

Abstract
Studies of human immunodeficiency virus (HIV) vaccines in animal models suggest that it is difficult to induce complete protection from infection (sterilizing immunity) but that it is possible to reduce the viral load and to slow or prevent disease progression following infection. We have developed an age-structured epidemiological model of the effects of a disease-modifying HIV vaccine that incorporates the intrahost dynamics of infection, a transmission rate and host mortality that depend on the viral load, the possible evolution and transmission of vaccine escape mutant viruses, a finite duration of vaccine protection, and possible changes in sexual behavior. Using this model, we investigated the long-term outcome of a disease-modifying vaccine and utilized uncertainty analysis to quantify the effects of our lack of precise knowledge of various parameters. Our results suggest that the extent of viral load reduction in vaccinated infected individuals (compared to unvaccinated individuals) is the key predictor of vaccine efficacy. Reductions in viral load of about 1 log10 copies ml−1 would be sufficient to significantly reduce HIV-associated mortality in the first 20 years after the introduction of vaccination. Changes in sexual risk behavior also had a strong impact on the epidemic outcome. The impact of vaccination is dependent on the population in which it is used, with disease-modifying vaccines predicted to have the most impact in areas of low prevalence and rapid epidemic growth. Surprisingly, the extent to which vaccination alters disease progression, the rate of generation of escape mutants, and the transmission of escape mutants are predicted to have only a weak impact on the epidemic outcome over the first 25 years after the introduction of a vaccine.
Several different human immunodeficiency virus (HIV) vaccination strategies have been proposed. These include therapeutic vaccination (administered to those who are already infected) and prophylactic vaccination (administered prior to infection) that either prevents infection (sterilizing vaccination) or ameliorates disease (disease-modifying vaccination). Thus far, both therapeutic and sterilizing vaccination strategies for HIV have proved elusive (35). However, recent studies in primates suggest that it is possible to produce disease-modifying vaccines that significantly reduce viral loads and AIDS mortality (1, 11, 22, 51). These vaccines appear to work primarily by inducing cytotoxic T lymphocyte (CTL) immunity. If these results can be reproduced in human infections, it seems likely that in the short to medium term a human vaccine for HIV may emerge that significantly increases the survival time of infected individuals. Although such disease-modifying vaccines would clearly benefit infected individuals by delaying disease, their long-term impact on the epidemic as a whole is less clear. In particular, immunological escape and subsequent disease progression have been seen in several vaccination studies (9, 10). If a vaccine reduces viral loads, then patients may experience a longer latent period of infection but still ultimately progress through the same advanced stages of disease seen in a “natural” infection. Thus, overall transmission may be increased by the addition of this extra asymptomatic period of transmission on top of the normal transmission period (4). In addition, there is concern that optimism over the effects of vaccination may lead to an increase in high-risk sexual behavior and an increase in HIV transmission (18), as has been observed following antiretroviral therapy (25; T. Kellogg, W. McFarland, and M. Katz, Letter, AIDS 13:2303-2304, 1999).
The aims of any vaccination campaign utilizing a disease-modifying vaccine should be to maximize the survival time of each infected individual and to minimize the number of new infections. Understanding how disease-modifying vaccines will affect the spread of HIV requires the incorporation of both intrahost effects (such as how viral loads and mortality change with time) and population effects (such as how vaccination affects the sexual transmission of HIV and how sexual activity changes with age). Important questions to be addressed regarding the potential use of disease-modifying vaccines are as follows: (i) what will be the likely impact of these vaccines on the epidemic as a whole, (ii) what vaccine effects are most important in determining this outcome, and (iii) in what settings will the vaccine be most effective?
Mathematical modeling allows a theoretical analysis of such questions based on our understanding of the intrahost and interhost dynamics of HIV. Previous models have provided insights into the effects of antiretroviral therapy, as well as partially effective vaccines (which induce sterilizing immunity in only a proportion of vaccinated individuals), live attenuated vaccines, and therapeutic vaccines or immunotherapies administered to infected individuals (3, 5, 15, 16, 18, 19, 45). The CTL-inducing vaccines currently being tested in monkeys appear unable to prevent infection, but instead appear able to modify the subsequent course of disease. Thus, although they are only partially effective in one sense, they differ substantially from previously studied partially effective vaccines, which blocked infection in only a fraction of vaccinees. For this reason, we prefer to refer to these new candidate HIV vaccines as disease-modifying vaccines. We have developed a model that allows us to investigate the likely outcomes of a disease-modifying vaccination strategy as well as to identify the factors that are most important to its success. The model incorporates uncertainty in the rate and duration of vaccination, the effects of vaccination on reducing viral loads and disease progression, the rate of emergence and transmission of escape mutants, and the extent to which vaccination may increase the rate of high-risk sexual activity (Table 1). We used this model to predict the outcome of disease-modifying vaccination for HIV type 1 (HIV-1) and the impact of factors such as the extent to which vaccination reduces the viral load or the disease progression rate of infected individuals, the duration of protection from vaccination, and the emergence of viral escape mutants.
TABLE 1.
Parameter valuesa
| Parameter | Range |
|---|---|
| Vaccination rate (V) (yr−1) | 0.1-0.2 |
| Loss of vaccine effectiveness (ω) (yr−1) (31, 44) | 0.1-0.2 |
| Viral load reduction (log10) (1, 11, 22, 51) | 0.1-0.5 or 0.5-1.5b |
| Disease progression rate of vaccinees (Pv) (1, 11, 22, 51) | 0-0.8 or 0.8-1.2b |
| Viral escape (ɛ) (yr−1) (28, 47) | 0.2-2 |
| Transmission of escape variants (HE) (%) (27) | 5-50 |
| Increase in risk behavior (R) (%) (52) | 0-30 |
Because of uncertainty in the effects of vaccination, ranges rather than exact values for parameters were estimated from the literature. At the start of each simulation, a value for each parameter was randomly sampled from the range. By performing many (1,000) simulations, we were able to observe the probability distribution of outcomes based on different parameter choices (risk analysis; Fig. 3). Similarly, it was possible to correlate the values of individual parameters with the outcome variables of HIV incidence and mortality (sensitivity analysis; Table 2).
Data are for progression-slowing and viral-load-reducing scenarios, respectively.
A structured model of HIV infection, transmission, and vaccination. (i) Disease-modifying vaccines.
In order to analyze the effects of a disease-modifying vaccine, one needs to consider how the vaccine may alter the viral load and mortality of an infected individual over the total duration of infection. In addition, because the duration of infection is long compared to the life span of an individual, one also needs to consider how aging may affect transmission of the virus. A vaccine that reduces disease progression, for example, will lead to disease occurring at an older age. The rate of sexual partner change (13, 34), the frequency of intercourse, and the risk of transmission decrease with age (48). Importantly, older individuals are not only less likely to transmit the virus, but they are more likely to have partnerships with other older people (8, 23), who are themselves less likely to transmit the virus. Our model, which is age structured, incorporates all of these effects and allows us to calculate the rate of HIV transmission and mortality in a population (24). We utilized epidemiological data on the changes in viral load (36, 43) and mortality (7, 50) with the duration of infection as well as social survey data on age-related partner change rates (13, 34), age-related risks of transmission (48), and sexual mixing between age groups (8, 23) to set parameter ranges in the model.
We use the term “behavioral infectiousness” to describe the changes in transmission that are dependent on the sexual behavior of different age groups, and we differentiate those changes from changes in transmission due to the disease stage, which we term “virological infectiousness.” The virological infectiousness at different times after infection is correlated with the viral load. Our model incorporates the observed epidemiological relationship that the probability of transmission increases 2.45-fold for every log10 increase in the viral load (48). We also assumed that the rates of male-to-female and female-to-male transmission are equal, as reported for a study in Rakai, Uganda (29, 48). The risk of transmission from an infected individual in our model is therefore a function of both his or her behavioral infectiousness and virological infectiousness (see reference 24 and Appendix).
Even though we utilized experimental and social survey data on both virological and behavioral infectiousness, both sources of data have their limitations. Sexual survey data, in particular, are notoriously unreliable, with male estimates of the number of sexual partners outnumbering those of females up to threefold (41). For our study, we utilized the female estimates and balanced the sexual mixing matrix to compute transmission probabilities (17). Moreover, some authors suggest that sexual behavior may be quite consistent between regions as diverse as the United States and Africa (56), while other data suggest that behaviors may be quite different across cultures and also over time (26, 32, 42). To avoid any dependence of our results on particular population variables, we considered the effects of vaccination in many different populations (with different epidemiologic and social parameters) that were randomly generated from our baseline population, which is based on the described empirical data (see Materials and Methods).
(ii) Intrahost dynamics.
In animal models of HIV infection, prior vaccination is able to reduce viral loads by ∼1 log10 copies ml−1 for acute infections and up to ∼3 log10 copies ml−1 for chronic infections (compared to unvaccinated control animals). This protection appears to be dependent largely on CTL-derived immunity, and most infectious challenges are performed within weeks or months of vaccination (1, 11, 20-22, 38, 51). Studies of highly exposed but uninfected individuals suggest that immunity may be relatively short-lived and that regular boosting with an antigen may be needed to maintain protection (31). Consistent with this observation, CTL numbers appear to decline rapidly after the treatment of HIV infections (44). However, more recent data suggest that the half-life of the CTL response to vaccination may be on the order of a decade (30). Therefore, it is likely that vaccine-elicited immunity may progressively be lost and that only recently vaccinated individuals would be expected to receive the full benefit of vaccination. The level of protection (measured by the reduction in viral load following infection compared to the viral load of an unvaccinated individual) will decline with time since the last vaccination until all immunity is lost. We have adopted the assumption that the total duration of vaccine protection will be between 5 and 10 years (Table 1). With such a duration of protection, a short average time between vaccinations (5 to 10 years) (Table 1) is required for widespread coverage. Note that if the duration of vaccine protection were significantly shorter (1 to 5 years), a higher frequency of revaccination (every 1 to 5 years) would be required.
Immune responses select for viral escape mutants, which are viruses that have mutated the regions of protein targeted by the host so that they are no longer recognized by T cells or antibodies (9, 10, 40, 46). Studies of natural infections suggest that strong CTL responses may be maintained for years without inducing escape mutations, although under other circumstances escape may occur rapidly (28, 47). The rate of viral escape from CTL immune responses is uncertain and probably varies with the strength of immune pressure, structural constraints of the viral protein, and the viral load. Based on experimental evidence, we adopted a rate for the evolution of escape mutant viruses of 0.2 to 2 year−1 (equivalent to an average time to evolution of an escape mutant of 6 months to 5 years; Table 1) (9, 10). Current evidence suggests that after mutants escape from vaccine-induced immune responses, disease progression is similar to that of a natural infection (9, 10). Thus, we assumed that once a vaccine escape mutant develops, any disease-modifying effects of the vaccine are lost and vaccinated individuals progress at the same rate as unvaccinated individuals with an equivalent viral load. In addition, the population transmission of vaccine escape mutants was taken into account (see Materials and Methods).
MATERIALS AND METHODS
The model of HIV infection, transmission, and vaccination was structured by sex, age, infection status, vaccination status, and duration of infection (Fig. 1; also see Appendix). The uninfected (susceptible) population (S) was structured according to sex and age (Fig. 1, vertical boxes). New susceptible individuals enter the youngest age group in the population (at the rate π for each sex). Uninfected individuals may become vaccinated (at the rate V) and enter the population, SV, of susceptible vaccinated individuals or may become infected with a wild-type or vaccine escape mutant virus (at rates λWT and λE, respectively) and enter the population I or IE, respectively, or may die at their demographically determined sex- and age-specific mortality rates (μs,a) (Berkeley Mortality Database [http://demog.berkeley.edu/wilmoth/mortality]). Individuals infected with wild-type virus (I) were structured according to sex, age, and time since they were infected (in yearly increments) (Fig. 1), and they progress through 1-year categories of duration of infection (at rate p), with empirically derived increases in viral loads and rates of HIV-associated (Ds,a,d [7]) and natural (μs,a [Berkeley Mortality Database]) mortality (24). Vaccinated susceptible individuals (SV) were structured according to sex, age (Fig. 1, vertical axis), and level of protection from vaccination (Fig. 1, horizontal axis). The level of protection in the vaccinated population was defined by the reduction in viral load (compared to unvaccinated individuals) that these individuals would experience after infection. Since it is assumed that vaccine-induced protection wanes with time (at the rate ω), individuals pass through progressively lower levels of protection until all protection is lost and they return to the unvaccinated population (S). Vaccinated individuals may become infected at a higher rate than unvaccinated individuals due to the potential effects of vaccination on increasing high-risk sexual activity (R). Vaccinated individuals who become infected move into different stages of the vaccinated infected population (IV) at the disease progression rate pV. The vaccinated infected population is divided into those with low viral loads and mortality rates (2, 39) due to vaccination (IV1) (Fig. 1, light gray boxes) and those who have progressed to have viral loads and mortality rates equivalent to those seen for natural infections (IV2) (Fig. 1, dark gray boxes). Only those who were recently vaccinated at the time of infection will attain the maximum protection from vaccination (and enter the lowest category of IV1). Those with waning vaccine-derived immunity will experience lower levels of protection with time.
FIG. 1.

Outline of the model. The schematic illustration of the model indicates the different compartments, movement between compartments, and the dynamics of aging and disease progression within compartments (see Appendix). As indicated on the right, squares indicate age classes. (See Materials and Methods for detailed description.)
The transmission of CTL escape mutant viruses has recently been documented (27). This has important implications for the long-term efficacy of a vaccine. If the transmission of viral escape mutants to another vaccinated individual occurs, then the vaccine-induced immune response may be unable to recognize the mutant virus, blunting the effect of the vaccine. However, the viral epitopes that are recognized by CTLs vary between individuals (largely on the basis of HLA restriction [40]). Infection of an HLA-mismatched (recipient) partner with an escape mutant restricted to an irrelevant (donor) HLA allele will often not affect CTL recognition of the virus. Thus, since most sexual partners will not share HLA alleles (and therefore CTL epitopes), only a fraction of transmissions (HE = 5 to 50%) from individuals infected with escape mutants will result in transmission of the escape mutant and evasion of the vaccine-induced immune response. The remainder of cases will result in the transmission of a virus that is mutated at an irrelevant CTL epitope and is therefore effectively a wild-type virus that can be controlled by the vaccine-induced immune response of the recipient.
Some vaccinated infected individuals (IV) may become “superinfected” with an escape mutant virus (at the rate RλE) or develop an escape mutant virus through mutation (at the rate ɛ) and therefore move from their current category (IV1 or IV2) into an equivalent category of infection with an escape mutant virus (IE1 or IE2). Individuals who are newly infected with an escape mutant virus (IE) are assumed to have similar mortality rates and disease progression to individuals infected with wild-type virus, and disease is assumed to progress at a normal rate. Individuals infected with an escape mutant virus can thus transmit either the mutant virus (with the probability HE) or wild-type virus (at the rate HWT [1 − HE]) (27).
Using this age and time-since-infection structured model based on population data, we were able to capture the effects of reducing the viral load and/or disease progression rate on mortality and transmission. We were also able to take into account factors such as the duration of vaccine-mediated immunity and the rate of viral escape. A vaccine that slows disease progression, for example, leads to advanced disease and high viral loads being delayed until an older age, that is, the high virological infectiousness experienced in late disease is delayed until the patient is older and has low behavioral infectiousness. On the other hand, viral-load-reducing vaccines lead to lower virological transmission early after infection (Fig. 2).
FIG. 2.

Effects of disease-modifying vaccine. After infection in an unvaccinated individual, both the viral load and mortality rise with time (solid line). Disease-modifying vaccines may act primarily by reducing the viral load after infection (dashed line) or by reducing disease progression (dashed-dotted line). A lower viral load in vaccinated individuals compared to unvaccinated individuals leads to reduced transmission (since transmission is dependent on the viral load) (top) and reduced mortality (bottom). If disease progresses at a normal rate, then viral loads and mortality rise at the same rate as in unvaccinated individuals, but starting at a lower baseline level (dashed line). Therefore, viral loads and mortality in a vaccinated individual will eventually rise to be equivalent to those seen in early natural infections, after a delay approximately equal to the reduction in viral load divided by the annual increase in viral load (0.09 log10 copies ml−1 year−1). If vaccination slows disease progression, then the viral load and mortality may be stable or rise slowly (dashed-dotted line). The reduction in viral load induced by vaccination will reduce the virological infectiousness of infected individuals, whereas a delay in the rise in viral load will reduce behavioral infectiousness since people do not achieve high viral loads until they are older and less sexually active.
Previous modeling has suggested that increased sexual risk behavior as a result of optimism over the effectiveness of vaccination (18) or antiretroviral therapy (14, 15) may increase the transmission of HIV. Factors such as increased sexual risk behavior as a result of vaccination can be incorporated into the model by increasing the rate of partner change among vaccinated individuals (18, 37). The extent to which optimism over vaccination may increase risky behavior is unknown. However, sexual behavior in the general population has changed relatively little as a result of HIV. Therefore, in accordance with published studies, we considered that the increase in risky behavior would vary between 0 and 30% (52, 55) (corresponding in the model to a 0 to 30% increase in the effective average partner change rate [Table 1]).
The model was used to investigate the effects of two different possible vaccines, one that predominantly reduced viral load and another that predominantly slowed disease progression. These analyses were initially performed on a baseline population, defined using parameters obtained from the literature (Tables 2 and 3). The initial prevalence of infection was approximately 0.5% of sexually active individuals, consistent with the current prevalence in most regions of the world outside sub-Saharan Africa (53). We performed 1,000 simulations of the effect of vaccination, using different vaccine parameters (Table 1), and then performed a risk analysis and sensitivity analysis (the technique is described in detail in reference 12) of these results.
TABLE 2.
Parameters governing changes in female sexual behavior with age used in the baseline modela
| Age | μ (yr−1) | T | Da* | c | Mixing matrix (%)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 | 55 | 60 | 65 | 70 | |||||
| 15 | 0.0004 | 1 | 0.54 | 4.78 | 60 | 28 | 6 | 3 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 20 | 0.0004 | 0.68 | 0.66 | 4.78 | 3 | 60 | 20 | 8 | 4 | 3 | 2 | 0 | 0 | 0 | 0 | 0 |
| 25 | 0.0006 | 0.68 | 0.81 | 3 | 1 | 9 | 50 | 20 | 10 | 5 | 3 | 2 | 0 | 0 | 0 | 0 |
| 30 | 0.0008 | 0.32 | 1.00 | 3 | 1 | 3 | 6 | 50 | 20 | 10 | 5 | 3 | 2 | 0 | 0 | 0 |
| 35 | 0.0011 | 0.32 | 1.23 | 2.4 | 0 | 2 | 3 | 7 | 50 | 20 | 9 | 5 | 3 | 1 | 0 | 0 |
| 40 | 0.0014 | 0.27 | 1.51 | 2.4 | 0 | 1 | 2 | 3 | 6 | 50 | 20 | 9 | 5 | 3 | 1 | 0 |
| 45 | 0.0022 | 0.27 | 1.85 | 0.37 | 0 | 0 | 1 | 2 | 4 | 8 | 50 | 20 | 8 | 4 | 2 | 1 |
| 50 | 0.0036 | 0.27 | 2.28 | 0.37 | 0 | 0 | 0 | 1 | 2 | 4 | 8 | 50 | 20 | 8 | 4 | 3 |
| 55 | 0.0061 | 0.27 | 2.80 | 1.21 | 0 | 0 | 0 | 0 | 1 | 2 | 4 | 8 | 50 | 20 | 8 | 7 |
| 60 | 0.0099 | 0.2 | 3.44 | 1.21 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 4 | 8 | 50 | 20 | 15 |
| 65 | 0.0161 | 0.2 | 4.22 | 1.21 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 4 | 8 | 50 | 35 |
| 70 | 0.1000 | 0.2 | 5.18 | 1.21 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 4 | 8 | 85 |
μ, age- and sex-specific natural mortality rate (Berkeley Mortality Database). T, relative rate of sexual transmission with age (48). Da, variation in HIV-specific death rate with age (relative to that at age 30) (50). c, age-specific effective average partner change rate (year−1) (13). For the mixing matrix for women, Ms,a1,a2 indicates the percentage of women of age a1 (rows, age indicated on far left) having partnerships with men of age a2 (columns, age indicated at top). The age-specific partner change rate for males was obtained by calculating the number of partners available to males of one age from females of all ages, i.e. population of women of a given age, times the age-specific partner change rate times the proportion of partners of women of age a1 with males of age a2. The mixing martix for males was then derived simply as the proportion of partnerships available to males of different ages, such that the numbers of sexual contacts were balanced between females and males.
TABLE 3.
Parameters governing changes in mortality and transmission with duration of infection used in the baseline modela
| Duration (yrs) | Dd | β |
|---|---|---|
| 1 | 0.003 | 0.082 |
| 2 | 0.011 | 0.068 |
| 3 | 0.017 | 0.074 |
| 4 | 0.028 | 0.080 |
| 5 | 0.043 | 0.087 |
| 6 | 0.058 | 0.094 |
| 7 | 0.068 | 0.102 |
| 8 | 0.085 | 0.110 |
| 9 | 0.099 | 0.120 |
| 10 | 0.106 | 0.130 |
| 11 | 0.141 | 0.141 |
| 12 | 0.147 | 0.152 |
| 13 | 0.170 | 0.165 |
| 14 | 0.194 | 0.179 |
| 15 | 0.216 | 0.194 |
Dd, HIV-associated mortality at different stages of infection (year−1) (7). β, transmission rate at different stages of infection (year−1) (36, 43, 48). The death rate of an infected individual of age a and the duration of infection d are related to both a duration-of-infection specific death rate (Dd) and an age-specific factor (Da). Therefore, HIV-associated mortality (D) is the product of Da and Dd.
To avoid any dependence of the results on particular population variables, we also performed a further analysis to investigate the effects of vaccination in different populations. To do so, we created 100 new “populations” by randomly varying the parameters used. The parameters for each new population were obtained by multiplying each baseline parameter value (i.e., each of the values in Table 2 and 3, independently) by a normally distributed random number with a mean of 1 and a variance of 0.2. Because the growth rate of the epidemic in the baseline population was relatively slow, with a doubling time of approximately 5 years, we also multiplied the transmission rate parameter (β) by a random number between 0.5 and 2, reflecting the possibility that transmission may be higher or lower in different populations. The effects of vaccination in different populations were then tested by simulating 100 vaccination scenarios for each of the 100 new populations. As a further control for our results, since the baseline population was in a relatively low-prevalence setting (0.5%), we also analyzed the impact of initiating vaccination at different stages of the epidemic (i.e., at a prevalence of 0.5, 1, 2, 4, 8, or 16%) in the different populations (100 populations × 100 vaccination scenarios × 6 prevalence rates = 60,000 simulations).
RESULTS
Reducing viral load.
The baseline population model was used to analyze a scenario in which vaccination resulted in a reduction in viral load of 0.5 to 1.5 log10 copies ml−1 after infection compared to unvaccinated controls. In order to test the effects of viral load reduction alone, we adopted the assumption that vaccination would have only minor effects on the normal rate of disease progression (80 to 120% of the normal rate) associated with a given viral load (39). This is referred to as a viral-load-reducing vaccine. Thus, a vaccinated individual who becomes infected will have a lower initial viral load than an unvaccinated individual, but their viral loads will increase from this level at approximately the same rate (0.09 log10 copies ml−1 year−1) (36, 43) (Fig. 2). This imposes a delay (approximately equal to the viral load reduction/0.09 years) until the viral load in the vaccinated individual reaches the level initially observed in the unvaccinated individual and thus increases survival by the same amount (Fig. 2). Because of the uncertainty in these and other effects of the vaccine, a simulation approach was used in which the value of each of the vaccination parameters was randomly assigned at the start of a simulation from within a range estimated from the literature (12) (Table 1). Without vaccination, the outcome of the model was exponential growth of the HIV incidence and mortality over the 25 years of the simulations (data not shown). By performing a large number of simulations, we obtained both a probability distribution of outcomes and an assessment of how sensitive the outcome is to changes in individual parameters. One thousand simulations with vaccination were performed, and the results were compared with those for no vaccination (Fig. 3) to quantify how many deaths or infections were averted by vaccination.
FIG. 3.

Predicting the effects of disease-modifying vaccination. (A) One thousand simulations were performed for each of two cases: (i) a population given a viral-load-reducing vaccine that reduced the viral load between 0.5 and 1.5 log10 copies ml−1 while the progression rate varied between 80 and 120% of normal (left) and (ii) a population given a progression-slowing vaccine that reduced the viral load between 0.1 and 0.5 log10 copies ml−1 and the disease progression rate from 0 to 80% of normal (right). The means (⧫), medians (solid bars), 25th and 75th percentiles (open bars), and outliers (lines) of the proportions of deaths averted (top) and infections averted (bottom) compared to the unvaccinated control are shown. Shaded areas indicate those simulations where more deaths and infections occurred with vaccination than without. Thus, the number of simulations in the shaded areas divided by the total number of simulations gives the fraction of simulations with worse outcome with vaccination; these numbers are quoted in the text. (B) The proportion of vaccinated individuals (top) and proportion of infections involving a vaccine escape mutant virus (bottom) at different times after the commencement of vaccination with the viral-load-reducing vaccine used for panel A.
For the baseline population, the mean number of deaths averted by vaccination reached 10.7% by 10 years and 35.2% by 25 years (Fig. 3). The number of deaths did not increase in any of the vaccination scenarios. Vaccination also averted a mean of 8.4% of new infections by 10 years and 26.7% of new infections by 25 years. However, HIV incidence rose in the first few years compared to simulations without vaccination (by 1% at 2 years, which is equivalent to −1% of the incidence averted) (Fig. 3) before commencing a decline in most scenarios. The fraction of simulations in which the cumulative HIV incidence increased compared to simulations with no vaccine dropped from 11.2% at 10 years to 3.5% by 25 years (Fig. 3, shaded areas). Thus, a viral-load-reducing vaccine has the potential to reduce HIV deaths, but it has a small risk of a long-term increase in HIV incidence.
The rise in new infections in the first few years after the introduction of vaccination, even in simulations that later led to a large decrease in HIV incidence, provides an interesting paradox. Why would an otherwise successful vaccine increase infection in the short term? The answer clearly lies in the increase in risky sexual behavior accompanying vaccination. The vaccine neither prevents infection nor affects individuals who are already infected. Therefore, those who were infected before vaccination was introduced are just as infectious, but vaccinated individuals become more susceptible if they increase their sexual risk behavior. Because early after the introduction of the vaccine in simulations, the majority of infected individuals were infected prior to vaccination, this leads to an overall increase in transmission. At later time points, however, many of the infected individuals were vaccinated prior to infection and thus had low viral loads and a lower rate of transmission. Provided that the reduction in viral load is at least 1.0 log10 copies ml−1, the reduced transmission due to vaccination would be sufficient to counterbalance a rise in risk behavior of up to 30% (Fig. 4).
FIG. 4.

Sensitivity analysis. The relationships between the viral load reductions (left), increases in sexually risky behavior (center), and proportions of individuals vaccinated at 10 years (right) and HIV mortality and incidence at 25 years for the viral-load-reducing (A) and progression-slowing (B) vaccines are shown. Each dot represents the outcome of one simulation with parameters chosen at random from the range given in Table 1. The results for 1,000 simulations of each scenario are shown. Cumulative incidence and mortality data are expressed as percentages of the unvaccinated control values (therefore, values of >100% indicate an increase in cumulative HIV incidence as a result of vaccination). The proportion of individuals vaccinated varied with the vaccination rate and the rate of loss of vaccine protection.
We investigated the sensitivity of the outcome to different parameter assumptions in the model. The results of this sensitivity analysis on HIV mortality and incidence at 25 years are summarized in Fig. 4 and Table 4. The proportion of individuals with vaccine protection, which is largely a function of the rate of vaccination and the duration of vaccine-induced immunity, varied from approximately 38 to 79% (Fig. 3B). Because vaccination reduced viral loads and thus the mortality rate, the proportion of the population with vaccine protection strongly influenced the mortality rate in the entire population (Fig. 4). Thus, both the vaccination rate and the rate of loss of vaccine protection were key factors in predicting the outcome. The level of viral load reduction and the increase in high-risk sexual behavior also had a strong influence on the outcome. Unexpectedly, neither the rate of viral escape nor the rate of transmission of escape variants was a key factor in determining incidence or mortality in the viral-load-reducing scenario. In addition, the rate of disease progression also appeared to have little effect on either incidence or mortality (Table 4).
TABLE 4.
Correlation between HIV incidence and mortality and model parametersa
| Parameter | Partial rank correlation coefficient
|
|||
|---|---|---|---|---|
| Viral-load-reducing vaccine
|
Progression-slowing vaccine
|
|||
| Incidence | Mortality | Incidence | Mortality | |
| Viral load drop (log10) | −0.958 | −0.965 | −0.920 | −0.926 |
| Loss of vaccine effectiveness (ω) | −0.589 | −0.739 | −0.161 | −0.392 |
| Progression rate of vaccinees (pv) | 0.176 | 0.204 | 0.440 | 0.525 |
| Viral escape rate (ɛ) | 0.093 | 0.109 | 0.539 | 0.655 |
| Increase in risky behavior | 0.928 | 0.809 | 0.979 | 0.928 |
| Transmission of escape (HE) | 0.469 | 0.554 | 0.439 | 0.455 |
| Vaccination rate (V) | −0.740 | −0.830 | −0.239 | −0.502 |
Parameter values for each simulation were chosen from a uniform probability distribution with the ranges shown in Table 1. The columns indicate the partial rank correlation coefficients of the model parameters with the outcome measures of mortality and incidence at 25 years for the two vaccination scenarios. There was no significant correlation between any of the individual parameters. The key factors affecting outcome are defined as those with a partial rank correlation coefficient of >0.5 (14).
Slowing disease progression.
The results presented above predict the effects of a vaccine that reduces viral loads by 0.5 to 1.5 log10 copies ml−1, without a significant change in the disease progression rate. We then asked whether a vaccine that reduces the viral load by <0.5 log10 copies ml−1 could reduce HIV incidence and mortality if it were accompanied by a significant reduction in disease progression. This question is interesting because it is currently unclear whether the reduced viral load seen when vaccinated monkeys are infected is stable or increases slowly (1, 11, 22, 51). The model was thus used to consider the effects of a progression-slowing vaccine that had a minimal effect on viral load (reducing viral loads by only 0.1 to 0.5 log10 copies ml−1) but a significant effect on reducing the disease progression rate (pV is 0 to 80% of the normal rate). Thus, the viral load and mortality rate for vaccinated infected individuals would increase more slowly than those for unvaccinated individuals (or not at all, in the case of pV = 0).
The mean number of deaths averted by the introduction of a progression-slowing vaccine increased from 6.4% at 10 years to 12.4% at 25 years. However, the number of deaths actually increased in 10.7% of the simulations at 25 years, suggesting an important risk of a rise in HIV-associated deaths as a result of vaccination (Fig. 3). Vaccination resulted in an increase in HIV incidence early after the introduction of vaccination, with a rise in incidence of 0.6% at 10 years. However, by year 25, vaccination reduced the mean incidence 3% compared to simulations with no vaccination. Thus, despite decreasing HIV-associated mortality in the short term, a progression-slowing vaccine has a high risk of increasing the number of infected individuals.
A sensitivity analysis suggested that the disease progression rate again had relatively little effect on either mortality or incidence at 25 years (Fig. 4; Table 4). The level of reduction in viral load and the increase in risky sexual behavior were again the key factors in determining HIV-associated mortality. However, the proportion of the population that was protected had a variable influence on the number of new infections that were prevented. In scenarios in which there were large increases in risky behavior associated with vaccination and in which vaccination only mildly reduced transmission, the higher the proportion of the population vaccinated, the higher the average rate of risky behavior and the higher the rate of transmission. Therefore, if increased risky sexual behavior after vaccination leads to an increase in HIV incidence, simply increasing the vaccination rate will not correct this and will instead lead to an increase in incidence. Thus, unless risky behavior can be controlled, there is a high risk that although vaccination may increase the survival of vaccinated individuals, it may lead to a long-term increase in HIV incidence and mortality.
Impact of vaccine in different populations.
In order to test the robustness of the conclusions presented above and to test whether these effects are likely to be seen across different populations, we randomly varied the baseline population parameters (as described in Materials and Methods) in order to generate 100 new populations. (Eleven of the populations were excluded from the analysis because the epidemics failed to reach a prevalence of 0.5% within 50 years of simulation.) We then simulated 100 viral-load-reducing vaccination scenarios (varying the vaccination parameters as in the simulations above) for each of these populations and assessed the impact of vaccination on HIV incidence and mortality at 25 years.
Figure 5 shows the mean proportions of deaths and infections averted at 10 (A) and 25 years (B) after the introduction of vaccination in different populations. The proportion of deaths averted and the proportion of infections averted were strongly dependent on the growth rate of the epidemic. A larger impact of a vaccine on a faster growing epidemic is to be expected. Since epidemic growth is nonlinear, any vaccine effects on the epidemic growth rate will have a larger impact on the overall number of deaths and new infections when the growth rate itself is high. The more slowly growing the epidemic, the longer it takes for the vaccine to affect the outcome. Thus, if we measure outcome at a fixed time (e.g., 10 years), the vaccine will have had less of an effect on a slowly growing epidemic than on a rapidly growing one. However, in extremely rapidly growing epidemics (doubling times of <2 to 3 years), the early benefits of vaccination in reducing HIV incidence (seen at 10 years) are reduced by 25 years. This occurs because although vaccination delays the peak of the epidemic, in very rapidly growing epidemics the peak in the vaccinated population will still occur within 25 years. The observed effects of vaccination in preventing new infections will not be as large as those seen in populations in which vaccination delays the peak of the epidemic beyond 25 years. Thus, the impact of vaccination is dependent on the timescale of our observation: although vaccination may slow the epidemic, it may not necessarily reduce the final size of it substantially (54).
FIG. 5.

Effects of vaccination on different populations. One hundred new populations were created by randomly varying the baseline population parameters, and the effects of vaccination were assessed by simulating 100 vaccination scenarios in each new population introduced at a 0.5% prevalence (as described in Materials and Methods). The average percentages of deaths (top) and infections (bottom) averted due to vaccination after 10 years (A) and 25 years (B) were plotted against the doubling times of the epidemics immediately prior to the introduction of vaccination. Each point represents the results for a different population. To investigate the effects of initial HIV prevalence, we studied vaccination introduced at an initial HIV prevalence of 0.5, 1, 2, 4, 8, or 16% (colored as indicated) (C). The average proportions of deaths (top) and infections (bottom) averted for each population at each initial HIV prevalence were plotted against the doubling time of the epidemic immediately prior to the introduction of vaccination.
Next, we studied the effect of different epidemic dynamics and the epidemic stage on the benefits of vaccination. For each of the 100 populations, we let the epidemic develop without vaccination until the infection prevalence reached 1, 2, 4, 8, or 16%. For each population, the time to reach these rates of prevalence was different because the epidemics had different growth rates (Fig. 5C). We thus could analyze the impact of vaccination on epidemics characterized by different prevalence and growth rates. At any given growth rate, the lower the initial prevalence, the larger the impact of a vaccine in terms of infections and deaths averted (that is, early intervention is advantageous). On the other hand, if epidemics with the same initial prevalence grow at different rates, there will be an epidemic with a specific growth rate for which the impact of the vaccine is maximal. For example, at an initial prevalence of 0.5%, the maximum proportion of infections averted occurs with a doubling time of about 3 to 4 years. In contrast, at an initial prevalence of 16%, the maximum proportion of infections averted is seen with doubling times of more than ∼7 years.
The mean number of deaths averted by vaccination at 25 years was more than ∼10% for all populations, regardless of the growth rate or initial prevalence of infection. However, an increased number of deaths was seen in a small number of vaccination scenarios, indicating a risk of increased HIV infection with vaccination in some epidemics. If we restricted the analysis to vaccination scenarios in which the drop in viral load after vaccination was at least 1 log10 copies ml−1, then there was a reduction in deaths in all of the populations and under all of the vaccination conditions considered. Thus, provided a vaccine can reduce the viral load of vaccinated infected individuals by at least 1 log compared with that of unvaccinated individuals, we believe there is an extremely low risk that it could increase HIV-associated mortality within the first 25 years.
DISCUSSION
Recent studies of HIV vaccination in monkeys suggest that future HIV vaccines may be unable to prevent infection but may improve the subsequent course of disease. We use the term disease modifying to describe these vaccines and to distinguish them from previous discussions of partially effective vaccines. Partially effective vaccines were assumed to prevent infection, but only in a proportion of vaccinated individuals (6, 19). In contrast, disease-modifying vaccines do not prevent initial infection but simply alter the subsequent course of infection. Such disease-modifying vaccines have not previously been used, and their implications for the spread of an epidemic are unclear. We employed a model of HIV epidemic spread that incorporated both intrahost and population dynamics to investigate the effects of a disease-modifying vaccine for HIV. Mathematical modeling involves a necessary simplification of the complexities of disease transmission. Thus, although our model is structured to account for the duration of infection and age, it does not account for the impact of high-risk groups such as intravenous drug users, commercial sex workers, or other core groups within a population. However, the model aims to guide rational vaccine design and development by addressing whether a disease-attenuating vaccine has the potential to reduce the burden of disease, what features of the vaccine are most likely to affect this, and under what circumstances the vaccine would be most effective.
The results of an uncertainty and sensitivity analysis suggest that the reduction in viral load in vaccinated infected individuals (compared to unvaccinated individuals) is the key factor to measure in attempting to predict the likely outcome of vaccination. Surprisingly, according to our model the extent to which a vaccine slows disease progression and the rate of immunological escape of vaccine-induced immune responses have relatively little effect on the long-term outcome. The level of increase in sexually risky behavior as a result of optimism over vaccination also correlated strongly with the epidemic outcome, as has been observed for antiretroviral therapy (25; Kellogg et al., letter) and predicted previously in other models (15, 18, 33). It is interesting to speculate that a disease-modifying vaccine, which does not prevent infection, may well induce less optimism and less increase in risky sexual behavior than a vaccine that promises protection from infection.
The baseline scenario is a low-prevalence epidemic (0.5%) that is growing relatively slowly (doubling time, ∼5 years). In order to investigate whether these results may be applicable to other populations, we also considered the impact of vaccination on different simulated populations, with vaccination being introduced at different stages of the epidemic. These results indicate that both the growth rate of the epidemic and the stage of the epidemic (in terms of the initial prevalence of infection) can influence the impact of vaccination. In general, vaccination has more influence on more rapidly growing epidemics and when it is introduced earlier in the course of the epidemic. However, the impact of vaccination also varies according to the time frame of analysis, i.e., the number of new infections may increase in the first few years after the introduction of vaccination (Fig. 3). Similarly, in a very fast growing epidemic, the maximal impact of vaccination may occur at about 10 years, with the subsequent effects of vaccination declining over time (Fig. 5) (45, 54). When considering the impact of a vaccine on different populations, the growth rate of the epidemic, the current prevalence of HIV, and the timescale over which we wish to assess the results are all important factors to consider. Disease-modifying vaccines would have a maximum effect if they were introduced into relatively early-stage epidemics (such as those seen in areas of Asia) but a smaller impact if they were introduced into late-stage, high-prevalence epidemics (such as those seen in areas of sub-Saharan Africa). However, our results demonstrate that if vaccination is able to reduce the viral load of vaccinated infected individuals by at least 1 log10 copies ml−1, then the number of HIV-associated deaths is predicted to be reduced at 25 years for all of the scenarios considered.
Importantly, the model also suggests that the success of any human vaccine trial should not be judged solely on short-term measures of how many infections were prevented in the vaccinated group. Indeed, it is likely that disease-modifying vaccines will lead to a short term (1 to 5 years) increase in HIV incidence if they lead to increased high-risk sexual activity. However, this does not preclude long-term benefits. Thus, rather than simply measuring HIV incidence in vaccinees and controls, vaccine trials should also aim to closely monitor viral loads of infected individuals in order to quantitate any reduction in viral load in vaccinated individuals. Campaigns promoting safe sex should also be continued to prevent vaccine-related increases in risky sexual behavior. The close monitoring of large numbers of patients may be necessary to measure the average reduction in viral load or to detect a slowing of disease progression in vaccinated individuals compared to controls. However, it is the reduction in viral load that appears to be the most important feature of a disease-modifying vaccine for predicting its ability to control the spread of HIV. The model suggests that reductions in viral load of about 1 log10 copies ml−1 may be sufficient to produce long-term reductions in HIV incidence and mortality. This reduction in viral load is relatively modest compared to that observed in vaccinated monkeys (∼3 log10 copies ml−1) (11, 51).
Acknowledgments
Portions of this work were done under the auspices of the U.S. Department of Energy and were supported under contract W-7405-ENG-36. We also acknowledge support from the James S. McDonnell Foundation 21st Century Research Awards/Studying Complex Systems (M.P.D.) and by NIH grants R01-RR06555 and R37-AI28433 (A.S.P.). R.M.R. was partially funded by a Marie Curie Fellowship of the European Community program “Quality of Life” under contract number QLK2-CT-2002-51691.
We thank Rob de Boer for helpful comments on the manuscript.
APPENDIX
A model of a heterosexual HIV epidemic structured by sex, age, and duration of infection (24) was modified by the incorporation of categories of susceptible vaccinated individuals (SV), infected vaccinated individuals (IV), and individuals infected with escape mutant viruses (IE). The last two categories were further divided into those who are currently benefiting from reduced viral loads due to vaccination (IV1 and IE1, respectively) and those whose disease has progressed to a stage at which their viral loads are equivalent to those seen in natural infections (IV2 and IE2) (Fig. 1). The mortality and transmission of these last groups were the same as those for an equivalent stage of natural infection (note that here we use stage of infection to denote the time since infection in a corresponding natural infection, not the usual CDC stage classification). The mortality and transmission for the categories of infection with reduced viral loads (IV1 and IE1) were estimated based on their relationship with viral load. That is, transmission was reduced 2.45-fold and mortality was reduced 2.22-fold for every log10 reduction in viral load (2, 39, 48).
The model populations that we studied contained ∼1 million individuals of each sex, with an age structure that was determined by allowing an influx of new susceptible individuals (π) into the youngest age group of 20,000 per year for each sex and by using published figures for natural mortality (Berkeley Mortality Database). The model was run for 200 years to generate a stable age distribution of uninfected individuals and a total population size of ∼2 million. The initial proportion of infected individuals was ∼0.1% of the sexually active population, and the age distribution was similar to that of the infected population of the United States in the early 1990s (49). This population was not chosen to simulate the AIDS epidemic in the United States but rather as a means to generate a baseline population with a well-defined age structure. The epidemic was allowed to expand, and vaccination was introduced when the prevalence reached a specified level (0.5% in the baseline analysis and 0.5, 1, 2, 4, 8, or 16% in the analysis of different populations).
The model was defined by a system of differential equations as outlined below (also see reference 24).
(i) For the unvaccinated susceptible population in the youngest age group,
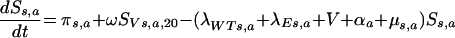 |
and for older age groups,
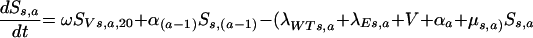 |
That is, new susceptible individuals enter the population at the youngest age group at the rate π or from the vaccinated population through the loss of vaccine protection at the rate ω. Individuals leave the population by aging (αa, which is zero for the oldest age group), vaccination (V), natural death (μs,a), or infection with a wild-type or escape mutant virus (λWT and λE, respectively).
(ii) For the recently vaccinated, uninfected population,
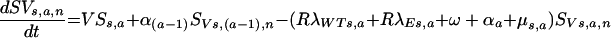 |
Thereafter, vaccinated individuals progress through stages of progressively lower levels of vaccine protection (indicated by the subscript “n”) at the rate ω
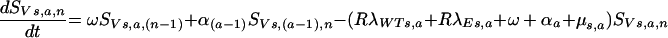 |
until they return to the unvaccinated uninfected population.
(iii) For newly infected unvaccinated individuals infected with wild-type virus,
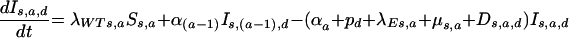 |
and for the progression through time since infection (indicated by the subscript “d”),
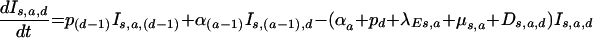 |
where the disease progression rate pd is zero for the most advanced disease stage.
(iv) For newly infected vaccinated individuals,
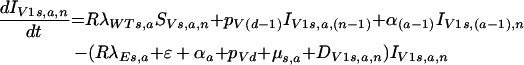 |
where RλWTs,a is the force of infection for vaccinated individuals. Individuals with the protection level n from vaccination enter the infected vaccinated population at the equivalent level and then progress through stages of infection at the rate pV.
Once viral loads rise to values equal to those seen after natural infections, individuals enter the category IV2:
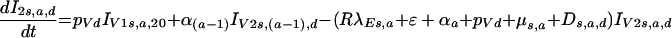 |
for the earliest category and thereafter progress through subsequent stages of infection, as follows:
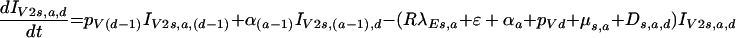 |
(v) For the vaccinated population infected with escape mutant viruses at early stages of infection (IE1),
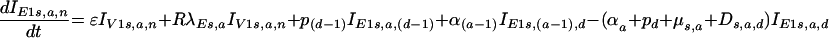 |
and once viral loads rise to values equal to those seen after natural infections (IE2),
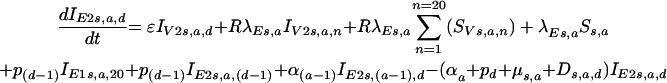 |
for the first category, and thereafter,
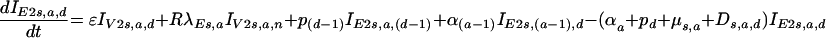 |
where the subscripts s and a correspond to gender and age, respectively, and n and d correspond to the duration of infection for those with lower than normal viral loads and normal viral loads, respectively.
The parameters are shown in Fig. 1 and explained in the text. The per capita forces of infection of wild-type and escape mutant viruses (λWT and λE) were calculated in two steps (24). First, the infectiousness of the population of age a and sex s was calculated, for both wild-type and escape mutant viruses (LWTs,a and LEs,a, respectively), as follows:
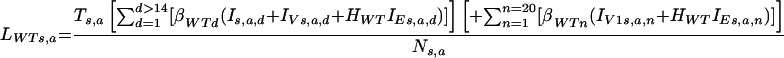 |
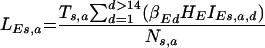 |
where we have taken into consideration the number of infected individuals in each category and their virological infectiousness (β), age-specific transmission rate (T) (48), and the probability that an individual infected with an escape mutant virus will transmit either a wild-type virus (HWT) or a virus that escapes the vaccine-induced immune response in the recipient (HE). We then calculated the force of infection for a given sex and age group (λWTs,a, λEs,a) by using the following formulas:
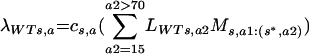 |
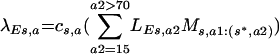 |
where the empirical effective average rate of partner change is c (13, 34) and the rate of sexual mixing between age groups is given by the variable Ms,a1:(s*,a2), the mixing of people of age a1 and sex s with people of age a2 and sex s* (8, 23) This force of infection was then multiplied by the increase in high-risk sexual activity of vaccinated individuals (R) in the vaccinated population (see the text).
REFERENCES
- 1.Amara, R. R., F. Villinger, J. D. Altman, S. L. Lydy, S. P. O'Neil, S. I. Staprans, D. C. Montefiori, Y. Xu, J. G. Herndon, L. S. Wyatt, M. A. Candido, N. L. Kozyr, P. L. Earl, J. M. Smith, H. L. Ma, B. D. Grimm, M. L. Hulsey, J. Miller, H. M. McClure, J. M. McNicholl, B. Moss, and H. L. Robinson. 2001. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292:69-74. [DOI] [PubMed] [Google Scholar]
- 2.Anastos, K., L. A. Kalish, N. Hessol, B. Weiser, S. Melnick, D. Burns, R. Delapenha, J. DeHoVitz, M. Cohen, W. Meyer, J. Bremer, and A. Kovacs. 1999. The relative value of CD4 cell count and quantitative HIV-1 RNA in predicting survival in HIV-1-infected women: results of the women's interagency HIV study. AIDS 13:1717-1726. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, R. M., and G. P. Garnett. 1996. Low-efficacy HIV vaccines: potential for community-based intervention programmes. Lancet 348:1010-1013. [DOI] [PubMed] [Google Scholar]
- 4.Anderson, R. M., S. Gupta, and R. M. May. 1991. Potential of community-wide chemotherapy or immunotherapy to control the spread of HIV-1. Nature 350:356-359. [DOI] [PubMed] [Google Scholar]
- 5.Anderson, R. M., and R. M. May. 1991. Infectious diseases of humans. Oxford University Press, Oxford, United Kingdom.
- 6.Anderson, R. M., J. Swinton, and G. P. Garnett. 1995. Potential impact of low efficacy HIV-1 vaccines in populations with high rates of infection. Proc. R. Soc. Lond. B Biol. Sci. 261:147-151. [DOI] [PubMed] [Google Scholar]
- 7.Anonymous. 2000. Time from HIV-1 seroconversion to AIDS and death before widespread use of highly-active antiretroviral therapy: a collaborative re-analysis. Lancet 355:1131-1137. [PubMed] [Google Scholar]
- 8.Aral, S. O., J. P. Hughes, B. Stoner, W. Whittington, H. H. Handsfield, R. M. Anderson, and K. K. Holmes. 1999. Sexual mixing patterns in the spread of gonococcal and chlamydial infections. Am. J. Public Health 89:825-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barouch, D. H., J. Kunstman, J. Glowczwskie, K. J. Kunstman, M. A. Egan, F. W. Peyerl, S. Santra, M. J. Kuroda, J. E. Schmitz, K. Beaudry, G. R. Krivulka, M. A. Lifton, D. A. Gorgone, S. M. Wolinsky, and N. L. Letvin. 2003. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J. Virol. 77:7367-7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barouch, D. H., J. Kunstman, M. J. Kuroda, J. E. Schmitz, S. Santra, F. W. Peyerl, G. R. Krivulka, K. Beaudry, M. A. Lifton, D. A. Gorgone, D. C. Montefiori, M. G. Lewis, S. M. Wolinsky, and N. L. Letvin. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415:335-339. [DOI] [PubMed] [Google Scholar]
- 11.Barouch, D. H., S. Santra, J. E. Schmitz, M. J. Kuroda, T. M. Fu, W. Wagner, M. Bilska, A. Craiu, X. X. Zheng, G. R. Krivulka, K. Beaudry, M. A. Lifton, C. E. Nickerson, W. L. Trigona, K. Punt, D. C. Freed, L. M. Guan, S. Dubey, D. Casimiro, A. Simon, M. E. Davies, M. Chastain, T. B. Strom, R. S. Gelman, D. C. Montefiori, et al. 2000. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 290:486-492. [DOI] [PubMed] [Google Scholar]
- 12.Blower, S., and H. Dowlatabadi. 1994. Sensitivity and uncertainty analysis of complex models of disease transmission: an HIV model, as an example. Int. Stat. Rev. 62:229-243. [Google Scholar]
- 13.Blower, S. M. 1993. Exploratory data analysis of three sexual behaviour surveys: implications for HIV-1 transmission in the U.K. Philos. Trans. R. Soc. Lond. B Biol. Sci. 339:33-51. [DOI] [PubMed] [Google Scholar]
- 14.Blower, S. M., A. N. Aschenbach, H. B. Gershengorn, and J. O. Kahn. 2001. Predicting the unpredictable: transmission of drug-resistant HIV. Nat. Med. 7:1016-1020. [DOI] [PubMed] [Google Scholar]
- 15.Blower, S. M., H. B. Gershengorn, and R. M. Grant. 2000. A tale of two futures: HIV and antiretroviral therapy in San Francisco. Science 287:650-654. [DOI] [PubMed] [Google Scholar]
- 16.Blower, S. M., K. Koelle, D. E. Kirschner, and J. Mills. 2001. Live attenuated HIV vaccines: predicting the tradeoff between efficacy and safety. Proc. Natl. Acad. Sci. USA 98:3618-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blower, S. M., and A. R. McLean. 1991. Mixing ecology and epidemiology. Proc. R. Soc. Lond. B Biol. Sci. 245:187-192. [DOI] [PubMed] [Google Scholar]
- 18.Blower, S. M., and A. R. McLean. 1994. Prophylactic vaccines, risk behavior change, and the probability of eradicating HIV in San Francisco. Science 265:1451-1454. [DOI] [PubMed] [Google Scholar]
- 19.Bogard, E., and K. M. Kuntz. 2002. The impact of a partially effective HIV vaccine on a population of intravenous drug users in Bangkok, Thailand: a dynamic model. J. Acquir. Immune Defic. Syndr. 29:132-141. [DOI] [PubMed] [Google Scholar]
- 20.Borrow, P., H. Lewicki, B. H. Hahn, G. M. Shaw, and M. B. Oldstone. 1994. virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68:6103-6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brander, C., and B. D. Walker. 1999. T lymphocyte responses in HIV-1 infection: implications for vaccine development. Curr. Opin. Immunol. 11:451-459. [DOI] [PubMed] [Google Scholar]
- 22.Chen, X., G. Scala, I. Quinto, W. Liu, T. W. Chun, J. S. Justement, O. J. Cohen, T. C. vanCott, M. Iwanicki, M. G. Lewis, J. Greenhouse, T. Barry, D. Venzon, and A. S. Fauci. 2001. Protection of rhesus macaques against disease progression from pathogenic SHIV-89.6PD by vaccination with phage-displayed HIV-1 epitopes. Nat. Med. 7:1225-1231. [DOI] [PubMed] [Google Scholar]
- 23.Darroch, J. E., D. J. Landry, and S. Oslak. 1999. Age differences between sexual partners in the United States. Fam. Plann. Perspect. 31:160-167. [PubMed] [Google Scholar]
- 24.Davenport, M. P., and J. J. Post. 2001. Rapid disease progression and the rate of spread of the HIV epidemic. AIDS 15:2055-2057. [DOI] [PubMed] [Google Scholar]
- 25.Dukers, N., J. Goudsmit, J. B. F. de Wit, M. Prins, G. J. Weverling, and R. A. Coutinho. 2001. Sexual risk behaviour relates to the virological and immunological improvements during highly active antiretroviral therapy in HIV-1 infection. AIDS 15:369-378. [DOI] [PubMed] [Google Scholar]
- 26.Fylkesnes, K., R. M. Musonda, M. Sichone, Z. Ndhlovu, F. Tembo, and M. Monze. 2001. Declining HIV prevalence and risk behaviours in Zambia: evidence from surveillance and population-based surveys. AIDS 15:907-916. [DOI] [PubMed] [Google Scholar]
- 27.Goulder, P. J. R., C. Brander, Y. H. Tang, C. Tremblay, R. A. Colbert, M. M. Addo, E. S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, M. Altfeld, S. Q. He, M. Bunce, R. Funkhouser, S. I. Pelton, S. K. Burchett, K. McIntosh, B. T. M. Korber, and B. D. Walker. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412:334-338. [DOI] [PubMed] [Google Scholar]
- 28.Goulder, P. J. R., R. E. Phillips, R. A. Colbert, S. McAdam, G. Ogg, M. A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, A. J. McMichael, and S. Rowland-Jones. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212-217. [DOI] [PubMed] [Google Scholar]
- 29.Gray, R. H., M. J. Wawer, R. Brookmeyer, N. K. Sewankambo, D. Serwadda, F. Wabwire-Mangen, T. Lutalo, X. B. Li, T. vanCott, and T. C. Quinn. 2001. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet 357:1149-1153. [DOI] [PubMed] [Google Scholar]
- 30.Hammarlund, E., M. W. Lewis, S. G. Hansen, L. I. Strelow, J. A. Nelson, G. J. Sexton, J. M. Hanifin, and M. K. Slifka. 2003. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 9:1131-1137. [DOI] [PubMed] [Google Scholar]
- 31.Kaul, R., S. L. Rowland-Jones, J. Kimani, T. Dong, H. B. Yang, P. Kiama, T. Rostron, E. Njagi, J. J. Bwayo, K. S. MacDonald, A. J. McMichael, and F. A. Plummer. 2001. Late seroconversion in HIV-resistant Nairobi prostitutes despite pre-existing HIV-specific CD8(+) responses. J. Clin. Investig. 107:341-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagarde, E., B. Auvert, M. Carael, M. Laourou, B. Ferry, E. Akam, T. Sukwa, L. Morison, B. Maury, J. Chege, I. N′Doye, and A. Buve. 2001. Concurrent sexual partnerships and HIV prevalence in five urban communities of sub-Saharan Africa. AIDS 15:877-884. [DOI] [PubMed] [Google Scholar]
- 33.Law, M. G., G. Prestage, A. Grulich, P. Van de Ven, and S. Kippax. 2001. Modelling the effect of combination antiretroviral treatments on HIV incidence. AIDS 15:1287-1294. [DOI] [PubMed] [Google Scholar]
- 34.Leigh, B. C., M. T. Temple, and K. F. Trocki. 1993. The sexual behavior of U.S. adults: results from a national survey. Am. J. Public Health 83:1400-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Letvin, N. L., D. H. Barouch, and D. C. Montefiori. 2002. Prospects for vaccine protection against HIV-1 infection and AIDS. Annu. Rev. Immunol. 20:73-99. [DOI] [PubMed] [Google Scholar]
- 36.Lyles, R. H., A. Munoz, T. E. Yamashita, H. Bazmi, R. Detels, C. R. Rinaldo, J. B. Margolick, J. P. Phair, and J. W. Mellors. 2000. Natural history of human immunodeficiency virus type 1 viremia after seroconversion and proximal to AIDS in a large cohort of homosexual men. Multicenter AIDS Cohort Study. J. Infect. Dis. 181:872-880. [DOI] [PubMed] [Google Scholar]
- 37.McLean, A. R., and S. M. Blower. 1993. Imperfect vaccines and herd immunity to HIV. Proc. R. Soc. Lond. B Biol. Sci. 253:9-13. [DOI] [PubMed] [Google Scholar]
- 38.McMichael, A., and T. Hanke. 2002. The quest for an AIDS vaccine: is the CD8 T-cell approach feasible? Nat. Rev. Immunol. 2:283-291. [DOI] [PubMed] [Google Scholar]
- 39.Mellors, J. W., C. R. Rinaldo, Jr., P. Gupta, R. M. White, J. A. Todd, and L. A. Kingsley. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 272:1167-1170. (Erratum, 275:14, 1977.) [DOI] [PubMed] [Google Scholar]
- 40.Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439-1443. [DOI] [PubMed] [Google Scholar]
- 41.Morris, M. 1993. Telling tails explain the discrepancy in sexual partner reports. Nature 365:437-440. [DOI] [PubMed] [Google Scholar]
- 42.Munguti, K., H. Grosskurth, J. Newell, K. Senkoro, F. Mosha, J. Todd, P. Mayaud, A. Gavyole, M. Quigley, and R. Hayes. 1997. Patterns of sexual behaviour in a rural population in north-western Tanzania. Soc. Sci. Med. 44:1553-1561. [DOI] [PubMed] [Google Scholar]
- 43.O'Brien, T. R., P. S. Rosenberg, F. Yellin, and J. J. Goedert. 1998. Longitudinal HIV-1 RNA levels in a cohort of homosexual men. J. Acquir. Immune Defic. Syndr. 18:155-161. [DOI] [PubMed] [Google Scholar]
- 44.Ogg, G. S., X. Jin, S. Bonhoeffer, P. Moss, M. A. Nowak, S. Monard, J. P. Segal, Y. Cao, S. L. Rowland-Jones, A. Hurley, M. Markowitz, D. D. Ho, A. J. McMichael, and D. F. Nixon. 1999. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J. Virol. 73:797-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owens, D. K., D. M. Edwards, and R. D. Shachter. 1998. Population effects of preventive and therapeutic HIV vaccines in early- and late-stage epidemics. AIDS 12:1057-1066. [PubMed] [Google Scholar]
- 46.Phillips, R. E., S. Rowland-Jones, D. F. Nixon, F. M. Gotch, J. P. Edwards, A. O. Ogunlesi, J. G. Elvin, J. A. Rothbard, C. R. M. Bangham, C. R. Rizza, and A. J. McMichael. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453-459. [DOI] [PubMed] [Google Scholar]
- 47.Price, D. A., P. J. R. Goulder, P. Klenerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. M. Bangham, and R. E. Phillips. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94:1890-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinn, T. C., M. J. Wawer, N. Sewankambo, D. Serwadda, C. Li, F. Wabwire-Mangen, M. O. Meehan, T. Lutalo, and R. H. Gray. 2000. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N. Engl. J. Med. 342:921-929. [DOI] [PubMed] [Google Scholar]
- 49.Rosenberg, P. S. 1995. Scope of the AIDS epidemic in the United States. Science 270:1372-1375. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg, P. S., J. J. Goedert, and R. J. Biggar. 1994. Effect of age at seroconversion on the natural AIDS incubation distribution. Multicenter Hemophilia Cohort Study and the International Registry of Seroconverters. AIDS 8:803-810. [DOI] [PubMed] [Google Scholar]
- 51.Shiver, J. W., T. M. Fu, L. Chen, D. R. Casimiro, M. E. Davies, R. K. Evans, Z. Q. Zhang, A. J. Simon, W. L. Trigona, S. A. Dubey, L. Huang, V. A. Harris, R. S. Long, X. Liang, L. Handt, W. A. Schleif, L. Zhu, D. C. Freed, N. V. Persaud, L. Guan, K. S. Punt, A. Tang, M. Chen, K. A. Wilson, K. B. Collins, G. J. Heidecker, V. R. Fernandez, H. C. Perry, J. G. Joyce, K. M. Grimm, J. C. Cook, P. M. Keller, D. S. Kresock, H. Mach, R. D. Troutman, L. A. Isopi, D. M. Williams, Z. Xu, K. E. Bohannon, D. B. Volkin, D. C. Montefiori, A. Miura, G. R. Krivulka, M. A. Lifton, M. J. Kuroda, J. E. Schmitz, N. L. Letvin, M. J. Caulfield, A. J. Bett, R. Youil, D. C. Kaslow, and E. A. Emini. 2002. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 415:331-335. [DOI] [PubMed] [Google Scholar]
- 52.Smith, T. 1994. American sexual behaviour: trends, sociodemographic differences, and risk behaviour. National Opinion Research Centre, Chicago, Ill.
- 53.UNAIDS/W.H.O. 2003. AIDS epidemic update 2003. Joint United Nations Programme on HIV/AIDS (UNAIDS)/World Health Organization, Geneva, Switzerland.
- 54.van Ballegooijen, M., J. A. Bogaards, G. J. Weverling, M. C. Boerlijst, and J. Goudsmit. 2003. AIDS vaccines that allow HIV-1 to infect and escape immunologic control: a mathematic analysis of mass vaccination. J. Acquir. Immune Defic. Syndr. 34:214-220. [DOI] [PubMed] [Google Scholar]
- 55.van der Straten, A., C. A. Gomez, J. Saul, J. Quan, and N. Padian. 2000. Sexual risk behaviors among heterosexual HIV serodiscordant couples in the era of post-exposure prevention and Viral suppressive therapy. AIDS 14:F47-F54. [DOI] [PubMed] [Google Scholar]
- 56.Webb, D. 1997. HIV and AIDS in Africa. Pluto Press, London, United Kingdom.