

Abstract
Gender influences Papillary Thyroid Cancer (PTC) with an incidence of 3:1 when comparing women to men with different aggressiveness. This gender discrepancy suggests some role of sex hormones in favoring the malignant progression of thyroid tissue to cancer. Estrogens are known to promote Stem Cell self-renewal and, therefore, may be involved in tumor initiation. The goals of these studies are to investigate the underlying causes of gender differences in PTC by studying the specific role of estrogens on tumor cells and their involvement within the Cancer Stem Cell (CSC) compartment. Exposure to 1 nmol l−1 Estradiol for 24 h promotes growth and maintenance of PTC Stem Cells, while inducing dose-dependent cellular proliferation and differentiation following Estradiol administration. Whereas mimicking a condition of hormonal imbalance led to an opposite phenotype compared to a continuous treatment. In vivo we find that Estradiol promotes motility and tumorigenicity of CSCs. Estradiol-treated mice inoculated with Thyroid Cancer Stem Cell-enriched cells developed larger tumor masses than control mice. Furthermore, Estradiol-pretreated Cancer Stem cells migrated to distant organs, while untreated cells remained circumscribed. We also find that the biological response elicited by estrogens on Papillary Thyroid Cancer in women differed from men in pathways mediated. This could explain the gender imbalance in tumor incidence and development and could be useful to develop gender specific treatment of (PTC).
Keywords: Estrogen, Thyroid cancer, Cancer stem cells, Gender medicine, Cancer signaling
1. Introduction
Gender bias occurs across a wide-variety of seemingly unrelated diseases. From a clinical management standpoint, why do various pathologies affect women and men in a differently with respect to incidence, progression and clinical outcomes? This phenomenon could be easily explained for pathologies involving gender specific reproductive organs, but not those disorders originating in organs, such as Thyroid gland, which are common to both. The goal of these studies is to focus on gender differences that impact the thyroid gland with specific focus on cancer.
Thyroid cancer (TC) incidence is on the rise worldwide. In Italy it is the second most common cancer in women, after breast cancer, and the fifth most common in men [8]. Specifically, Papillary Thyroid Cancer (PTC) incidence is three times higher in women compare to men. Moreover, women are more likely to be affected at the beginning of the reproductive age, with a peak between 40 and 49 years, whereas men are affected later in life, around at 60–69 years and have a lower disease-free survival [17]. Whilst the principal causes of TC development, such as nutritional factors (i.e., Iodine uptake), ionized radiation and genetic changes in BRAF, RET, and NTRK, seem not to be involved in this gender discrepancy [17] some studies have reported a correlation between the number of ovulatory cycles, high number of pregnancies, and lactation suppressant and TC incidence [5,25]. Furthermore, other studies have demonstrated that long exposure to exogenous estrogens is associated with the occurrence of TC [1,6,21]. Collectively, these studies suggest a specific role for sex hormones, and in particular for Estrogen, in regulating thyroid function. In recent years different researchers have begun to examine the estrogen role in the development of thyroid pathologies [9,28]. Estrogen is known to be involved in cellular processes such as growth, cell motility and organ function. Consistent with this, different research groups have reported Estrogen in the modulation of TC proliferation and migration [10,12,15,18,23,30]. Estradiol (E2) is the most potent form of estrogen being that it has the highest affinity to its receptors ERα, ERβ, and GPER1 [4,19]. In particular, ERαstimulates proliferation with an anti-apoptosis effect, while ERβ is associated with apoptosis and growth inhibition. For this reason, the ERα/ERβ ratio is helpful to elucidate the TC pathophysiology [13,19]. Studies in mice have demonstrated that circulating estrogens are directly responsible for increased susceptibility of female mice to thyroid disease. Specifically, E2 activate PI3K pathway, inhibit p27, and affect the transcriptional regulation of thyroid genes (i.e., TPO, DUOX1, and NIS) [3]. Despite this and other studies demonstrating a strong direct effect by estrogens on thyroid growth and function, the specific dynamics that move the development and the initiation of proliferative and neoplastic disorders still remains to be clarified.
Recently it has been reported that estrogens are involved in increasing hematopoietic SC self-renewal in female subjects and more specifically during pregnancy [16]. Based on the fetal and CSC carcinogenesis models for the thyroid gland [29], the interaction between SCs and progenitors with hormonal pathways could be potential mechanisms to explain estrogen involvement. Xu and colleagues were among the first to report the estrogenic effect on SCs in thyroid disease isolated from goiter tissue [26]. They observed that thyroid SCs enhance their sphere-forming ability in presence of E2 and showed ERα mRNA levels, eight times higher than those of more differentiated thyrocytes.
The impact on estrogen levels in TC has not been fully explored. Hence, the goal of these studies were to focus on the interaction between estrogen and PTC with specific focus on the SC involvement, in order to contribute to the understanding of hormones mechanism of action on thyroid pathogenesis and its gender differences. In detail, we studied the effect of continuous and unbalanced presence of estrogens on thyroid tissues and their effect on tumor development.
2. Materials and methods
Five-week-old non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice from Charles River Laboratories (Wilmington, Massachusetts) were maintained in accordance to the institutional guidelines of the University of Padova Animal Care Committee.
2.1. Sample collection and cell isolation
TC tissues were obtained at the time of total thyroidectomy from 39 patients affected by PTC (33 women and 6 men, age range 19–74 years, mean age 45 ± 14.8 years), operated at Surgical Clinic 2, University of Padova, in accordance with the ethical standards of the institutional committee on human experimentation. The diagnosis was based on the histological analysis of thyroid specimens determining size, tumor progression, and the involvement of regional lymph nodes. Staging was established according to the UICC TNM classification of malignant tumor [20]. The study protocol was reviewed and approved by the local Ethics Committee (protocol number 448P) and each patient provided a written informed consent.
Tumor tissues were washed 3–5 times in a PBS solution containing Antibiotic-Antimycotic (Euroclone®, Milan, Italy), Penicillin-Streptomycin (Euroclone®, Milan, Italy), Gentamicin (Euroclone®, Milan, Italy), and Metronidazole. An enzymatic and mechanical digestion was performed, using Collagenase 2 (0.15 g/100 mL, Gibco®, Waltham, Massachusetts) and Hyaluronidase (2 mg/100 mL, Sigma Chemicals, Saint Louis, Missouri) in Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma-Aldrich®, Saint Louis, Missouri) in agitation for 25 min at 37 °C.
2.2. Cell cultures and estrogen treatments
Human TC cell lines used in this study are K1 (ECACC, Saint Louis, Missouri) and BCPAP (Leibniz-Institut DSMZ, Braunschweig, Germany) at passage 10, both authenticated by STR DNA typing. To obtain primary cultures enriched in thyroid CSCs, cells were plated in attachment or in ultra-low adhesion in Stem Cell Medium (SCM) composed of Advanced DMEM/F-12 (Gibco®, Waltham, Massachusetts), B-27 Supplement without Vitamin A (Gibco®, Waltham, Massachusetts), N-2 Supplement (Gibco®, Waltham, Massachusetts), N-acetyl-L-cysteine (1 mmol l−1, Sigma-Aldrich®, Saint Louis, Missouri), Nicotinamide (10 mmol l−1, Sigma-Aldrich®, Saint Louis, Missouri), HEPES (10 mmol l−1, Sigma-Aldrich®, Saint Louis, Missouri), L-glutamine (2 mM, Euroclone®, Milan, Italy), Penicillin-Streptomycin (Euroclone®, Milan, Italy), supplemented with 5% (v/v) Fetal Bovine Serum (Euroclone®, Milan, Italy), Recombinant Human EGF (2 ug/100 mL, PeproTech, Rocky Hill, New Jersey), and Recombinant Human FGF-basic (1 ug/100 mL, PeproTech, Rocky Hill, New Jersey). All cell culture was carried out at 37 °C in a 5% (v/v) CO2 humidified incubator.
For estrogen treatment, cells were starved overnight in DMEM/F-12, no phenol red (Gibco®, Waltham, Massachusetts) without serum, and then treated for different 17B-Estradiol (Sigma-Aldrich®, Saint Louis, Missouri) concentrations and administration time, in the same medium supplemented with 5% (v/v) of charcoal stripped ultra centrifugated FBS. The different experimental tests were carried out using Recombinant Human NOGGIN (10 ug/100 mL, R&D systems™; Minneapolis, Minnesota) and FGF2 at different concentrations (1 ug/100 mL and 4 ug/100 mL).
2.3. Flow cytometry and ALDH activity assay
Cells were stained with conjugated OCT3-PE (#561556 PE Mouse monoclonal anti-OCT3, BD, East Rutherford, New Jersey) and SOX2-FITC (#560301 Alexa Fluor® 488 Mouse monoclonal anti-SOX2, BD, East Rutherford, New Jersey), or with primary ERA (NCL-L-ER-6F11; mouse IgG1; Novocastra™, Wetzlar, Germany), ERB (PPG5/10; mouse IgG2a; Dako, Glostrup, Denmark), GPER1 (NBP1-31239; rabbit IgG; Novus Biologicals, Littleton, Colorado), PAX8 (ab13611; goat IgG; abcam®, Cambridge, United Kingdom), TTF1 (NCL-L-TTF-1; mouse IgG1, kappa; Novocastra™, Wetzlar, Germany) antibodies conjugated with anti-mouse or anti-rabbit Alexa Fluor® 488 (Life Technologies™, Carlsbad, California). Alternatively, cells were stained with isotype-matched control. Cells were rinsed and analyzed by flow citometry using BD FACSAria™ 2 (East Rutherford, New Jersey).
Purification of thyroid cells with a high ALDH enzymatic activity was performed using the ALDEFLUOR™ Kit (Stemcell Technologies, Vancouver, Canada). Cells obtained from freshly dissociated TC tissues were re-suspended in the ALDEFLUOR assay buffer containing the ALDH substrate BODIPY®- aminoacetaldehyde (BAAA, 1 umol l−1 ×106 cells) and incubated for 40 min at 37 °C. As negative control, an aliquot of cells of each sample was treated with 50 mmol l−1 of diethylaminobenzaldehyde (DEAB), a specific ALDH inhibitor. The intracellular fluorescent product was measured by flow cytometry.
2.4. Immunofluorescence and histochemistry
For immunofluorescence assay, cells were plated on coverslips and cultured in SCM complemented with 10 ug/100 mL NOGGIN and 1 ug/100 mL FGF2. After treatment, adhesive cells were washed with PBS, fixed with 2% (w/v) paraformaldehyde at 37 °C for 20 min, permeabilized by 0.1% (v/v) Triton X-100 (Sigma-Aldrich, Saint Louis, Missouri) in 0.1% (w/v) sodium citrate for 15 min in ice, and then rinsed with PBS. The coverslips were exposed overnight at 4 °C to antibodies against BETA-CATENIN (H-102; rabbit polyclonal IgG; Santa Cruz Biotechnology, Inc., Dallas, Texas), ERA (NCL-L-ER-6F11; mouse monoclonal IgG1; Novocastra™, Wetzlar, Germany), ERB (PPG5/10; mouse monoclonal IgG2a; Dako, Glostrup, Denmark), NANOG (N-17; goat polyclonal IgG; Santa Cruz Biotechnology, inc., Dallas, Texas), OCT3 (C-10; mouse monoclonal IgG2b; Santa Cruz Biotechnology, Inc., Dallas, Texas), PAX8 (ab13611; goat polyclonal IgG; abcam®, Cambridge, United Kingdom), NIS (SPM186; mouse monoclonal IgG1; abcam®, Cambridge, United Kingdom), SOX2 (09-0024; rabbit polyclonal IgG; Stemgent®, San Diego, California), TG (EPR9730; rabbit monoclonal IgG; abcam®, Cambridge, United Kingdom), TPO (EPR5380; rabbit monoclonal IgG; abcam®, Cambridge, United Kingdom), TSHR (4C1; mouse monoclonal IgG2a; Santa Cruz Biotechnology, Inc., Dallas, Texas), TTF1 (NCL-L-TTF-1; mouse monoclonal IgG1, kappa; Novocastra™, Wetzlar, Germany), or isotype-matched control. Unbound antibodies were removed with a washing buffer composed by PBS with 0.05% (v/v) Tween-20 and 3% (w/v) BSA, followed by incubation with conjugated secondary antibodies (Life Technologies™, Carlsbad, California) plus RNAse (20 mg/100 mL, Sigma-Aldrich, Saint Louis, Missouri). Nuclei counterstaining was performed using Toto-3 iodide at room temperature for 10 min (642/660, Molecular Probes, Invitrogen, Carlsbad, California). A confocal analysis was performed in order to acquire fluorescence imaging.
Histochemistry was performed on 5 um-thick paraffin-embedded sections of thyroid specimens. For H&E staining, slides were stained in hematoxilin for 1 min, water washed, and then exposed to eosin for 30 s. H&E- and Alcian bleu-stained section were dehydrated and mounted in synthetic resin.
2.5. Animals and subcutaneous tumor model
Subcutaneous xenografts were established by the injection of CSC-enriched primary culture cells (1.5 × 106) in immunocompromised NOD/SCID Il2rg-/- female mice, alone or in combination with estrogen therapy (17B-Estradiol SE-121, 1.7 mg/pellet 60-day release for Immunodeficient Mice, Innovative Research of America, Sarasota, Florida). Tumor size was calculated weekly according to the formula: (π/6) × (larger diameter) × (smaller diameter)2.
The migratory potential was evaluated through an intrasplenic injection in NOD/SCID female mice of 3 ×105 luciferase (LUC)/GFP-transduced cells, untreated and treated with 1 nmol l−1 E2 for 24 h. In order to localize and quantify dynamically the optical signal-bioluminescence in a non-invasive localization of the luciferase-marked cell population, D-luciferin (150 mg/kg, Promega, Fitchburg, Massachusetts) was injected (i.p.) at time 0 and up to 6 weeks, 5 min before the bioluminescence analysis. An image detection was performed by Biospace® instrument (San Francisco, California).
2.6. Real-Time PCR
The total RNA extraction and transcription were performed using RNeasy® Mini kit (Qiagen®, Venlo, Netherlands) and RT2 First Strand Kit (Qiagen®, Venlo, Netherlands) following manufacturing instructions. An evaluation of estrogens, cancer and cancer stem cell target genes was performed using RT2 profiler PCR arrays (PAHS-005Z, PAHS-033Z, and PAHS-176Z, Qiagen®, Venlo, Netherlands) with the Rotor-Gene Q (Qiagen®, Venlo, Netherlands). Arrays were run for E2 treated and untreated samples. Cycle thresholds were normalized using ACTB, B2 M, GAPDH, and RPLP0 housekeeping gene, using a threshold of 0.02 in a logarithmic scale for data analyses.
2.7. Statistical and data analysis
Data were expressed as mean value ± standard deviation. Statistical significance was determined using a T-test Student. Results were considered significant, when p values were less than 0.05.
The pathway analysis was performed using the Qiagen® online software (www.qiagen.com/geneglobe). Data were examined with Qiagen card analyzer available at http://pcrdataanalysis.sabio-sciences.com/pcr/arrayanalysis.php. The scatter plot compares the normalized expression of every gene on the array between two groups by plotting them against one another to visualize quickly large gene expression changes. The central line indicates unchanged gene expression. The boundary (fold regulation cut-off) was set at 2.
3. Results
3.1. Thyroid tumor bulk contains stem-like cells
We evaluated ALDH activity in two different PTC cell lines, K1 and BCPAP, in order to validate the presence of thyroid stem-like cells. It has been reported that K1 cells have their origin in the PTC cell line GLAG-66, established from a male patient, while BCPAP derives from a PTC of a woman. We found a different amount of cells harboring the relative fluorescence: 23.6% in K1, 4.87% in BCPAP (Fig. 1A).
Fig. 1.

Thyroid tumor bulk contains stem-like cells. (A) Percentage of ALDHhigh cells within K1 and BCPAP cell lines: 23.6% in K1, 4.87% in BCPAP. (B) Immunofluorescence anatlysis of SOX2, OCT3, BETA-CATENIN, and TSHR on primary cell cultures (zoom 40×; confocal microscope). (C) Optimization of isolation methods. Cell culture maintained in attachment and ultra-low adhesion, in the presence or absence of FBS (magnitude 10×; optical microscope).
Thyroid CSC isolation and enrichment was optimized measuring cell survival on different laboratory plastics in the presence or absence of serum (Fig. 1C). Using ultra-low adhesion and attachment conditions, thyroid cells formed spheres in both culture. We observed that over time, non-adherent cells had a better survival rate in the presence of adherent cells. These results suggest that a complex microenvironment allows the secretion of certain factors needed for their growth. In contrast, under ultra-low conditions in presence of 10% (v/v) of serum, the cells tend to agglomerate to take the form of a primordial follicle, containing a colloidal-like substance. In order to identify the optimal balance between the in vitro niche maintenance and the undifferentiated state preservation, we chose a 5% (v/v) serum medium for the culture conditions in attachment, able to maintain the adherent population without inducing to differentiation of the stem counterpart. We observed a clear expression of SOX2, OCT3, and BETA-CATENIN on these cells, in particular OCT3, confirming their stem characteristic. The thyroid tissue derivation was confirmed by the presence of TSHR (Fig. 1B).
3.2. BMP pathway inhibition acts with FGF-2 in maintenance of stemness in vitro
The previous method was used to isolate CSC-enriched cultures from PTC patients, after enzymatic and mechanical digestion. We carried out the experiments on tissues derived from male and female PTC patients, at early and late stage (Fig. 2A). Primary cell lines enriched in thyrospheres, were able to maintain the complex microenvironment containing both non- and adherent cell populations in culture (Fig. 2B).
Fig. 2.

BMP pathway inhibition acts with FGF2 in maintenance of stemness in vitro. (A) Histological examination of paraffined PTC samples (magnitude 40×, 63×, 40×, respectively; optical microscope). (B) CSC-enriched cell cultures isolated from the same PTC samples, able to maintain the double (adherent and floating) population in culture (magnitude 20×; optical microscope). (C) FACS analysis on CSC-enriched cell cultures for SOX2, OCT3, PAX8, TTF1, ERA, and ERB after treatment with FGF2 and NOGGIN. Conditions: control; 1 ug/100 mL FGF2; 10 ug/100 mL NOGGIN + 1 ug/100 mL FGF2; 10 ug/100 mL NOGGIN + 4 ug/100 mL FGF2. 48 h treatment.
Due to challenges maintaining an undifferentiated state for a prolong time in culture, we incorporated components of media and complements more standardly used for embryonic SCs. Wang et al. found that BMP antagonist NOGGIN and FGF2 were sufficient to keep a prolonged growth of embryonic SCs retaining their features [24]. Based on this and other studies [14], we tested the role of BMP inhibitor and FGF2 on cultured cells, looking for the right balance between undifferentiated state, growth, and thyroid derivation. We treated tumor cells with 1 ug/100 mL FGF2, 10 ug/100 mL NOGGIN + 1 ug/100 mL FGF2, or 10 ug/100 mL NOGGIN + 4 ug/100 mL FGF2. After 48 h, the cells were analyzed at FACS instrument for SC (SOX2 and OCT3) and progenitors (PAX8 and TTF1) markers. We found that the presence of NOGGIN and FGF2 in culture enhances the stem/progenitor cells markers expression, in particular at concentration of 10 ug/100 mL for NOGGIN and 1 ug/100 mL for FGF2 (Fig. 2c). Interesting, the combination of factors that enhances stemness features in vitro, also led to an increase of ERA expression. In other words, it favors aggressiveness leading to a greater ERA/ERB ratio towards growth and anti-apoptosis function. We optimized a culture medium composed of SCM with the addition of 10 ug/100 mL NOGGIN and 1 ug/100 mL FGF2, which was used for all subsequent experiments.
3.3. Estrogen treatment enhances stem markers expression in a time-and dose-dependent manner
To-date, there have been conflicting results within the literature on the concentrations of E2 and time- dependent impacts in TC proliferation and migration. Consequently, we performed an immunofluorescence panel in order to analyze the estrogen effect on thyroid cells studying the presence and the localization of SC (OCT3, SOX2, NANOG, BETA-CATENIN), progenitors (PAX8, TTF1), differentiation (TSHR, TPO, TG), and estrogen (ERA, ERB) markers. This panel has been designed to analyze CSC-enriched primary cultures and BCPAP cell line as control after 1 nmol l−1 and 10 nmol l−1 E2 treatment for 24 h and for 48 h repeated administration (Fig. 3A and B). Stem cell markers, including SOX2, OCT3 and NANOG are enhanced following treatment, concurrent with localizing at nuclear or perinuclear level, in particular with 1 nmol l−1 E2 administration. Following 48 h treatment, OCT3 expression is not present in human TC cells (Fig. 3B). Similarly, β-CATENIN changes their localization, being nuclear with 1 nmol l−1 E2 and citoplasmatic at 10 nmol l−1 E2. Comparing BCPAP and CSC-enriched patients culture cells, β-CATENIN appears more limited to the membrane in differentiated tumor cells. Progenitor marker TTF1 increases its expression with 10 nmol l−1 E2, in particular after treatment for 48 h, in patient cells (Fig. 3B). Even PAX8 expression enhances after 48 h, as we can observe in particular in BCPAP (Fig. 3A). Differentiation markers (TSHR, TPO, and TG) increase their expression after 10 nmol l−1 treatment and in particular after 48 h. Finally, ERA appears more evident in untreated cells and after 1 nmol l−1 24 h E2 treatment, while it decreases its expression in 48 h E2 CSC-enriched patient cells. Contrarily, ER β passes from perinuclear localization in untreated cells and 1 nmol l−1 24 h E2-treated cells to a more nuclear localization after treatment for 48 h E2 treatment.
Fig. 3.
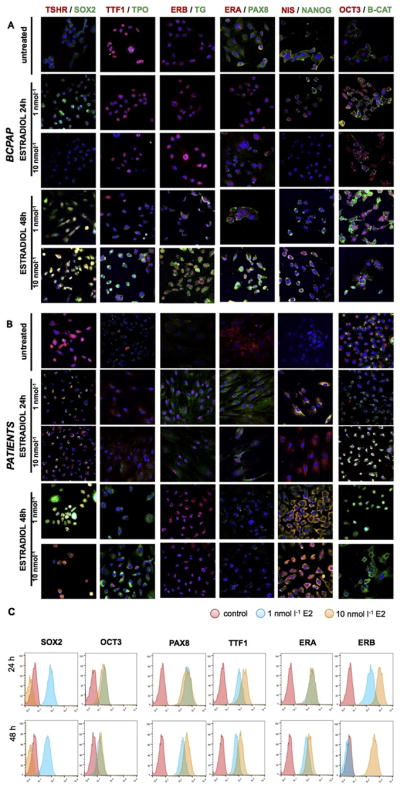
Estrogen treatment enhances stem markers expression in a time- and dose-dependent manner. (A) Immunofluorescence analysis on BCPAP for SOX2, OCT3, NANOG, PAX8, TTF1, TSHR, TG, TPO, NIS, BETA-CATENIN, ERA, and ERB after E2 treatment at 1 and 10 nmol l−1 for 24 h and 48 h (magnitude 40×, confocal microscope). (B) Same immunofluorescence analysis on CSC-enriched patient cell cultures. (C) FACS analysis on CSC-enriched cell cultures for SOX2, OCT3, PAX8, TTF1, ERA, and ERB after E2 treatment.
In summary, we observed that 1 nmol l−1 24 h E2 treatment leads to a marked expression of SC markers, with a higher aggressiveness given by ERα expression and then its pro-proliferation and anti-apoptotic function. Contrariwise,10 nmol l−1 E2 treatment for 48 h promotes differentiation, increasing the expression of progenitors and differentiated thyrocytes markers, and enforcing protectiveness improved by ERβ, which enhances its expression and passes from perinuclear at nuclear localization. This implies a more active form of ERβ and then a major inclination for anti-proliferation and pro-apoptosis effect. All these data are confirmed with the FACS analysis (Fig. 3C).
3.4. A continuous presence of estradiol has an opposite effect compared to a hormonal imbalance simulation
We enhanced the effect of the continuous exposure of estrogens prolonging the 1 nmol l−1 and 10 nmol l−1 E2 treatment for 7 days, and observing the modulation of different markers after further 7 days after stopping the treatment. Moreover, given that clinical and epidemiological studies reported an unbalanced estrogen metabolism in TC [17,27], we simulated an in vitro condition of hormonal imbalance. After 7 days E2 treatment, the morphology of 1 nmol l−1 and 10 nmol l−1 treated cells remained essentially unchanged. In unbalanced conditions, instead, many cells detached from the flask and survived in suspension (Fig. 4A).
Fig. 4.

A continuous presence of Estradiol has an opposite effect compared to a hormonal imbalance simulation. (A) Morphological evaluation of CSC-enriched cell cultures after 1 and 10 nmol l−1 continuous E2 treatment, and with hormonal imbalance simulation (magnitude 10×; optical microscope). (B) Mean Fluoresce Index of FACS analysis for SOX2, OCT3, NANOG, ERA, ERB, and GPER1. UNTREAT = control; E2 TREAT = cells after 7-days treatment; STOPPING TREAT = cells after stopping treatment for further 7 days. * = P < 0.005.
Comparing the Mean Fluorescence Index (MFI) of SOX2, OCT3, and NANOG, we noted that these conditions led to an opposite behavior: continuous treatment repressed SOX2 and OCT3, while unbalanced treatment enhanced their expression (Fig. 4B). Observing ERs expression, we noted that the ERα/ERβ ratio changed, giving a more protective effect when receiving continuous E2 administration, but was more prone to proliferation in imbalanced conditions, playing out an anti-apoptosis function. In particular, ERβ and GPER1 showed the same trend, strongly decreasing in expression with hormonal imbalance and increasing in continuous presence of E2. ERβ and GPER1 expression continues to decline 7 days after discontinuing the treatment.
3.5. Host estrogen therapy enhances the tumorigenic potential of Thyroid CSCs
We tested the tumorigenic capacity through subcutaneous inoculation of 1.5 ×106 CSC-enriched cells in immunocompromised NOD/SCID Il2rg-/- mice, alone or in combination with estrogen therapy (6 mice for cohort). In both cases, injected cells gave rise to a tumor mass, confirming the presence of tumorigenic cells. After 6 weeks, TC cells injected into mice receiving estrogen therapy developed tumors at 9 folds higher rate than those developed in untreated mice (4.24 vs. 0.47 cm3 mean, P < 0.05, Fig. 5A–C). Xenograft-derived cells were harvested and propagated in spheroid cultures, validating their CSC-like features (Fig. 5D). Histological examinations suggested that the E2 therapy received by the hosts has led to a more differentiated tumor compared to the tumor mass grown in untreated mice (Fig. 5E).
Fig. 5.

Host estrogen therapy enhances the tumorigenic potential of thyroid CSCs. (A) Diagram of tumor volume growth with or without host E2 therapy. 4,24 vs. 0.47 cm3 mean, * = P < 0.05. (B) Macroscopic analysis of xenograft tumors. Untreated vs E2-treated. (C) Subcutaneous xenografts obtained by the injection of CSC-primary culture cells (1.5 × 106) in immunocompromised NOD/SCID Il2rg -/- female mice, alone or in combination with estrogen therapy. Untreated vs E2-treated. (D) Thyroidspheres in culture after xenograft tumor digestion (magnitude 20×, optical microscope). Untreated vs E2-treated. (E) H&E staining of paraffined xenograft tumors (magnitude 10×, optical microscope). Untreated vs E2-treated.
3.6. E2-treated CSCs empower their migratory activity in vivo
We analyzed the migratory capacity of CSC-enriched cells culture using a model in vivo. This assay was performed through intrasplenic injection in NOD/SCID mice of 3 × 105 luciferase (LUC)/GFP-transduced cells, untreated or pretreated in vitro with 1 nmol l−1 E2 for 24 h.
This in vivo study highlighted a substantial migratory capacity of the inoculated E2 treated cells. 30 min after injection, the spleen was removed and observed using a Biospace instrument: the untreated cells were retained for the most part in the spleen, while pretreated cells were almost entirely migrated. After 5 weeks, E2 pretreated cells migrated to distant organs, such as lung, brain, and liver. Also untreated cells migrated, but in organs circumscribed to the abdomen and peritoneum (Fig. 6).
Fig. 6.

E2-treated CSCs empower their migratory activity in vivo. Intrasplenic injection in NOD/SCID female mice of 3 ×105 luciferase (LUC)/GFP-transduced cells, untreated or treated with 1 nmol l−1 E2 for 24 h (magnitude 10×, EVOS™, Thermo Fischer, Waltham, Massachussetts). Analysis of migratory capacity at time: 0 min; 30 min; 5 weeks. Biospace.
3.7. Estrogens perform their function in a different way in women and men
To investigate the mechanism of action of estrogen, we performed a pathway analysis for factors involved in estrogen and cancer signaling, with a focus on the SC compartment. To unravel insights toward the gender disparity in incidence and aggressiveness of TC, we compared female and male samples for 252 genes involved in estrogen, cancer, and human cancer stem cells pathways. Estrogen treatment resulted in a substantial altered expression (Fig. 7, Table 1).
Fig. 7.

Estrogens perform their function in a different way among women and men. Estrogen, cancer, and cancer stem cell pathway analysis for female and male CSC-enriched cell cultures. The scatter plot compares the normalized expression of 252 genes on the array between two groups (untreated and E2-treated).
Table 1.
Under- and over-expressed genes after E2 treatment in CSC-enriched female and male TC samples.
| Male | Female | |
|---|---|---|
| Estrogen Receptors | ↑ERB | ↑ERA |
| Growth Factors | ↑FGFR2 | ↑EGF |
| ↑FGFR2 | ||
| Cancer Stem Cell Markers | ↑ABCG2 | ↑ABCB5 |
| ↑ALDH1A1 | ||
| ↑GATA3 | ||
| ↑ITGA6 | ||
| Loss of Stemness | ↓DACH1 | |
| ↓PECAM | ||
| Pluripotency | ↑NANOG | |
| ↑OCT3 | ||
| ↑SOX2 | ||
| Self-Renewal | ↓HEDGEHOG | ↑HEDGEHOG |
| ↑HIPPO | ||
| ↑NOTCH | ||
| ↑WNT | ||
| Cell-Cycle | ↑WEE1 | ↑AURKA |
| ↑CCND3 | ||
| ↑CDC20 | ||
| ↑MKI67 | ||
| ↑SKP2 | ||
| ↑STMN1 | ||
| ↓WEE1 | ||
| Angiogenesis | ↑VEGFR2 | ↓ANGPT1 |
| ↓ANGPT2 | ||
| ↑SERPINF1 | ||
| ↓VEGFR1 | ||
| ↓VEGFR2 | ||
| Epithelial-Mesenchymal Transition | ↓KLF17 | ↑KLF17 |
| ↑SNAI1 | ↑OCLN | |
| ↓SNAI1 | ||
| ↓TWIST1 | ||
| ↓TWIST2 |
Specifically, we noted that in cells derived from males the receptor expression was as follows: ERα> GPER1 > ERβ. This was in striking contrast to those patterns observed In cells derived from females GPER1 > ERβ> ERα. These data suggest that estrogenic signaling in protective in protective in females at basal level. After E2 treatment, males showed a down-regulation of both receptors, while female reported an over-expression of ERα. Most important, what changes is the ratio ERα/ERβ, leading to a stronger propensity in females than in males towards proliferation and anti-apoptosis.
Estrogen interacts in both women and men with growth factors pathway, as evidenced by EGF, ERβ-B3, and FGFR2 over-expression in patient samples, and with proliferation through LIN28A and LIN28B. Most importantly, in both genders estrogen treatment led to a strong increase of CSC markers expression. In particular, both showed an up-regulation of ABCB5, ABCG2, ALDH1A1, GATA3, and ITGA6, and a down-regulation of adhesion molecules, such as PECAM1.
The up-regulation of DNMT1 and the absence of BMP family members indicate the propensity of cells for self-renewal. The pluripotency is maintained by the increase of OCT3, SOX2, and NANOG expression, evidenced principally in females. In these patients, we observed the hyper-activation of Hippo (with LATS1 and TAZ), Hedgehog (PTCH1 and SMO), and Notch (DLL1, JAG1, MAML1, and NOTCH1) signaling. In contrast, in male patients we observed a down-regulation of Hedgehog signaling proteins, suggesting a gender-dependent impact on these signaling pathway.
Despite E2 enhances within CSC compartment in both genders, data imply that females and males modulate the estrogen effect distinctly. Indeed, deepening the hallmarks of cancer, we noted that the angiogenic behavior is different among patients. In particular, we observed an increase in angiogenic factors in males and an opposite decrease in females of VEGFA, VEGFR1, and VEGFR2). Moreover, only in females there is a down-regulation of IL8 and an up-regulation of SERPINF1, a potent angiogenesis inhibitor, and of ANGPT1 and ANGPT2, which could have a role in blocking angiogenesis.
The cell cycle is also differently regulated in women and in men. While in male patients we observed a down-regulation of CCND2 and an up-regulation of WEE1, female patients showed an opposite attitude, with enhancement of AURKA, CCND3, CDC20, MKI67, SKP2, STMN1, and decrease of WEE1. This suggests that, if the entry in S phase is inhibited and the G2-M progression is negatively regulated in males, the E2 treatment led to a promotion of cell cycle phase in females. In females, otherwise, it is evidenced an alteration in DNA repair activity, with down-regulation of DDIT3, GADD45, and PPP1R15A. Similarly, the telomerase activity is enhanced by DKC1 and TEP1 up-regulation in female.
Migration, metastasis, and the Epithelial-Mesenchymal Transition, showed gender dependent differences in our model system: ID1, OCLN, SNAI1, TWIST1, TWIST2, and ZEB1 are inversely regulated. In fact, data suggested that the EMT is enhanced in males following E2 treatment, but not in female. In particular, KLF17, a negative regulator of EMT through the binding to ID1, is down-regulated in male and up-regulated in female. Similarly, we observed a reduction of SNAI1, TWIST1, and TWIST2 expression in females, but not in males, where we found a decrease of DACH1, a TGFB signaling inhibitor.
In summary, estrogen treatment enhances CSC markers in TC in women and in men, however the impact on signaling pathways regulating cell cycle and migration were distinct. Males displayed enhanced angiogenesis and EMT activation, confirming the more aggressive behavior of TC and the lower disease free survival in men. Females showed the activation of all cell cycle phases, making them more inclined to an effective response to the chemotherapy. On the other hand, the variety of markers and their modulation require further validation.
4. Discussion
Thyroid diseases are widespread among the population. However, they are often undervalued by the scientific point of view, since they rarely end with fatal outcome. The incidence of these pathologies affects mainly the female gender, with a frequency of 3–5 times higher depending on the severity degree of disease, which may not involve severe physical discomforts, but has a strong impact on life quality. The origin of the disorders may be attributed to the loss of the balance between the many factors that regulate cellular homeostasis. On the contrary, it has been observed that excessive or unregulated exposure to various hormones, and in particular to estrogen, may affect the course of thyroid diseases. Recent studies also demonstrate that sex hormones could exert a supportive role in the propagation of SCs and progenitors, as suggested by the cross talk between estrogen signaling, growth factors, and Wnt pathway. In the present study, we investigated the fine mechanisms regulating the interconnection between estrogens, thyroid, and SCs. We confirmed the hypothesis of the CSC tumorigenic model for TC, identifying a CSC niche in thyroid microenvironment able to reconstitute the tumor in vivo. Moreover, this study strengthens the role of the microenvironment on the tumor bulk, also in in vitro conditions.
Dose- and time-dependent effects of estrogen observed in our experiments may explain the severity of the consequences to an intermittent or non-physiological presence of estrogens in our body, illustrating the possible scenarios related to hormonal imbalance in female and male patients. These bring social implications for steroid/hormone use to the forefront. We propose that an improper use of the emergency contraception pill could be more harmful than a constant and continuous hormonal treatment, which instead seems to have a protective role on TC. Likewise, an impaired use of anabolic steroids often supplemented in athletes may lead to their transformation in E2 by aromatase, causing the peripheral aromatization of androgens and then to an abnormal exposition to female sex hormones. Indeed, we observed that the collateral estrogen effects could be shown both in presence of local cell interaction with estrogen but also when administered exogenous hormones, resulting in a 9-fold more tumorigenic malignancy and with distant metastasis. Hence, the constant activation of ERs (enhanced also by estrogen-like substances and endocrine disrupting chemicals) could have a heavy impact on the tumor development. In view of that, the metastasis risk could be bounded using ERs-inhibitors or antagonists.
What is surprising is that estrogens affect TC both in women and in men, but with different and often opposing impact. This modulation could contribute to the highly aggressive progression in males, potentially acting through the EMT and angiogenesis, while the activation of the cell-cycle in women could explain the propensity to apoptosis and a better prognosis.
The goal of these studies was to evaluate factors contributing to progression and metastatic load in thyroid malignancies, and in particular to focus on the causes that underlie of gender discrepancy. Deepening this interaction could explain the gender imbalance in tumor incidence and development for the purpose of prevent and cure thyroid pathologies with a more targeted approach.
Acknowledgments
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
We would like to thank Clara Benna and Eric Casal Ide for their collaboration in the tissue samples collection, Patrizia Porto and Aichi Msaki for their carefulness in editing this paper.
Nomenclature
Acronyms and abbreviations
- ABCB5
ATP binding cassette subfamily B member 5
- ABCG2
ATP binding cassette subfamily G member 2
- ACTB
Actin beta
- ALDH (ALDH1A1)
Aldehyde dehydrogenase 1 family member A1
- ANGPT1
Angiopoietin 1
- ANGPT2
Angiopoietin 2
- AURKA
Aurora kinase A
- B2M
Beta-2-microglobulin
- BETA-CATENIN
Catenin beta-1
- BMP
Bone morphogenetic protein
- BRAF
B-Raf proto-oncogene, serine/threonine kinase
- BSA
Bovine Serum Albumins
- CCND2
Cyclin D2
- CCND3
Cyclin D3
- CDC20
Cell division cycle 20
- CSC
Cancer stem cell
- DACH1
Dachshund family transcription factor 1
- DDIT3
DNA damage inducible transcript 3
- DEAB
Diethylaminobenzaldehyde
- DKC1
Dyskerin pseudouridine synthase 1
- DLL1
Delta like canonical Notch ligand 1
- DMEM
Dulbecco’s Modified Eagle Medium
- DNMT1
DNA methyltransferase 1
- DUOX1
Dual oxidase 1
- E2
Estradiol
- EGF
Epidermal growth factor
- EMT
Epithelial-Mesenchymal Transition
- ERs
Estrogen Receptors
- ERA
Estrogen Receptor Alpha
- ERB
Estrogen Receptor Beta
- ERBB3
Erb-b2 receptor tyrosine kinase 3
- FBS
Fetal bovine serum
- FGF2
Fibroblast growth factor 2
- FGFR2
Fibroblast growth factor receptor 2
- GADD45
Growth arrest and DNA damage inducible alpha
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GATA3
GATA binding protein 3
- GPER1
G protein-coupled estrogen receptor 1
- H&E
Haematoxylin and eosin
- ID1
Inhibitor of DNA binding 1, HLH protein
- IL8
Interleukin-8
- ITGA6
Integrin subunit alpha 6
- JAG1
Jagged 1
- KLF17
Kruppel like factor 17
- LATS1
Large tumor suppressor kinase 1
- LIN28A
Lin-28 homolog A
- LIN28B
Lin-28 homolog B
- MAML1
Mastermind like transcriptional coactivator 1
- MKI67
Marker of proliferation Ki-67
- NANOG
Homeobox protein NANOG
- NIS
Sodium/iodide cotransporter
- NOD/SCID
Non-obese diabetic/severe combined immunodeficiency
- NOTCH1
Neurogenic locus notch homolog protein 1
- NTRK
High affinity nerve growth factor receptor
- OCLN
Occludin
- OCT3
POU class 5 homeobox 1
- PAX8
Paired box 8
- PBS
Phosphate buffered saline
- PECAM1
Platelet and endothelial cell adhesion molecule 1
- PI3K
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PPP1R15A
Protein phosphatase 1 regulatory subunit 15A
- PRLP0
Ribosomal protein lateral stalk subunit P0
- PTC
Papillary Thyroid Cancer
- PTCH1
Patched 1
- RET
Ret proto-oncogene
- SCM
Stem cell medium
- SERPINF1
Pigment epithelium-derived factor
- SKP2
S-phase kinase associated protein 2
- SNAI1
Snail family transcriptional repressor 1
- SMO
Smoothened, frizzled class receptor
- SOX2
SRY-box 2
- STMN1
Stathmin 1
- TAZ
Tafazzin
- TC
Thyroid Cancer
- TEP1
Telomerase associated protein 1
- TG
Thyroglobulin
- TGFB
Transforming growth factor beta-1
- TPO
Thyroid peroxidase
- TSHR
Thyroid stimulating hormone receptor
- TTF1
Transcription termination factor 1
- TWIST1
Twist family bHLH transcription factor 1
- TWIST2
Twist family bHLH transcription factor 2
- UICC
Union for International Cancer Control
- VEGFA
Vascular endothelial growth factor A
- VEGFR1
Vascular endothelial growth factor receptor 1
- VEGFR2
Vascular endothelial growth factor receptor 2
- WEE1
WEE1 G2 checkpoint kinase
- ZEB1
Zinc finger E-box binding homeobox 1
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- 1.Ahmed RA, Aboelnaga EM. Thyroid cancer in Egypt: histopathological criteria, correlation with survival and oestrogen receptor protein expression. Pathol Oncol Res. 2015;21:793–802. doi: 10.1007/s12253-014-9892-5. [DOI] [PubMed] [Google Scholar]
- 3.Antico-Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene. 2010;29:5678–5686. doi: 10.1038/onc.2010.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antico Arciuch VG, Di Cristofano A. Estrogen signaling and thyrocyte proliferation. In: Ward DL, editor. Thyroid and Parathyroid Diseases-New insights into Some Old and Some New Issues. 2012. [Google Scholar]
- 5.Braganza MZ, de Gonzalez AB, Schonfeld SJ, Wentzensen N, Brenner AV, Kitahara CM. Benign breast and gynecologic conditions, reproductive and hormonal factors, and risk of thyroid cancer. Cancer Prev Res (Phila) 2014;7:418–425. doi: 10.1158/1940-6207.CAPR-13-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caini S, Gibelli B, Palli D, Saieva C, Ruscica M, Gandini S. Menstrual and reproductive history and use of exogenous sex hormones and risk of thyroid cancer among women: a meta-analysis of prospective studies. Cancer Causes Control. 2015;26:511–518. doi: 10.1007/s10552-015-0546-z. [DOI] [PubMed] [Google Scholar]
- 8.Dal Maso L, Lise M, Zambon P, Falcini F, Crocetti E, Serraino D, Cirilli C, Zanetti R, Vercelli M, Ferretti S, et al. Incidence of thyroid cancer in Italy, 1991–2005: time trends and age-period-cohort effects. Ann Oncol. 2011;22:957–963. doi: 10.1093/annonc/mdq467. [DOI] [PubMed] [Google Scholar]
- 9.Derwahl M, Nicula D. Estrogen and its role in thyroid cancer. Endocr Relat Cancer. 2014;21:T273–T283. doi: 10.1530/ERC-14-0053. [DOI] [PubMed] [Google Scholar]
- 10.Dong W, Zhang H, Li J, Guan H, He L, Wang Z, Shan Z, Teng W. Estrogen induces metastatic potential of papillary thyroid cancer cells through estrogen receptor alpha and beta. Int J Endocrinol. 2013;2013:941568. doi: 10.1155/2013/941568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee ML, Chen GG, Vlantis AC, Tse GM, Leung BC, van Hasselt CA. Induction of thyroid papillary carcinoma cell proliferation by estrogen is associated with an altered expression of Bcl-xL. Cancer J. 2005;11:113–121. doi: 10.1097/00130404-200503000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Leitman DC, Paruthiyil S, Vivar OI, Saunier EF, Herber CB, Cohen I, Tagliaferri M, Speed TP. Regulation of specific target genes and biological responses by estrogen receptor subtype agonists. Curr Opin Pharmacol. 2010;10:629–636. doi: 10.1016/j.coph.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Song Z, Zhao Y, Qin H, Cai J, Zhang H, Yu T, Jiang S, Wang G, Ding M, et al. A novel chemical-defined medium with bFGF and N2B27 supplements supports undifferentiated growth in human embryonic stem cells. Biochem Biophys Res Commun. 2006;346:131–139. doi: 10.1016/j.bbrc.2006.05.086. [DOI] [PubMed] [Google Scholar]
- 15.Manole D, Schildknecht B, Gosnell B, Adams E, Derwahl M. Estrogen promotes growth of human thyroid tumor cells by different molecular mechanisms. J Clin Endocrinol Metab. 2001;86:1072–1077. doi: 10.1210/jcem.86.3.7283. [DOI] [PubMed] [Google Scholar]
- 16.Nakada D, Oguro H, Levi BP, Ryan N, Kitano A, Saitoh Y, Takeichi M, Wendt GR, Morrison SJ. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature. 2014;505:555–558. doi: 10.1038/nature12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahbari R, Zhang L, Kebebew E. Thyroid cancer gender disparity. Future Oncol. 2010;6:1771–1779. doi: 10.2217/fon.10.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajoria S, Suriano R, Shanmugam A, Wilson YL, Schantz SP, Geliebter J, Tiwari RK. Metastatic phenotype is regulated by estrogen in thyroid cells. Thyroid. 2010;20:33–41. doi: 10.1089/thy.2009.0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santin AP, Furlanetto TW. Role of estrogen in thyroid function and growth regulation. J Thyroid Res. 2011;2011:875125. doi: 10.4061/2011/875125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sobin LH, Gospodarowicz MK, Wittekind C. International Union Against Cancer (UICC) TNM Classification of Malignant Tumors. 7. Wiley–Blackwell; 2009. [Google Scholar]
- 21.Sungwalee W, Vatanasapt P, Kamsa-Ard S, Suwanrungruang K, Promthet S. Reproductive risk factors for thyroid cancer: a prospective cohort study in Khon Kaen, Thailand. Asian Pac J Cancer Prev. 2013;14:5153–5155. doi: 10.7314/apjcp.2013.14.9.5153. [DOI] [PubMed] [Google Scholar]
- 23.Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A, Musti AM, Picard D, Ando S, Maggiolini M. 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol Pharmacol. 2006;70:1414–1423. doi: 10.1124/mol.106.026344. [DOI] [PubMed] [Google Scholar]
- 24.Wang G, Zhang H, Zhao Y, Li J, Cai J, Wang P, Meng S, Feng J, Miao C, Ding M, et al. Noggin and bFGF cooperate to maintain the pluripotency of human embryonic stem cells in the absence of feeder layers. Biochem Biophys Res Commun. 2005;330:934–942. doi: 10.1016/j.bbrc.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 25.Xhaard C, Rubino C, Clero E, Maillard S, Ren Y, Borson-Chazot F, Sassolas G, Schvartz C, Colonna M, Lacour B, et al. Menstrual and reproductive factors in the risk of differentiated thyroid carcinoma in young women in France: a population-based case-control study. Am J Epidemiol. 2014;180:1007–1017. doi: 10.1093/aje/kwu220. [DOI] [PubMed] [Google Scholar]
- 26.Xu S, Chen G, Peng W, Renko K, Derwahl M. Oestrogen action on thyroid progenitor cells: relevant for the pathogenesis of thyroid nodules? J Endocrinol. 2013;218:125–133. doi: 10.1530/JOE-13-0029. [DOI] [PubMed] [Google Scholar]
- 27.Zahid M, Goldner W, Beseler CL, Rogan EG, Cavalieri EL. Unbalanced estrogen metabolism in thyroid cancer. Int J Cancer. 2013;133:2642–2649. doi: 10.1002/ijc.28275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zane M, Catalano V, Scavo E, Bonanno M, Pelizzo MR, Todaro M, Stassi G. Estrogens and stem cells in thyroid cancer. Front Endocrinol (Lausanne) 2014;5:124. doi: 10.3389/fendo.2014.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zane M, Scavo E, Catalano V, Bonanno M, Todaro M, De Maria R, Stassi G. Normal vs cancer thyroid stem cells: the road to transformation. Oncogene. 2016;35:805–815. doi: 10.1038/onc.2015.138. [DOI] [PubMed] [Google Scholar]
- 30.Zeng Q, Chen GG, Vlantis AC, van Hasselt CA. Oestrogen mediates the growth of human thyroid carcinoma cells via an oestrogen receptor-ERK pathway. Cell Prolif. 2007;40:921–935. doi: 10.1111/j.1365-2184.2007.00471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]