environment (Fig. 1). These results are consistent with previous findings from our laboratory using collagen-coated substrates (25) but extended the results to consider fibronectin and laminin. Cell viability remained consistent at approximately 50% after 24 hours of culture, and it was slightly higher (~60%) on fibronectin-coated substrates (Fig. 1A). Low cell viability likely resulted from the absence of fetal bovine serum (FBS) in our culture medium, which consisted of commercially available base medium supplemented with cytokines (SCF, Flt3L, and TPO) known to support HSC cultures.
Fig. 1. Biophysical changes in cultured HSCs in response to matrix biophysical signals.
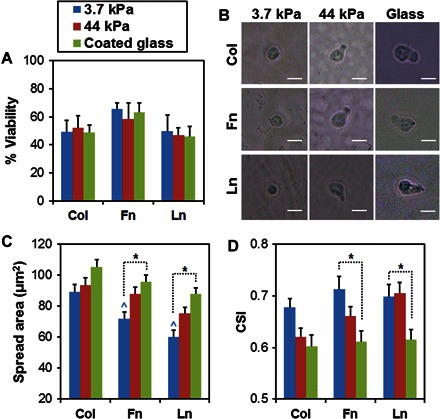
(A) After 24 hours of culture, cell viability remained consistent at approximately 50% for all conditions, although it was slightly higher on fibronectin (Fn)–coated substrates. (B) Phase-contrast images show changes in cell morphology, quantified as spreading (C) and CSI (D). On soft PA gels (3.7 kPa), HSCs remained rounded, with very little cytoplasm and few, if any, cellular protrusions. On stiffer substrates (>44 kPa), enhanced HSC spreading and polarity were observed. Scale bars, 10 μm. n = 24 to 45 cells. *P < 0.05. ^P < 0.05 with respect to spreading on collagen (Col)–coated substrates at 3.7 kPa. Ln, laminin.
Overall, HSCs spread more with increasing substrate stiffness (Fig. 1, B and C). Here, we use the term “spread” to describe the observed morphological changes in cultured HSCs, but we recognize that the spreading of HSCs may be mechanistically different from the spreading activities of typical anchorage-dependent cells because HSCs are only very weakly adherent cells, sometimes even regarded as nonadherent (26). This trend of increased spreading activity was observed on all ECM protein–coated substrates, although only the results on fibronectin- and laminin-coated substrates were statistically meaningful (P < 0.05; Fig. 1C). Increased spreading was correlated with more irregular (less circular) morphology (Fig. 1D). On soft PA gels (3.7 kPa), HSCs remained largely round with very little cytoplasm and just a few, if any, cellular protrusions (Fig. 1B). On stiffer (>44 kPa) substrates, their cytoplasm extended along one direction, effectively polarizing the cell body. We quantified changes in cell morphology by calculating cell shape index (CSI), whose values range from 1 to 0 to represent a shape between a perfect circle and a linear line. CSI values of the cultured HSCs decreased with increasing substrate stiffness, indicating increased irregularity (less circularity) in cell morphology (Fig. 1D).
Matrix contact alters cell cycle and proliferation response within 24 hours
Whereas most (>93%) of the cultured HSCs were still negative for lineage surface markers (Fig. 2A), ~50% no longer retained Sca-1+c-Kit+ phenotype by the end of the 24-hour culture period (Fig. 2B), indicating that lineage specification had begun. In addition, a flow cytometric proliferation assay using CellTrace Violet showed that cultured cells were actively dividing and proliferating (Fig. 2 and fig. S1), although the extent to which this occurred depended on the specific ECM protein used to coat the substrates (Fig. 2, C and D). Specifically, the highest cell division activity was observed on collagen-coated substrates followed by fibronectin-coated and then laminin-coated substrates (Fig. 2, C and D; P < 0.05), and these responses were independent of the substrate stiffness. Here, the percentage of original cells dividing represents the number of divided cells per total cells detected (Fig. 2C), whereas proliferation index indicates the number of divided cells per originally seeded cells that were detected (Fig. 2D).
Fig. 2. Surface antigen expression and proliferation profiles of cultured HSCs.
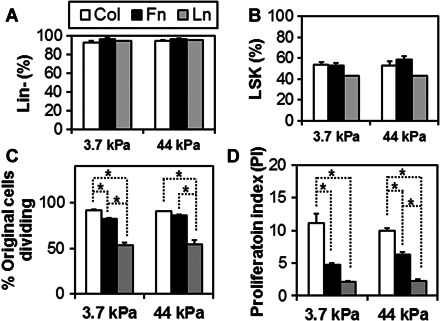
Changes in surface antigen expressions of the cultured HSCs were analyzed for lineage (Lin) surface markers (A) and LSK markers (B). Although most (>93%) of the cultured HSCs were still negative for lineage surface markers, ~50% no longer retained LSK phenotype by the end of the 24-hour culture period, indicating that substantial early lineage specification had begun. Analysis of the fraction of dividing HSC (C) and overall HSC proliferation index (D) after 24 hours indicates that matrix chemistry affects cell division. The highest fraction of dividing cells and the greatest proliferation index were observed on collagen-coated substrates followed by fibronectin-coated and then laminin-coated substrates, with these effects largely independent of the substrate stiffness. n = 3 from independent experiments. *P < 0.05.
Matrix stiffness and ligand type selectively affect HSC lineage specification
We subsequently examined the impact of matrix biophysical cues on HSC lineage specification via colony-forming unit (CFU) assay. We harvested cultured cells from the PA substrates after 24 hours of culture for clonal expansion of HSPCs in methylcellulose medium. After 11 to 14 days of incubation, HSPCs gave rise to colonies corresponding to different stages of myeloid lineage specification, identifiable by specific morphological features. Quantifying the number of colonies associated with discrete fate specification events allowed us to assess the degree of myeloid specification as a function of matrix environment. The colonies correspond to three stages of myeloid specification: CFU-GEMM colonies represent only early stages of myeloid specification, with colonies arising from primitive myeloid progenitors with multilineage potential that were retained after culture on the functionalized PA substrates. CFU-GM colonies represent further myeloid specification, with colonies arising because of the presence of myeloid progenitors restricted to granulocyte and monocyte lineages. CFU-G/E/M/Mk colonies arose because of LSK cells that differentiated to the point of myeloid progenitors committed to a single lineage, corresponding to granulocyte (G), erythrocyte (E), monocyte/macrophage (M), and megakaryocyte (Mk) lineage, respectively (fig. S2A).
Comparing the number of colonies arising from cultured versus freshly isolated [LSK cells immediately after fluorescence-activated cell sorting (FACS), hence no matrix contact] HSCs, we noted changes in myeloid specification upon matrix engagement (fig. S2). These changes were complex and reflected a selective impact of matrix stiffness and ligand cues. To better resolve their impact on myeloid specification of the cultured HSCs, we normalized the CFU assay results to those from freshly isolated HSCs (control, represented as dashed lines at the value of 1) (Fig. 3).
Fig. 3. Lineage specification state of cultured HSCs.

CFU assay was used to assess myeloid lineage specification of the cultured HSCs on collagen-coated (A), fibronectin-coated (B), and laminin-coated (C) substrates, with all results normalized versus colonies formed by a control group of freshly isolated HSCs (dashed line). The resulting CFU assay data showed changes in myeloid specification as a result of matrix engagement. The most primitive CFU-GEMM colonies increased on stiff (>44 kPa) versus soft (3.7 kPa) fibronectin-coated substrates but decreased on collagen- or laminin-coated substrates. The number of CFU-E and CFU-M colonies, indicative of late-stage myeloid specification, increased with matrix stiffness, whereas matrix contact hindered CFU-G colony formation. CFU-GEMM, multipotential myeloid progenitor cells; CFU-GM, granulocyte-macrophage progenitors; CFU-G, granulocyte progenitors; CFU-E, erythrocyte progenitors; CFU-M, monocyte/macrophage progenitors; CFU-Mk, megakaryocyte progenitors. n = 3 from independent experiments. *P < 0.05. ^P < 0.05 and ^^P < 0.001 with respect to colony numbers from freshly isolated HSCs represented as dashed lines (control).
HSC specification events were strongly influenced by matrix stiffness, although HSC fate specification events also reflected a selective influence of matrix ligand cues. The prevalence of the most primitive CFU-GEMM colonies increased on stiff (>44 kPa) versus soft (3.7 kPa) fibronectin-coated substrates (P < 0.05; Fig. 3B) but decreased on collagen- or laminin-coated substrates (P < 0.05 compared to control; Fig. 3, A and C). The numbers of CFU-E and CFU-M colonies, indicative of late-stage myeloid specification, were also strongly associated with matrix biophysical cues, increasing with increasing matrix stiffness (P < 0.05; Fig. 3). Whereas numbers of CFU-E colonies on fibronectin-coated substrates or CFU-M colonies on laminin-coated substrates did not show statistically meaningful stiffness dependence (Fig. 3, B and C), CFU-E and CFU-M colony numbers otherwise increased on matrix-functionalized glass substrates versus soft (3.7 kPa) PA substrates (P < 0.05). CFU-Mk colonies were influenced by substrate stiffness on fibronectin-coated substrates (P < 0.05; Fig. 3B) but not on collagen- or laminin-coated substrates (Fig. 3, A and C). Regardless of substrate stiffness or matrix ligand type, matrix contact hindered CFU-G colony formation, although these changes were not statistically meaningful (Fig. 3, A to C).
By comparing HSC specification to substrates resembling physiologically relevant stiffness (3.7 and 44 kPa) only, colony variations were less pronounced but remained notable. Increases in CFU-GEMM colonies on fibronectin substrates and CFU-E colonies on laminin substrates were observed for 44-kPa versus 3.7-kPa PA substrates (P < 0.05; Fig. 3, B and C). These results suggest a pronounced impact of matrix ligand type on myeloid lineage specification across a range of biophysical environments encountered by HSPCs within the bone marrow. Moreover, it also suggests that stiffness may be a particularly important factor in the regulation of early-stage (CFU-GEMM) and specific later-stage myeloid progenitors (CFU-E).
Actomyosin contractility modulates early HSC fate specification events
We then sought to abrogate the effect of matrix biophysical cues on HSC fate specification and targeted changes in actomyosin contractility, hence intracellular tension, as potentially important during early HSC fate specification. To assess the role of actomyosin contractility in early HSC fate specification, we first cultured primary HSCs on ECM protein–coated substrates for 24 hours with blebbistatin, a known inhibitor of myosin II. During this culture period, blebbistatin did not have statistically meaningful effect on cell spreading (3.7 kPa versus 44 kPa), although it resulted in cells maintaining more circular morphology even at a stiffer matrix of 44 kPa (P < 0.001; fig. S3).
Notably, adding blebbistatin induced pronounced functional changes in lineage specification in cultured HSCs (Fig. 4 and fig. S4). Previously noted stiffness-induced changes were abrogated, with decreases in CFU-GEMM numbers on fibronectin-coated substrates (P < 0.05; Fig. 4B) and CFU-E/M/Mk on collagen- and fibronectin-coated substrates (P < 0.05; Fig. 4, A and B). A pronounced impact of matrix ligand cues was observed on later-stage CFU colonies (CFU-GM and CFU-G/E/M/Mk), with laminin substrates promoting better myeloid specification compared to collagen or fibronectin. For example, CFU-GM and CFU-M/Mk increased on laminin-coated substrates after blebbistatin treatment (P < 0.05; Fig. 4C), but either remained the same or decreased on collagen- and fibronectin-coated substrates regardless of substrate stiffness (P < 0.05; Fig. 4, A and B). In addition, although CFU-E colony numbers dropped on collagen or fibronectin (P < 0.05; Fig. 4, A and B), the effect was largely muted on laminin substrates (P < 0.05; Fig. 4C), suggesting that specification toward erythrocyte on laminin under tension-compromised settings may be favored at soft (3.7 kPa) or extremely stiff (glass) conditions but not at osteoid stiffness (44 kPa). Whereas matrix engagement reduced the number of CFU-G colonies across all matrix conditions, addition of blebbistatin rescued CFU-G colonies, with a multiple-fold increase on laminin-coated substrates (P < 0.001) compared to collagen- or fibronectin-coated substrates (P < 0.05; Fig. 4).
Fig. 4. Actomyosin contractility alters HSC matrix engagement and lineage specification.

Blebbistatin (100 μM; BB) added to the culture medium altered HSC response to matrix biophysical cues. CFU assay results (± BB) were normalized to colonies formed by a control group of freshly isolated HSCs (dashed lines). Normalized colony counts for HSCs cultured with and without blebbistatin on collagen-coated (A), fibronectin-coated (B), and laminin-coated (C) substrates are shown. Blebbistatin abrogated previously noted stiffness-induced changes, with decreases in CFU-GEMM numbers on fibronectin-coated substrates and CFU-E/M/Mk colonies on collagen- and fibronectin-coated substrates. A pronounced impact of matrix ligand cues was observed on later-stage CFU colonies, with laminin substrates combined with blebbistatin promoting better myeloid specification compared to collagen or fibronectin. Whereas matrix engagement reduced the number of CFU-G colonies for all matrix conditions, addition of blebbistatin rescued CFU-G colonies, with a multiple-fold increase on laminin-coated substrates compared to collagen- or fibronectin-coated substrates. n = 3 from independent experiments. *P < 0.05, **P < 0.001.
To further assess the role of myosin II–mediated actin contractility and cytoskeletal tension in HSC fate decisions, we subsequently cultured primary HSCs on fibronectin- and laminin-coated substrates for 24 hours with Y-27632 added to the culture medium before performing the CFU assay (fig. S5). Y-27632 is a known inhibitor of Rho-associated protein kinase (ROCK) involved in myosin activation, and ROCK is the downstream effector of guanosine triphosphatase RhoA that plays an essential role in actin dynamics to modulate cell proliferation, adhesion, survival, and differentiation (27–29). Unlike blebbistatin, no pronounced impact of matrix ligand content was observed with Y-27632, because almost identical patterns of CFU colonies arose on both fibronectin- and laminin-coated substrates (fig. S5). Nonetheless, previously noted stiffness-induced changes in HSC fate decisions were again abrogated, with decreases in CFU-GEMM on fibronectin-coated (P < 0.001; fig. S5A) and laminin-coated substrates (P < 0.05; fig. S5B), and decreases in CFU-E/M/Mk on fibronectin- and laminin-coated substrates (P < 0.05; fig. S5, A and B). Although CFU-G numbers increased slightly with Y-27632 exposure, the results were not statistically significant. Together with the results from blebbistatin exposure, these findings suggest that interfering with myosin activation and myosin-generated cytoskeletal tension via ROCK inhibition has a substantial impact on maintaining early myeloid progenitors and later-stage myeloid progenitors, particularly for erythrocyte, monocyte/macrophage, and megakaryocyte lineages. These findings also suggest that, although blebbistatin and Y-27632 both obliterate matrix stiffness–related changes, selective effects of matrix cues remain with blebbistatin but not with Y-27632. These findings suggest that intracellular tension may enhance the effect of collagen or fibronectin engagement on maintaining primitive HSPCs but also reduce the effect of laminin engagement on promoting differentiation toward myeloid lineages. Moreover, CFU-G results suggest that commitment to granulocyte lineage in vitro may be compromised with matrix engagement, consistent with our results showing overall down-regulation of CFU-G formation upon matrix contact (Fig. 3).
Direct integrin activation rapidly alters HSC fate specification
Binding to ECM ligands activates specific integrin-mediated adhesion. Our findings from blebbistatin-added HSC cultures point to the selective impact of specific integrin activation in the early fate specification of HSCs. Because our CFU assay results suggest that fibronectin is capable of maintaining early hematopoietic progenitors, providing extrinsic cues that have a divergent impact on myeloid specification (CFU-GEMM versus CFU-E/M/Mk), we chose to examine the HSC response from adhesion to fibronectin. Therefore, we investigated how rapidly HSCs respond to discrete fibronectin binding motifs known to interact with α5β1 and ανβ3, respectively, on fibronectin-coated PA substrates.
To examine the role of select fibronectin-associated integrins (α5β1 and ανβ3) in early hematopoietic specification, we cultured primary HSCs on top of fibronectin-coated PA substrates for 24 hours with either α5β1 antibody (2 μg/ml) or ανβ3 antibody (2 μg/ml) solution added to the culture medium. Introducing either of these antibodies to the HSC cultures caused changes in HSC specification toward multiple myeloid lineages (Fig. 5 and fig. S6). In particular, CFU results revealed a decrease in CFU-GEMM (P < 0.05) colonies (Fig. 5, A and B), consistent with a loss of the fibronectin substrate’s earlier observed capacity to promote the maintenance of primitive CFU-GEMM colonies (Fig. 3B), whereas other colony numbers remained similar to control levels of CFU numbers from freshly isolated HSCs (Fig. 5A). The effects resulting from α5β1 antibody versus ανβ3 antibody were more subtle. Although both antibodies led to a decrease in CFU-GEMM formation compared to HSCs cultured on full fibronectin-coated substrates (Fig. 5B) and freshly isolated HSCs (Fig. 5A), blocking α5β1 engagement led to a much greater decrease (P < 0.001) compared to ανβ3 (P < 0.05). Whereas CFU-GM numbers were greater on substrates where α5β1 was blocked (versus ανβ3 inhibition; Fig. 5B), other later-stage CFUs (G, E, M, and Mk) did not show statistically meaningful differences between conditions (Fig. 5B and fig. S6). These results suggest that α5β1 activation may have a more selective role in maintaining early myeloid progenitors compared to ανβ3.
Fig. 5. Role of integrins α5β1 and ανβ3 during myeloid lineage specification.
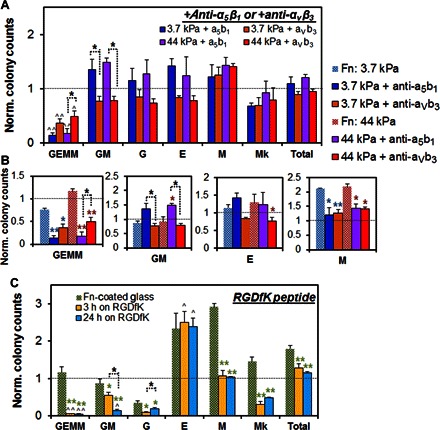
(A and B) HSC lineage specification was subsequently traced on fibronectin substrates blocked with an antibody to α5β1 or ανβ3, with results normalized against freshly isolated HSCs. Although both antibodies led to a decrease in CFU-GEMM formation compared to HSCs cultured on full fibronectin-coated substrates and freshly isolated HSCs, α5β1 antibody led to a much greater decrease (P < 0.001) compared to ανβ3 antibody (P < 0.05). Although CFU-GM numbers were higher with α5β1 antibody compared to ανβ3 antibody, later-stage CFUs (G, E, M, and Mk) did not show statistically meaningful difference between the two conditions. These results suggest that α5β1 activation may have a more selective role in maintaining early myeloid progenitors compared to ανβ3. (C) HSC lineage specification was also traced on glass substrates coated with cyclic RGD (RGDfK) to assess the role of ανβ3 in early HSC fate specification. It has been reported that ανβ3 is one of the major integrin receptors involved in cell-RGDfK interactions. CFU results suggest that myeloid lineage specification occurs rapidly, with no difference in results after 3 or 24 hours of exposure. Consistent with previous findings, CFU-GEMM, CFU-GM, and CFU-G decreased, whereas CFU-E increased, compared to control as a result of RGDfK engagement. n = 3 to 4 from independent experiments. *P < 0.05 and **P < 0.001 with respect to no-antibody conditions on full fibronectin-coated substrates. ^P < 0.05 and ^^P < 0.001 with respect to freshly isolated HSCs.
To further explore the influence of ανβ3 activation on hematopoietic specification, we cultured primary HSCs on glass substrates coated with cyclic RGD (RGDfK) for up to 24 hours. Although not exclusive, ανβ3 is one of the major integrin receptors involved in cell-RGDfK interactions (30, 31). CFU assay results showed that myeloid lineage specification occurred rapidly, within 3 hours of culture, with the effect maintained through 24 hours of culture (Fig. 5C). Consistent with previous findings, CFU-GEMM, CFU-GM, and CFU-G decreased, whereas CFU-E increased, compared to freshly isolated HSCs as a result of RGDfK engagement (P < 0.05; Fig. 5C). Some notable differences were observed in HSC specification on fibronectin-coated versus RGDfK-coated surfaces. Whereas CFU-E formation was greater in cultured (fibronectin or RGDfK surface) versus freshly isolated HSCs, additional myeloid specification was largely reduced on RGDfK substrates (GEMM, GM, G, M, and Mk) versus fibronectin-coated substrates (P < 0.05; Fig. 5C). Because ανβ3 is a major integrin associated with binding RGDfK and adding ανβ3 antibody, but not α5β1 antibody, led to decreased CFU-E formations (P < 0.05; Fig. 5, A and B), we suspect that ανβ3 may be particularly important for HSC specification to erythroid lineages. Although these results clearly point to the need for a more extensive study examining the role of different integrins and possible overlap in their functional capacity on regulating HSC differentiation activities, our findings suggest that specific integrin activation provides important biophysical signals that selectively modulate HSC lineage specification toward myeloid lineages (Fig. 6).
Fig. 6. Biophysical explanation for niche-mediated regulation of HSC fate decisions.

Our results suggest a model for marrow-inspired matrix cues, notably stiffness and ligands, altering early HSC fate decisions. Substrates resembling endosteal regions (44 kPa; fibronectin) promoted proliferation of primitive myeloid progenitors, whereas those resembling vascular zones (laminin) favored differentiation toward erythroid lineages. These results suggest that the biophysical elements of the putative HSC niches modulate HSC fate decisions directly. Marrow schematic is modified with permission from Choi et al. (22).
DISCUSSION
Here, we report a direct link between in vitro matrix biophysical properties and early HSC fate specification events. Our findings indicate that HSC morphological properties (spreading and shape), proliferation, and functional capacity (myeloid lineage specification) can be regulated by matrix signals in the form of matrix stiffness and ligand type. We find that these fate decisions occur rapidly, within 24 hours ex vivo, and in some cases as early as within 3 hours of ex vivo cultures. Morphological changes in cultured HSCs may be explained by how matrix stiffness regulates intracellular tension. On stiff substrates, the stiff matrix likely resists contractile forces exerted by cells to help cluster integrins, where integrins act as anchoring points to strengthen integrin-mediated adhesion and facilitate actin polymerization (32). On soft substrates, the soft matrix fails to resist the exerted forces and instead deforms, thereby hindering cell spreading. Hence, our results from imaging analyses showed that HSCs at soft matrix stiffness (3.7 kPa) remain round, whereas HSCs at stiff matrix stiffness (>44 kPa) spread out to form an elongated, polarized cell body, to a degree similar on all substrates and independent of the type of ligand cues presented. These results are consistent with those by Chowdhury et al. (33), who observed that changes in matrix biophysical cues affect the softness and differentiation state of embryonic stem cells.
Integrin-mediated adhesion progresses through discrete stages, from initial attachment to nascent, growing, and mature adhesion, where integrin clustering, recruitment of adhesion-associated proteins, and the generation of focal adhesion complexes successively strengthen the adhesive bond between the cell and the surrounding matrix (34). These, in turn, initiate a complex cascade of intracellular signaling events that may lead to endogenous changes altering cellular activity. During the early stages of adhesion, integrin activation and clustering induce actin polymerization, although these processes may be independent of myosin II–mediated contraction because of the very low forces involved (35). In later stages of adhesion, myosin II binds and cross-links actin to exert contractile forces to induce cell spreading and migration (36). Unlike many anchorage-dependent cells that form distinct focal adhesions indicative of strong myosin II–mediated contraction and adhesion, HSCs do not. However, previous reports have implicated HSC intracellular tension via changes in actomyosin contractility in several lymphoid- and myeloid lineage–associated processes, including neutrophil chemotaxis, macrophage phagocytosis, and enucleation in erythroid lineages in suspension cultures (37). Recent reports also suggest that HSC nuclear tension (lamin A and B expressions) (38) and myosin II regulation (A and B isoform switching and polarization of myosin IIB during asymmetric division of HSCs) (26) are important determinants of HSC lineage specification, acting via intracellular tension events.
The explicit role matrix ligands play in modulating HSC fate decisions has remained largely unknown because of the complexity of the matrix surrounding HSCs in vivo (22, 39). Previous reports have suggested that ECM components, such as matrix proteins (for example, fibronectin, collagen, laminin, and osteopontin) and proteoglycans, may affect hematopoiesis and HSC function in vivo; further, matrix proteins, such as fibronectin and collagen, have been shown to induce adhesion-mediated morphological changes in HSCs in vitro (22, 39). HSCs engage matrix components via integrins, and several integrins involved in HSC-matrix engagement have been identified (for example, α4β1, α5β1, αLβ2, and αMβ2) (40). The expressions of these integrins are found to be differentially regulated during the development and regeneration of the bone marrow (41), suggesting that the adhesive interactions between HSCs and the surrounding matrix ligands during discrete stages of hematopoiesis may be critical for HSC maintenance and to orchestrate HSC fate decisions. Our findings demonstrate that matrix stiffness and ligand type can induce complex, combinatorial effects on proliferation and lineage specification of HSCs by modulating integrin activation and actomyosin contractility.
Proliferation of cultured HSCs based on flow cytometric analyses showed strong correlations to matrix ligand cues, with proliferation heightened on collagen-coated substrates and reduced on laminin-coated substrates regardless of substrate stiffness. Critical for future efforts aimed at in vitro expansion of primary HSCs, we observed a direct effect of matrix stiffness and ligand type on the maintenance of a primitive hematopoietic fraction and subsequent myeloid lineage specification. Discrete combinations of these factors were found to selectively affect disparate stages of myeloid specification. Combining the results from our CFU assays, we observe that stiff (≥44 kPa) fibronectin-coated substrates best promote early hematopoietic myeloid progenitors, likely enhanced via α5β1 integrin activation, whereas laminin-coated substrates promote myeloid differentiation particularly when actomyosin contractility is inhibited with blebbistatin. The complexity of the data makes it difficult to parse out exact combinations of biophysical signals that trigger discrete specification events toward particular hematopoietic lineages. However, our results suggest that intracellular tension plays a major role in specifying HSC lineage and that specification to granulocyte and erythrocyte lineages is highly correlated with lower and higher ends of myosin II–mediated HSC intracellular tension, respectively. In addition, activation of ανβ3 integrin, but not α5β1 integrin, supports erythroid lineage specification. These observations are consistent with reports in the literature that describe increased cellular deformability during granulocyte development, which likely facilitates their migration across the endothelial barrier (42), and that F-actin bundles are detected in maturing but not in immature erythroblasts (43).
Until now, a wide range of bioengineering research efforts have focused on identifying the influence of biophysical signals on other stem cell populations, notably MSCs, that adhere to substrates more strongly (44–48). Specifically, MSC interactions with substrates with varying mechanical properties, surface chemistry, and topographical features were found to directly affect their growth, differentiation, and regenerative capacity (45–48). However, much less effort has explored how matrix-based biophysical cues affect early HSC fate decisions (49). Our work shows that marrow-inspired matrix cues directly affect early HSC fate decisions. Further, these results are important for formulating design criteria for biomaterial environment to promote ex vivo control of HSC fate specification events for stem cell biomanufacturing and disease model development. These results also suggest important avenues for continued progress. A relative lack of functional metrics for continuous monitoring of single HSC activity in situ makes it difficult to assess whether our observations are a direct result of cues inducing endogenous changes across the population or within selective cellular subsets. Development of new tools to trace single HSC fate decisions with minimal sample manipulation and increased sensitivity is highly desirable to better resolve nuances of HSC-matrix interactions. Recent efforts in the area of time-lapse HSC tracing (14), Raman spectroscopy–based single HSC classification tools (50), and label-free photonic crystal imaging tools that can trace cell-matrix engagement (51) offer exciting opportunities for functionally phenotyping HSC fate specification events across a range of engineered bone marrow microenvironments. The observed gradations in matrix ligand composition and mechanical properties across the bone marrow may be directly linked to the differential regulation of HSC fate and motivate ongoing approaches using graded material platforms to support hematopoietic homeostasis ex vivo (52, 53).
CONCLUSIONS
In summary, our work provides a possible biophysical explanation of the niche-mediated regulation of HSC fate decisions. Specifically, we show that biophysical features of putative endosteal niches (high fibronectin content, stiffer microenvironment) are capable of supporting early-stage HSPCs, whereas those of putative perivascular niches (high laminin content) promote rapid commitment toward myeloid lineages, particularly to erythrocyte lineage at conditions inducing higher intracellular tension. Our results highlight the importance and relevance of marrow-inspired matrix cues as regulators of HSC fate decisions. These findings provide the basis for developing guidelines for an in vitro biomaterial culture and screening platform for HSCs. They also suggest continued need for new analytical tools to dynamically resolve real-time functional changes in HSCs at the single-cell level.
MATERIALS AND METHODS
Preparation of PA gel and glass substrates
PA gels were prepared in a 14-mm glass-bottom culture dish (In Vitro Scientific), as previously reported (25, 54, 55). In brief, a stock solution of 19:1 acrylamide/N,N′-methylenebisacrylamide was diluted in deionized water, and an appropriate amount of 30% acrylamide solution was added to obtain the desired concentrations of acrylamide and bis-acrylamide. Tetramethylethylenediamine (1:2000, v/v) and 10% ammonium persulfate (1:200, v/v) were added to the mixture to initiate cross-linking via free radical formation. This solution was promptly placed onto the glass portion of the glass-bottom dish pretreated with 3-aminopropyltrimethoxysilane and 0.5% glutaraldehyde. A 12-mm glass coverslip treated with dichlorodimethylsilane was placed on top of the solution to induce the formation of a thin gel (~75 μm) with an even surface. Once polymerization was complete, the coverslip was carefully removed and the gel was washed twice with deionized water and stored at 4°C until needed. Unless specified, all reagents were purchased from Thermo Fisher Scientific.
PA gel surface was functionalized with type I collagen, fibronectin, or laminin via photocrosslinking with sulfo-SANPAH (25, 54). PA gel surface was first covered with 50 mM sulfo-SANPAH and was exposed to a 365-nm ultraviolet (UV) source (UVP) for 6 min. Upon rinsing the gel twice with 50 mM Hepes (pH 8.5), the procedure was repeated once more with fresh 50 mM sulfo-SANPAH. Protein solution of type I collagen (from rat tail, Corning Inc.), fibronectin (from human plasma, Corning Inc.), or laminin (from Engelbreth-Holm-Swarm murine sarcoma basement membrane, Sigma-Aldrich) at 100 μg/ml was placed on top and was left to react overnight at 4°C. The immobilization of matrix ligands on PA substrates was verified using immunofluorescence of protein-coated substrates, as previously reported (25). Protein-coated substrates were incubated first with primary antibodies (collagen I antibody from Abcam, fibronectin antibody H-300 from Santa Cruz Biotechnology, and laminin antibody from Thermo Fisher Scientific) and then with the appropriate secondary antibodies (Alexa Fluor 568 or 488 antibody, Life Technologies). Subsequently, the fluorescently tagged substrates were imaged using an inverted confocal microscope (Zeiss LSM 710, Carl Zeiss Inc.) to measure the relative fluorescence intensity. The images showed that PA gel surfaces were evenly decorated with immobilized ligands (fig. S7). Coated PA gels were used within 2 days of coating and sterilized with UV before use.
Coated glass substrates were prepared first by cleaning glass-bottom dishes with 1 M HCl and rinsing three times with sterile phosphate-buffered saline (PBS) followed by sterile deionized water. Appropriate protein solution (100 μg/ml) was then placed on top and incubated for 1 hour at 37°C. Protein-coated glass substrates were gently washed with warmed PBS and were immediately used. For RGDfK-coated glass, glass-bottom 96-well plates (In Vitro Scientific) were functionalized according to previously reported procedures (30), with slight modifications. In brief, glass-bottom well plates were cleaned and rendered hydrophilic by oxygen plasma treatment for 5 min. Next, glass surfaces were aminosilanized with 3-aminopropyltrimethoxysilane, heated to 80°C for 4 hours and cooled, and then reacted with mPEG-SVA and biotin-mPEG-SVA (mixed at 10:1 ratio; molecular weight, 5000; Laysan Bio), followed by NeutrAvidin (Thermo Fisher Scientific), and finally biotin-RGDfK (Peptides International) to immobilize RGDfK onto the glass surface.
HSC isolation
C57BL6 mice (The Jackson Laboratory) between 4 and 10 weeks of age were euthanized with carbon dioxide in compliance with the University of Illinois Institutional Animal Care and Use Committee guidelines. Their femurs and tibias were collected, cleaned, crushed, and filtered through a 40-μm cell strainer (BD Falcon) in PBS supplemented with 2% FBS (buffer) to collect WBM cells. Red blood cells were removed with ACK lysis buffer (Thermo Fisher Scientific Inc.). WBM cells were first incubated with Fc receptor blocking antibody (CD16/CD32) to prevent nonspecific binding and then fluorescently labeled with a cocktail of fluorescein isothiocyanate–conjugated lineage antibodies [CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), 7/4, Ter-119], phycoerythrin-conjugated Sca-1, and allophycocyanin-conjugated c-Kit. All antibodies were purchased from eBioscience. Propidium iodide (Thermo Fisher Scientific Inc.) was used to exclude dead cells. Labeled cells were sorted via FACS (BD FACSAria cell sorter, BD Biosciences) to isolate LSK cell population (fig. S8). This consistently yielded 4 × 104 to 6 × 104 LSK cells per mouse. Freshly isolated LSK cells were either immediately analyzed (“freshly isolated cells”) or seeded on top of the prepared substrates and cultured for a specified time period.
Cell seeding and culture conditions
Isolated LSK cells were seeded on top of coated substrates at a cell seeding density of 13,000 cells/ml per substrate. This seeding density minimized cell-cell interactions (25). StemPro-34 serum-free medium (Thermo Fisher Scientific Inc.) supplemented with the complementary StemPro-Nutrient Supplement, SCF (100 ng/ml), Flt3L (100 ng/ml), and TPO (100 ng/ml) (PeproTech) was used as the culture medium. Seeded LSK cells were cultured for a specified time period (3 or 24 hours) and then either analyzed in situ for imaging analyses or harvested with TrypLE (Thermo Fisher Scientific Inc.) for subsequent analyses.
Cell spreading quantifications
Spread area and perimeter of the cells were calculated using ImageJ from phase-contrast images of the cultured HSCs fixed with 3.7% paraformaldehyde and imaged with an inverted microscope (Leica DM IL LED, Leica Microsystems). CSI, a dimensionless measure of the circularity of a cell, was defined as (4·π·(cell area)·(cell perimeter)−2), where 0 is the theoretical minimum for a cell infinitely elongated in one direction and 1 represents a perfectly circular cell (25). Cells in direct contact with one another were excluded from analysis.
CFU assay and subsequent flow cytometric analysis of CFU cells
Cultured HSCs were lifted with TrypLE and resuspended in methylcellulose medium supplemented with murine cytokines (MethoCult GF M3434; Stem Cell Technologies) according to the manufacturer’s protocols. In brief, lifted cells were mixed with methylcellulose medium supplemented with growth factors, dispensed to 35-mm culture dishes using a 3-cm3 syringe, and incubated for 11 to 14 days at 37°C and 5% CO2, at which point the colonies that arose (CFU-GEMM, CFU-GM, CFU-M, CFU-G, CFU/BFU-E, CFU-Mk) were enumerated with an inverted microscope (DMI4000, Leica Microsystems). CFU colonies of more than 30 cells were enumerated. Representative images of the CFU colonies are shown in fig. S9. For the subsequent analysis of colony-forming cells, CFU colonies were harvested in bulk from the methylcellulose medium according to the previously reported protocol (56). Harvested colony-forming cells were incubated with CD11b (Mac-1), Gr-1 (Ly-6G/C), or a cocktail of LSK antibodies (eBioscience) and analyzed by flow cytometry (BD LSR II analyzer, BD Biosciences). Flow cytometric analysis of the colony-forming cells indicates that our CFU colony enumeration techniques are consistent with known changes in cell surface antigen expressions (fig. S10).
Inhibition studies
For antibody blocking experiments, α5β1 (Millipore) or ανβ3 (Santa Cruz Biotechnology) (2 μg/ml) antibody solution was added to the culture medium at the start of culture. For myosin II inhibition studies, 100 μM blebbistatin (Sigma-Aldrich), a known inhibitor of myosin II, was added to the cell culture medium at the start of culture. For ROCK inhibition studies, 10 μM Y-27632 (Tocris Bioscience), a known inhibitor of ROCK, was added to the cell culture medium at the start of culture.
Proliferation analysis with CellTrace Violet
For proliferation analysis, cells were stained with CellTrace Violet cell proliferation kit (Life Technologies) according to the manufacturer’s protocol before cell seeding and analyzed by flow cytometry after the 24-hour culture period (BD LSR II Analyzer, BD Biosciences). For each cell division that the parental cell went through, CellTrace Violet was distributed equally to its daughter cells. Cell proliferation indexes were calculated using De Novo FCS Express software.
Statistical analysis
Sample size was chosen so that each data point would have values from at least three independent experiments (n = 3 or more) to compute statistically meaningful averages and SDs. Because we used FACS-isolated primary HSCs whose numbers fluctuate from mouse to mouse, certain data points ended up with more or less technical replicates than others (n = 1 to 7). One-way analysis of variance (ANOVA) with Tukey post hoc testing was used to determine statistical differences between data groups at a significance level of P < 0.05. All errors are reported as SEM unless otherwise noted.
Supplementary Material
Acknowledgments
We acknowledge B. Pilas for assistance with flow cytometry and M. Sivaguru for assistance with fluorescence imaging. Funding: This material is based on work supported by the NSF under grant 1254738 (B.A.C.H.). Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH under award number R01 DK099528 and by the National Institute of Biomedical Imaging and Bioengineering of the NIH under award number R21 EB018481. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was partially supported by grants 160673 and 189782 from the American Cancer Society of Illinois (B.A.C.H.). Additional funding was provided by the Department of Chemical and Biomolecular Engineering and Carl R. Woese Institute for Genomic Biology at the University of Illinois at Urbana-Champaign (B.A.C.H.). Author contributions: J.S.C. and B.A.C.H. designed the project. J.S.C. performed experiments and analyzed data. J.S.C. and B.A.C.H. interpreted the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1600455/DC1
Experimental design
fig. S1. HSC proliferative index is increased on collagen substrates.
fig. S2. CFU colony counts of cultured HSCs compared to those from freshly isolated HSCs.
fig. S3. Morphology of cultured HSCs with blebbistatin added to the culture medium.
fig. S4. Actomyosin contractility alters HSC matrix engagement and lineage specification.
fig. S5. Myosin-mediated actin cytoskeletal tension alters HSC lineage specification.
fig. S6. Role of integrins α5β1 and ανβ3 in myeloid lineage specification.
fig. S7. Immunofluorescence of protein-coated substrates.
fig. S8. FACS isolation of LSK cell population.
fig. S9. CFU colony enumeration.
fig. S10. Flow cytometric analysis of the colony-forming cells harvested from the CFU assay.
fig. S11. Mechanical characterization of PA gels.
table S1. Elastic modulus of PA gels.
REFERENCES AND NOTES
- 1.Wilson A., Trumpp A., Bone-marrow haematopoietic-stem-cell niches. Nat. Rev. Immunol. 6, 93–106 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Morrison S. J., Scadden D. T., The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nilsson S. K., Debatis M. E., Dooner M. S., Madri J. A., Quesenberry P. J., Becker P. S., Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J. Histochem. Cytochem. 46, 371–377 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Hines M., Nielsen L., Cooper-White J., The hematopoietic stem cell niche: What are we trying to replicate?. J. Chem. Technol. Biotechnol. 83, 421–443 (2008). [Google Scholar]
- 5.Jansen L. E., Birch N. P., Schiffman J. D., Crosby A. J., Peyton S. R., Mechanics of intact bone marrow. J. Mech. Behav. Biomed. Mater. 50, 299–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh I.-H., Humphries R. K., Concise review: Multidimensional regulation of the hematopoietic stem cell state. Stem Cells 30, 82–88 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Ehninger A., Trumpp A., The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 208, 421–428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silberstein L. E., Lin C. P., A new image of the hematopoietic stem cell vascular niche. Cell Stem Cell 13, 514–516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunisaki Y., Bruns I., Scheiermann C., Ahmed J., Pinho S., Zhang D., Mizoguchi T., Wei Q., Lucas D., Ito K., Mar J. C., Bergman A., Frenette P. S., Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acar M., Kocherlakota K. S., Murphy M. M., Peyer J. G., Oguro H., Inra C. N., Jaiyeola C., Zhao Z., Luby-Phelps K., Morrison S. J., Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526, 126–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Y., Yin T., Wiegraebe W., He X. C., Miller D., Stark D., Perko K., Alexander R., Schwartz J., Grindley J. C., Park J., Haug J. S., Wunderlich J. P., Li H., Zhang S., Johnson T., Feldman R. A., Li L., Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 457, 97–101 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Nombela-Arrieta C., Pivarnik G., Winkel B., Canty K. J., Harley B., Mahoney J. E., Park S.-Y., Lu J., Protopopov A., Silberstein L. E., Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell Biol. 15, 533–543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H. J., Li N., Evans S. M., Diaz M. F., Wenzel P. L., Biomechanical force in blood development: Extrinsic physical cues drive pro-hematopoietic signaling. Differentiation 86, 92–103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kokkaliaris K. D., Loeffler D., Schroeder T., Advances in tracking hematopoiesis at the single-cell level. Curr. Opin. Hematol. 19, 243–249 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Holst J., Watson S., Lord M. S., Eamegdool S. S., Bax D. V., Nivison-Smith L. B., Kondyurin A., Ma L., Oberhauser A. F., Weiss A. S., Rasko J. E. J., Substrate elasticity provides mechanical signals for the expansion of hemopoietic stem and progenitor cells. Nat. Biotechnol. 28, 1123–1128 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Lee-Thedieck C., Rauch N., Fiammengo R., Klein G., Spatz J. P., Impact of substrate elasticity on human hematopoietic stem and progenitor cell adhesion and motility. J. Cell Sci. 125, 3765–3775 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Kurth I., Franke K., Pompe T., Bornhäuser M., Werner C., Extracellular matrix functionalized microcavities to control hematopoietic stem and progenitor cell fate. Macromol. Biosci. 11, 739–747 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Müller E., Grinenko T., Pompe T., Waskow C., Werner C., Space constraints govern fate of hematopoietic stem and progenitor cells in vitro. Biomaterials 53, 709–715 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Muth C. A., Steinl C., Klein G., Lee-Thedieck C., Regulation of hematopoietic stem cell behavior by the nanostructured presentation of extracellular matrix components. PLOS ONE 8, e54778 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prewitz M. C., Seib F. P., von Bonin M., Friedrichs J., Stißel A., Niehage C., Müller K., Anastassiadis K., Waskow C., Hoflack B., Bornhäuser M., Werner C., Tightly anchored tissue-mimetic matrices as instructive stem cell microenvironments. Nat. Methods 10, 788−794 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Csaszar E., Kirouac D. C., Yu M., Wang W., Qiao W., Cooke M. P., Boitano A. E., Ito C., Zandstra P. W., Rapid expansion of human hematopoietic stem cells by automated control of inhibitory feedback signaling. Cell Stem Cell 10, 218–229 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Choi J. S., Mahadik B. P., Harley B. A. C., Engineering the hematopoietic stem cell niche: Frontiers in biomaterial science. Biotechnol. J. 10, 1529–1545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peyton S. R., Putnam A. J., Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J. Cell. Physiol. 204, 198–209 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Oliver W. C., Pharr G. M., An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments. J. Mater. Res. 7, 1564–1583 (1992). [Google Scholar]
- 25.Choi J. S., Harley B. A. C., The combined influence of substrate elasticity and ligand density on the viability and biophysical properties of hematopoietic stem and progenitor cells. Biomaterials 33, 4460–4468 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Shin J.-W., Buxboim A., Spinler K. R., Swift J., Christian D. A., Hunter C. A., Léon C., Gachet C., Dingal P. C. D. P., Ivanovska I. L., Rehfeldt F., Chasis J. A., Discher D. E., Contractile forces sustain and polarize hematopoiesis from stem and progenitor cells. Cell Stem Cell 14, 81–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McBeath R., Pirone D. M., Nelson C. M., Bhadriraju K., Chen C. S., Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 6, 483–495 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Sit S.-T., Manser E., Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 124, 679–683 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Kilian K. A., Bugarija B., Lahn B. T., Mrksich M., Geometric cues for directing the differentiation of mesenchymal stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 4872–4877 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X., Ha T., Defining single molecular forces required to activate integrin and notch signaling. Science 340, 991–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danhier F., Breton A. L., Préat V., RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol. Pharm. 9, 2961–2973 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Humphrey J. D., Dufresne E. R., Schwartz M. A., Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdhury F., Na S., Li D., Poh Y.-C., Tanaka T. S., Wang F., Wang N., Material properties of the cell dictate stress-induced spreading and differentiation in embryonic stem cells. Nat. Mater. 9, 82–88 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolfenson H., Lavelin I., Geiger B., Dynamic regulation of the structure and functions of integrin adhesions. Dev. Cell 24, 447–458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu C.-h., Law J. B. K., Suryana M., Low H. Y., Sheetz M. P., Early integrin binding to Arg-Gly-Asp peptide activates actin polymerization and contractile movement that stimulates outward translocation. Proc. Natl. Acad. Sci. U.S.A. 108, 20585–20590 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vicente-Manzanares M., Ma X., Adelstein R. S., Horwitz A. R., Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin J.-W., Swift J., Ivanovska I., Spinler K. R., Buxboim A., Discher D. E., Mechanobiology of bone marrow stem cells: From myosin-II forces to compliance of matrix and nucleus in cell forms and fates. Differentiation 86, 77–86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shin J.-W., Spinler K. R., Swift J., Chasis J. A., Mohandas N., Discher D. E., Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 110, 18892–18897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi J., Harley B., Challenges and opportunities to harnessing the (hematopoietic) stem cell niche. Curr. Stem Cell Rep. 2, 85–94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klamer S., Voermans C., The role of novel and known extracellular matrix and adhesion molecules in the homeostatic and regenerative bone marrow microenvironment. Cell Adh. Migr. 8, 563–577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mikkola H. K. A., Orkin S. H., The journey of developing hematopoietic stem cells. Development 133, 3733–3744 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Lichtman M. A., Kearney E., Cellular deformability during maturation of the myeloblast. Possible role in marrow egress. N. Engl. J. Med. 283, 943–948 (1970). [DOI] [PubMed] [Google Scholar]
- 43.Koury S. T., Koury M. J., Bondurant M. C., Cytoskeletal distribution and function during the maturation and enucleation of mammalian erythroblasts. J. Cell Biol. 109, 3005–3013 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trappmann B., Gautrot J. E., Connelly J. T., Strange D. G. T., Li Y., Oyen M. L., Cohen Stuart M. A., Boehm H., Li B., Vogel V., Spatz J. P., Watt F. M., Huck W. T. S., Extracellular-matrix tethering regulates stem-cell fate. Nat. Mater. 11, 642–649 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Engler A. J., Sen S., Sweeney H. L., Discher D. E., Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Curran J. M., Stokes R., Irvine E., Graham D., Amro N. A., Sanedrin R. G., Jamil H., Hunt J. A., Introducing dip pen nanolithography as a tool for controlling stem cell behaviour: Unlocking the potential of the next generation of smart materials in regenerative medicine. Lab Chip 10, 1662–1670 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Gilbert P. M., Havenstrite K. L., Magnusson K. E. G., Sacco A., Leonardi N. A., Kraft P., Nguyen N. K., Thrun S., Lutolf M. P., Blau H. M., Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 329, 1078–1081 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMurray R. J., Gadegaard N., Tsimbouri P. M., Burgess K. V., McNamara L. E., Tare R., Murawski K., Kingham E., Oreffo R. O. C., Dalby M. J., Nanoscale surfaces for the long-term maintenance of mesenchymal stem cell phenotype and multipotency. Nat. Mater. 10, 637–644 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Dalby M. J., Gadegaard N., Oreffo R. O. C., Harnessing nanotopography and integrin–matrix interactions to influence stem cell fate. Nat. Mater. 13, 558–569 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Ilin Y., Choi J. S., Harley B. A. C., Kraft M. L., Identifying states along the hematopoietic stem cell differentiation hierarchy with single cell specificity via Raman spectroscopy. Anal. Chem. 87, 11317–11324 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen W., Long K. D., Lu M., Chaudhery V., Yu H., Choi J. S., Polans J., Zhuo Y., Harley B. A. C., Cunningham B. T., Photonic crystal enhanced microscopy for imaging of live cell adhesion. Analyst 138, 5886–5894 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Mahadik B. P., Pedron Haba S., Skertich L. J., Harley B. A. C., The use of covalently immobilized stem cell factor to selectively affect hematopoietic stem cell activity within a gelatin hydrogel. Biomaterials 67, 297–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahadik B. P., Wheeler T. D., Skertich L. J., Kenis P. J. A., Harley B. A. C., Microfluidic generation of gradient hydrogels to modulate hematopoietic stem cell culture environment. Adv. Healthc. Mater. 3, 449–458 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Tse J. R., Engler A. J., Preparation of hydrogel substrates with tunable mechanical properties. Curr. Protoc. Cell Biol. Chapter 10, Unit 10.16 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Pelham R. J. Jr, Wang Y.-L., Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. U.S.A. 94, 13661–13665 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller C. L., Dykstra B., Eaves C. J., Characterization of mouse hematopoietic stem and progenitor cells. Curr. Protoc. Immunol. Chapter 22, Unit 22B.2 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Li Q. S., Lee G. Y. H., Ong C. N., Lim C. T., AFM indentation study of breast cancer cells. Biochem. Biophys. Res. Commun. 374, 609–613 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Domke J., Radmacher M., Measuring the elastic properties of thin polymer films with the atomic force microscope. Langmuir 14, 3320–3325 (1998). [Google Scholar]
- 59.Engler A., Bacakova L., Newman C., Hategan A., Griffin M., Discher D., Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 86, 617–628 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin D. C., Dimitriadis E. K., Horkay F., Robust strategies for automated AFM force curve analysis—I. Non-adhesive indentation of soft, inhomogeneous materials. J. Biomech. Eng. 129, 430–440 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1600455/DC1
Experimental design
fig. S1. HSC proliferative index is increased on collagen substrates.
fig. S2. CFU colony counts of cultured HSCs compared to those from freshly isolated HSCs.
fig. S3. Morphology of cultured HSCs with blebbistatin added to the culture medium.
fig. S4. Actomyosin contractility alters HSC matrix engagement and lineage specification.
fig. S5. Myosin-mediated actin cytoskeletal tension alters HSC lineage specification.
fig. S6. Role of integrins α5β1 and ανβ3 in myeloid lineage specification.
fig. S7. Immunofluorescence of protein-coated substrates.
fig. S8. FACS isolation of LSK cell population.
fig. S9. CFU colony enumeration.
fig. S10. Flow cytometric analysis of the colony-forming cells harvested from the CFU assay.
fig. S11. Mechanical characterization of PA gels.
table S1. Elastic modulus of PA gels.