

To the editor
With the advent of biosimilar version of brand biologics, regulatory authorities in all major jurisdictions throughout the world have developed guidance documents to facilitate their approval1–4. The common theme in the new guidance documents is the stipulation that sponsors must demonstrate biosimilarity between their proposed product and an approved reference product by using state-of-the-art analytical technologies to reduce the size of clinical studies. In the US guidance documents a “totality of evidence” approach is described that can be used to establish the degree of similarity and guide regulatory decision making3, 4.
Higher order structure is an important quality attribute of biosimilars that must be assessed in a thorough comparability exercise. To date, the higher order structure has been evaluated with low-resolution techniques, such as circular dichroism, Fourier transform infrared and Raman spectroscopies, and indirectly with several biological and stability assays5. In 2008, a two dimensional nuclear magnetic resonance (2D-NMR) spectroscopy approach was first applied to the high-resolution assessment of the higher order structure of a native recombinant protein therapeutic6. The technique resolves in a 2D frequency map the positions of proton–nitrogen atom pairs from each amide and amino group in a biologic. Each signal correlates to the specific local chemical and structural environment of the atom pair and, in total, provides a comprehensive read-out of the drug substance conformation along the polypeptide chain at atomic resolution providing a potentially useful tool to establish drug substance consistency across manufacturing changes or comparability of the higher order structure of biosimilars.
In this work, an inter-laboratory comparative study was performed on filgrastim (methionyl granulocyte colony stimulating factor; Met-G-CSF) to demonstrate the precision and robustness of the NMR approach. Filgrastim was selected because of its therapeutic importance, the drug had been characterized by NMR7 and the fact that a number of filgrastim biosimilars have already been approved in Europe, the USA and other jurisdictions. Here, a US approved originator product (Neupogen) and three unapproved, non-US sourced filgrastim products were used in their fully formulated state (Supplementary Table 1) to prepare a set of four samples for analysis using heteronuclear 2D-NMR correlation at 15N natural isotopic abundance. Spectra were acquired on the same samples on six different spectrometers, at four different field strengths ranging from 500 MHz to 900 MHz in four different laboratories (for instrument specifications and experimental parameters, see Supplementary Tables 2–9). All the NMR data were analyzed and evaluated using the same software packages for visual evaluation, chemical shift analysis and principal component analysis (PCA) to assess spectral similarity through multiple approaches.
The inter-laboratory study data show that the resolution of the NMR method allows one to quickly visualize the high degree of similarity of the spectral patterns obtained from the 2D-NMR experiments run on different spectrometers. In this respect, visual comparison of spectral overlays allows an operator to directly assess sample similarity (Fig. 1a, Supplementary Table 10, and Supplementary Figs. 1–4)
Figure 1.
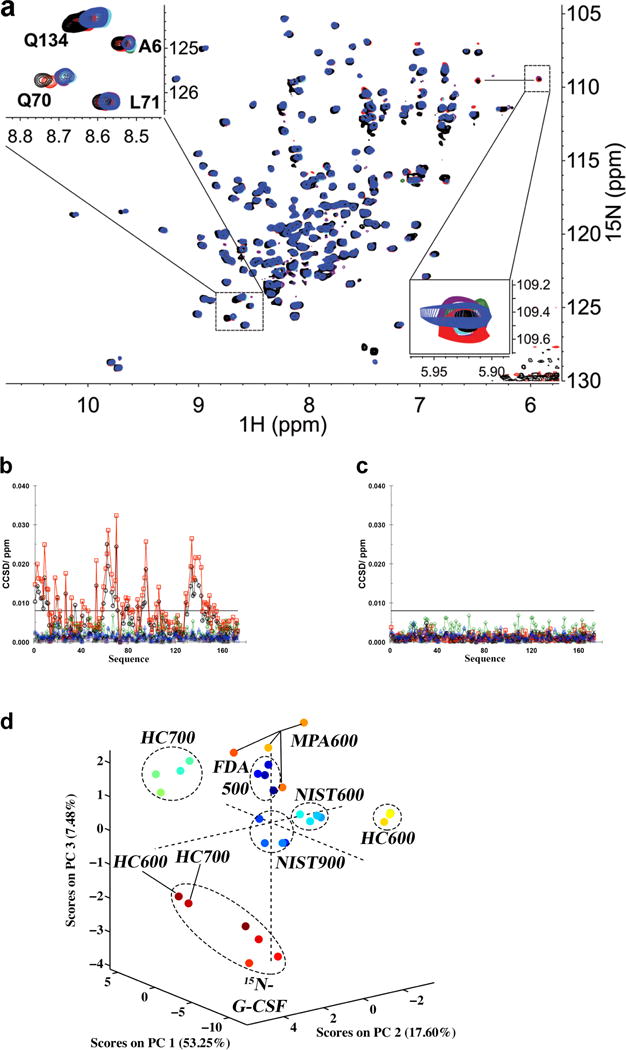
Spectral and Statistical Comparisons of NMR data. (a) Overlay plot of the same region on the same sample from Amgen at four fields and six instruments: NIST 900 (purple), NIST 600 (cyan), HC 700 (black), HC 600 (red), FDA 500 (green) and MPA 600 MHz (blue). Note that all resonances except data from HC 700 and HC 600 are perfectly overlaid under the blue peaks. This is highlighted in the upper left side inset showing an expansion of the regions containing signals from the amides of Q134, Q70, L71 and L6. In the lower right side inset, a weaker peak is plotted using different contours to show that it is observed in all spectra. Additional peaks in the lower right corner of the spectra arise from unsuppressed residual signal from H2O in some spectra. (b) An example of the combined chemical shift difference (CCSD) plots for the system suitability sample, 15N-met-G-CSF, as a function of sequence (NIST 900 (plus), NIST 600 (star), HC 700 (circle), HC 600 (square), FDA 500 (diamond) and MPA 600 (triangle)) with the sample temperature difference on the HC instruments (Extended Data). (c) Same as ‘b’ after temperature calibration. The reference chemical shift was based on average shift from NIST, FDA and MPA. The horizontal bar indicates the measured experimental precision limit of 8 ppb (Extended Data). (d) A plot of the result of the principal component analysis performed on data of the 4 drug products recorded at the 4 laboratories on six instruments, FDA 500 (dark blue); NIST 900 (blue, the cluster at ORIGIN); NIST 600 (turquoise); HC 700 (green); HC 600 (yellow); MPA 600 (orange) (Extended Data). Dashed ellipses show the relative clustering of data from each laboratory. For this panel, the temperatures during collection of HC 600 and HC 700 data were not calibrated.
Differences between two spectral patterns can result from variation in solution conditions (e.g., pH or ionic strength), and/or the temperature of the sample, and/or difference in the higher order structure of the protein. As an example, in our study, the overlay of spectra acquired for the same filgrastim sample on two spectrometers revealed a number of peak signal deviations that could be mapped to solvent exposed residues (Fig. 1a, inset). Four representative residues in loop regions that exhibited these deviations are shown in Supplementary Fig. 2b. We found that these differences could be explained by a difference in temperature of 2°C and 4°C for the HC 700 and HC 600 spectrometers, respectively. The change of temperature does not impact all amide resonances equally7 and has been described by Baxter and Williamson8. Proper instrument calibration removed these discrepancies (Supplementary Fig. 2c).
For a more quantitative analysis, chemical shifts of each individual signal in the 2D map were compared using a root mean square deviation (RMSD) analysis or combined chemical shift difference (CCSD) methods (Fig. 1b,c Supplementary Figs. 5, 6 and Supplementary Tables 11–12; refs 9,10). The chemical shift of each nuclei is an absolute frequency position, and perturbations in the higher order structure, such as altered hydrogen bonding, can impact the local electronic environments of nuclei leading to changes in chemical shifts. Thus, amide 1HN and 15N shifts chemical shift changes can be assessed individually using RMSD from the average values or in aggregate by CCSD (CCSD involves taking a weighted average of the observed changes in 1HN and 15N shift values9). With either of these methods, the chemical shift assignment of the primary sequence allows atomic level mapping of spectral signals to a known protein structure but this is not required for the method to be useful. From RMSD and CCSD analyses, an experimental precision of 8 ppb was determined across the six spectrometers in the four laboratories. Notably, 8 ppb is close to the digital resolution of approximately 5 ppb of an individual spectrum (see Supplementary Methods) which establishes a threshold for the precision of the measurement. Similarly, an intra-laboratory precision of approximately 4 ppb was found that falls well within the digital resolution of a spectrum (Supplementary Table 12), and a related study found an experimental precision on the same sample of 2.4 ppb10. These precision limits are well below any chemical shift changes that could be induced by a significant structural change (e.g., a point mutation, a modification of a residue by oxidation or local conformational change). These approaches also readily identify the temperature deviation of the non-calibrated spectrometers (Fig. 1b), further demonstrating the need for matched experimental conditions if the method is to be used as a structure comparability tool. As expected, the recalibrated spectra were well within the determined experimental precision (Fig. 1c).
Although the comparison of spectral overlays and chemical shifts provides the most straightforward method for the analysis of two or three samples, it can be cumbersome when large numbers of datasets need to be compared, such as the monitoring of lot-to-lot consistency, a multivariate statistical analysis approach may be more appropriate. Here, we used principal component analysis (PCA) as it provides a single readout of variance in all 2D-NMR spectra collected for filgrastim. The PCA plot shown in Figure 1d was calculated with all peaks in the 2D-NMR spectrum with a defined intensity threshold (see Supplementary Methods). This analysis included peak shifts due to sample temperature differences, the presence of impurities (e.g., oxidized species in the system suitability sample) and variation of signal intensities resulting from spectrometer-dependent linewidths. For each spectrometer, except the MPA 600, all drug products are tightly clustered as a result of the high-similarity of the higher order structure. The slightly wider spread of the MPA 600 data is attributed to a difference in the performance of the pulsed NMR method chosen to obtain the 2D map on this spectrometer as well as aging of the samples over time (Supplementary Table 8). NIST 900 data recollected after one year on the same samples confirmed this interpretation (Supplementary Fig. 7). Similar to the chemical shift based analysis, the PCA test clearly distinguishes the two samples with temperature deviation; whereas the 15N-GCSF system suitability sample is found to cluster in its own region mainly because of the differences in protein impurities.
To our knowledge this is the first reported inter-laboratory study of a high-resolution 2D-NMR method to assess biotherapeutic higher order structure. The results clearly demonstrate both the precision and robustness of 2D-NMR as a multi-frequency-based higher order structure assessment tool. The data acquired during this study show minimal measurement drift attributable to the nine months period of the study, and across the various instruments and laboratories. As traceable reference values are established using this technique, a comparability exercise of the higher order structure of a recombinant protein therapeutic can be performed using the NMR signature of a comparator from a validated database without the need for recording its spectra again. Utilization of NMR spectral analyses by both sponsors of biosimilars and originators will provide greater assurance of drug product quality to the regulatory agencies.
Supplementary Material
Footnotes
FDA Disclaimer
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
NIST Disclaimer
Certain commercial equipment, instruments, and materials are identified in this paper in order to specify the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the material or equipment identified is necessarily the best available for the purpose.
References
- 1.Health Canada. GUIDANCE FOR SPONSORS: Information and Submission Requirements for Subsequent Entry Biologics (SEBs) Health Canada -Publications. 2010;17 2010/03/05. [Google Scholar]
- 2.EMEA Comparability of biotechnological/biological products subject to changes in their manufacturing process. CPMP/ICH/5721/03. 2006;13 [Google Scholar]
- 3.US FDA. Guidance for Industry: Quality Considerations in Demonstrating Biosimilarity to a Reference Protein Product. Office of Communications US FDA. 2012:1–20. UCM291134. [Google Scholar]
- 4.US FDA. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Produc. Office of Communications US FDA. 2012:1–25. UCM291128. [Google Scholar]
- 5.Gabrielson JP, Weiss WF. 4th Technical decision-making with higher order structure data: starting a new dialogue. J Pharm Sci. 2015;104:1240–1245. doi: 10.1002/jps.24393. [DOI] [PubMed] [Google Scholar]
- 6.Aubin Y, Gingras G, Sauve S. Assessment of the three-dimensional structure of recombinant protein therapeutics by NMR fingerprinting: demonstration on recombinant human granulocyte macrophage-colony stimulation factor. Anal Chem. 2008;80:2623–2627. doi: 10.1021/ac7026222. [DOI] [PubMed] [Google Scholar]
- 7.Aubin Y, Hodgson DJ, Thach WB, Gingras G, Sauvé S. Monitoring Effects of Excipients, Formulation Parameters and Mutations on the High Order Structure of Filgrastim by NMR. Pharmaceutical Research. 2015 doi: 10.1007/s11095-015-1713-3. [DOI] [PubMed] [Google Scholar]
- 8.Baxter NJ, Williamson MP. Temperature dependence of 1H chemical shifts in proteins. J Biomol NMR. 1997;9:359–369. doi: 10.1023/a:1018334207887. [DOI] [PubMed] [Google Scholar]
- 9.Williamson MP. Using chemical shift perturbation to characterise ligand binding. Progress in Nuclear Magnetic Resonance Spectroscopy. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Arbogast LW, Brinson RG, Marino JP. Mapping monoclonal antibody structure by 2D 13C NMR at natural abundance. Anal Chem. 2015;87:3556–3561. doi: 10.1021/ac504804m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.