

Abstract
Bioinformatic analyses have suggested that Mce proteins in diverse actinobacteria are components of complex ATP-binding cassette transporter systems, comprising more than eight distinct proteins. In Mycobacterium tuberculosis, these proteins are implicated in interactions of this deadly pathogen with its human host. Here, we provide direct evidence that the Mce4 system of Rhodococcus jostii RHA1 is a steroid uptake system. Transcriptional analyses indicate that the system is encoded by an 11-gene operon, up-regulated 4.0-fold during growth on cholesterol versus on pyruvate. Growth of RHA1 on cholesterol and uptake of radiolabeled cholesterol both required expression of genes in the mce4 operon encoding two permeases plus eight additional proteins of unknown function. Cholesterol uptake was ATP-dependent and exhibited Michaelis-Menten kinetics with a Km of 0.6 ± 0.1 μM. This uptake system was also essential for growth of RHA1 on β-sitosterol, 5-β-cholestanol, and 5-α-cholestanone. Bioinformatic analysis revealed that all mce4 loci in sequenced genomes are linked to steroid metabolism genes. Thus, we predict that all Mce4 systems are steroid transporters. The transport function of the Mce4 system is consistent with proposed roles of cholesterol and its metabolism in the pathogenesis of M. tuberculosis.
The mce genes of Mycobacterium tuberculosis have garnered much interest due to their demonstrated role in pathogenesis, although the function of these genes remains unclear. The first mce gene, now known as mce1A, was discovered when a DNA fragment cloned from M. tuberculosis into a noninvasive Escherichia coli strain enabled uptake of the latter bacterium by nonphagocytic mammalian epithelial (HeLa) cells and facilitated its phagocytosis by macrophages (1). A subsequent study showed that coating latex beads with the protein encoded by mce1A facilitated uptake of the beads by HeLa cells, and so the gene was designated mce for mammalian cell entry (2). The genome sequence of M. tuberculosis revealed four loci (mce1–4), each containing two yrbE genes followed by six mce genes (3). The Mce proteins are predicted to have similar structures and to be secreted (4), and Mce1A was localized to the surface of M. tuberculosis cells (2). The capacity to facilitate uptake by HeLa cells was further narrowed to a small region of the MceA1 protein (2, 5, 6). Coating beads with either Mce3A or Mce3E also facilitated such uptake (7), whereas coating beads with Mce2A failed to do so (2). Together, these studies suggest that some of the Mce proteins might interact with host cells and play a role in uptake of M. tuberculosis by host cells. However, direct evidence for this function of Mce proteins is lacking.
Mutational studies also support the involvement of Mce proteins in pathogenesis of M. tuberculosis. One genome-wide screen suggested that disruption of mce1 and mce4 genes causes early and late growth defects, respectively, in mice (8), and these phenotypes were confirmed with mce1D and yrbE4A deletion mutants, respectively (8–10). Another genome-wide screen suggested that disruption of yrbE4A impairs macrophage infection (11), and a yrbE4A deletion mutant was severely impaired in replication in interferon γ-activated macrophages (10). Disruption of any one of yrbE1B, mce2A, and mce3A genes in M. tuberculosis increased survival of intratrachaelly infected mice (12). Similarly, deletion of nearly entire mce3 or mce4 operons increased long term survival of mice following low dose aerosol infection, and the latter deletion also decreased bacterial burdens of the mice (13). By contrast, an mce1A deletion was hypervirulent in mice (14). Thus, mce mutant strains have phenotypes generally supporting the involvement of these genes in pathogenesis, but the mutants exhibited disparate virulence phenotypes in different assays.
Analysis of genome sequences indicated that diverse actinobacteria, including members of Mycobacterium, Nocardia, Rhodococcus, and Streptomyces, harbor mce loci (15, 16). Moreover, PCR-based assays revealed the existence of mce4 loci in 11 of 20 Mycobacterium spp. screened (17). The actinobacteria with mce loci include both pathogens and free-living bacteria, strongly suggesting that the function of these genes is not limited to virulence. Bioinformatic analyses (15, 18) indicated that the yrbE genes of all mce loci encode transmembrane proteins with similarity to permease components of ATP-binding cassette (ABC)2 transporters. The hallmark of ABC transporters is an ATPase with highly conserved features. Some mce loci include genes encoding such ATPases, which belong to the Mkl family, a subset of ABC transporter ATPases (15). Further, all actinobacteria containing mce loci were found to also contain genes encoding Mkl ATPases, usually not proximal to the mce loci. Based on genetic interaction mapping and mutagenesis, Joshi et al. (9) provided evidence that the Mce1 and Mce4 proteins in M. tuberculosis are both functionally linked to a single Mkl ATPase (MceG), encoded by a gene not linked to either the mce1 or the mce4 loci. These observations strongly suggest that mce loci encode a novel type of ABC transporter. However, direct evidence for this transport function has not been demonstrated.
Many mce loci also include two or four genes downstream of the yrbE-mce genes (14). The latter genes have been referred to as mce-associated (mas) genes (15); however, there is already precedent for using Mas to denote mycocerosic acid synthase of M. tuberculosis (19), so we will simply refer to the latter genes in mce loci as additional mce genes (i.e. mce4HI).
We previously found that the mce4 locus of Rhodococcus jostii RHA1 was among a large cluster of genes up-regulated during growth on cholesterol (20). Moreover, we found that RHA1 mutants with deletions of either the supAB genes (homologous to yrbE4AB genes; Δsup) or the mce4ABCDEF genes (Δmce4AF) lost the ability to grow on cholesterol. These findings led us to hypothesize that the Mce4 system of RHA1 functions in cholesterol uptake. Because the mce4 locus of M. tuberculosis is also clustered with cholesterol metabolism genes homologous to those in RHA1, we hypothesized the same function for the Mce4 systems in both organisms. Consistent with this, Pandey and Sassetti (10) recently showed that an M. tuberculosis yrbE4A disruption mutant is impaired in mineralization of C-4 of cholesterol, assimilation of C-26 of cholesterol, and growth with cholesterol as a substrate.
In the present study, we used mutational analysis and a direct assay of cholesterol uptake to conclusively determine the role of the Mce4 system of RHA1 in steroid uptake. We also used bioinformatic analysis to assess the role of Mce4 systems in other actinobacteria.
EXPERIMENTAL PROCEDURES
Bacterial Growth
R. jostii RHA1 was grown at 30 °C on a shaker on defined Goodies medium (21) plus either 10 mM pyruvate or 2 mM of a steroid substrate. Cholesterol was from Sigma-Aldrich, and other steroids were from Steraloids (Newport, RI). Biomass was estimated on the basis of total protein measured by disrupting cells with hot alkaline lysis and using the BCA protein assay (Pierce) with bovine serum albumin as the standard.
PCR
Nucleic acids were extracted, and RNA was purified as described previously (20). cDNA was synthesized with the SuperScript™ III reverse-transcriptase (Invitrogen) and random hexamers (Invitrogen). Quantitative PCR was done using TaqMan probes (Applied Biosystems), and gene expression differences were calculated as described previously (20). Primer and probe sequences are in supplemental Table S1. PCR conditions for reverse-transcription PCR (RT-PCR) and RT-quantitative-PCR assays were: 95 °C for 5 min and then 35 cycles of 95 °C for 40 s, 66 °C (PCR 1) or 59 °C (PCR 2–10) for 40 s, 72 °C for 1 min. A final extension at 72 °C was for 7 min before holding at 4 °C.
Gene Deletion and Complementation
The mce4H-mce4I-ro04706 genes of R. jostii RHA1 were deleted (Δmce4HI) using the sacB counter selection system essentially as described (22). Sequences of all oligonucleotides used are in supplemental Table S1. Oligonucleotides ro04706-F and ro04706-R were used to amplify and clone a 1.8-kb region downstream of the three genes of RHA1, comprised of the last 13 codons and the stop codon of ro04706, into SmaI-digested pK18mobsacB, yielding pK18-Ro04706. Oligonucleotides ro04704-F and ro04704-R were used to amplify the upstream region of the three genes. The obtained 1.8-kb PCR product, composed of the first 20 codons including the start codon of ro04704, was cloned into pGEM-T. A double digestion with NcoI (blunt-ended with Klenow) and SpeI was performed on the resulting plasmid, and the liberated fragment was cloned into BamHI (blunt-ended with Klenow) and SpeI double-digested pK18-Ro04706, resulting in pK18-Ro04704-06 used for mce4H-mce4I-ro04706 gene deletion. The deletion was verified by PCR using oligonucleotides ro04704-06-F with ro04704-06-R and ro04704contr-F with ro04706contr-R, matching sequences flanking the targeted three genes.
The supA-supB gene deletion (Δsup) and the mceH-mceI gene deletion (Δmce4HI) were complemented, resulting in the strains Δsup-C and Δmce4HI-C, respectively, as follows. The supA-supB genes were amplified using primers ro04696-F and ro04797-R. The resulting 1636-bp amplicon was digested with NdeI and HindIII and cloned in pTip-QC2 (23), yielding pTip-supAB. Similarly, the mce4H and mce4I genes were amplified using primers ro04704ex-F and ro04705ex-R. The resulting 1267-bp amplicon was subcloned, digested with BspHI and HindIII, and ligated into NcoI/HindIII-digested pTip-QC1 (23), yielding pTip-mce4HI. The pTip-supAB and pTip-mceHI plasmids were introduced into the Δsup strain and the Δmce4HI strain, respectively, by electroporation. A “vector control” strain without the supA-supB genes (ΔsupV) was also produced by electroporating Δsup with pTip-QC2.
Cholesterol Uptake Assay
Cholesterol uptake was measured in resting cell suspensions. Cells were grown to mid-log phase on pyruvate, washed twice, and suspended at a cell density of 900 mg of protein/liter (A600 = 13.3) in Goodies buffer. Aliquots of 0.05 ml Goodies medium were placed in 4-ml vials, and [4-14C]cholesterol (53 mCi/mmol, PerkinElmer Life Sciences) was added from an ethanol stock solution. Aliquots of 0.05 ml of cell suspensions were added to the vials for a final cell density of 450 mg of protein/liter. The vials were incubated with shaking for 5 min at room temperature. Cholesterol uptake was stopped by diluting the suspensions with 1.0 ml of ice-cold buffer, collecting the cells on a 0.22-μm Millipore nitrocellulose filter (Fisher Scientific, Mississauga, Ontario), washing the cells with 10 ml of 5% Tween 20 (Fisher) and then with 10 ml of 50% ethanol, and finally, with 10 ml of 5% Tween 20. The filters were placed in Beckman Ready-Safe scintillation mixture (Beckman Coulter) and counted in a Beckman LS-600IC scintillation counter to determine the amount of cholesterol taken up by the cells. The rate of uptake was found to be constant for greater than 5 min, and there was a linear relationship between uptake rate and cell biomass over a cell density range of 69–1375 mg of protein/liter. Where indicated, 60 mM sodium azide, 2.0 mM dicyclohexylcarbodiimide (DCCD), or 10 mM sodium orthovanadate was added to cell suspensions 10 min prior to the addition of the labeled cholesterol. Steady-state kinetic parameters were analyzed using the least squares and dynamic weighting options of LEONORA (24).
RESULTS
The mce4 Locus
The RHA1 mce4 locus has the typical gene arrangement of other mce loci and appears to be an operon (Fig. 1). As reported previously (15), orthologous mce4 loci occur in Nocardia farcinica (IFM 10152), Mycobacterium bovis (AF2122/97), Mycobacterium avium subsp. paratuberculosis (k-10), Mycobacterium smegmatis (MC2 155), and M. tuberculosis (H37Rv and other strains). We also found mce4 loci in M. avium (104), M. bovis BCG (Bacille de Calmette et Guèrin), Mycobacterium marinum (M), Mycobacterium ulcerans (Agy99), and Mycobacterium vanbaalenii (PYR-1). The mce4 gene products of RHA1 have the greatest similarity to their respective orthologs in N. farcinica. The sup genes of the RHA1 mce4 locus are orthologs of yrbE4 genes in other mce4 loci.
FIGURE 1. The mce4 locus of RHA1.

Three predicted transcriptional promoters are indicated (P1, P2, P3). Numbered solid bars (2–10) indicate transcribed regions detected in RT-PCR assays, and those numbers correspond to the electrophoresis gel lane numbers, showing RT-PCR products. All positive RT-PCR reactions were negative in controls without reverse-transcriptase (not shown). The dashed bar (1) indicates a nontranscribed control, with lane 1a showing the corresponding PCR product from genomic DNA and lane 1b showing the result of the RT-PCR assay. Bars at the top show regions deleted in three mutant strains (Δsup, Δmce4AF, Δmce4HI).
The RHA1 mce4 locus has the conserved configuration of two yrbE (sup) plus eight mce genes found in all known mce4 loci. However, the mce4 locus of RHA1 is unique in that mce4I is followed by two additional genes. The first of these genes, ro04706, is only two nucleotides downstream of mce4I and encodes a hypothetical protein with an N-terminal region similar to the conserved domain of the substrate-binding site of ketosteroid isomerases (Pfam family PF02136). The second of these genes, ro04707, encodes a protein with similarity to the conserved domain of the 3-β-hydroxysteroid dehydrogenase/isomerase family (Pfam family PF01073). Both of these genes appear to have orthologs in all of the above strains, but none of those orthologs is closely linked to mce4 loci. The orthologs in H37Rv are Rv2042c and Rv0139, respectively, which are not closely linked to the large steroid metabolism gene cluster of H37Rv. However, Rv0139 is part of a large regulon that also includes the steroid metabolism genes and mce4 genes of H37Rv (25). The four genes upstream of supA (yrbE4A) are conserved in all of the above organisms and putatively encode a 3-ketoacyl-(acyl-carrier-protein) reductase, a ferredoxin and two acyl-CoA dehydrogenases. The fifth gene upstream of supA (yrbE4A) is conserved in all the above organisms, except N. farcinica, and putatively encodes an acyl-CoA synthetase.
In RHA1, the 11 genes from supA through ro04706 are closely spaced (Fig. 1), with maximum intergenic regions of 20 bases and with six of the predicted genes overlapping the following ones by four nucleotides. Putative transcriptional promoters were found upstream of hsd4A, supA, and ro04707, but none were found within the 11-gene mce4 cluster, suggesting co-transcription of that cluster. This conclusion was further supported by RT-PCR analysis, which detected transcripts including intergenic regions within this cluster. Seven of the 10 intergenic regions were assayed, and all were detected. Transcripts including the intergenic regions on either side of the 11-gene cluster were also detected, suggesting some level of transcription through those regions. We used expression of the supA gene as a proxy for expression of mce4 locus genes in an RT-quantitative-PCR assay. The supA gene was up-regulated 4.0-fold during growth on cholesterol, relative to during growth on pyruvate. Together, the above results strongly suggest that the 11-gene mce4 cluster is co-transcribed and regulated as an operon. The results do not preclude the existence of additional smaller transcripts within the region nor larger transcripts including additional flanking genes.
Like other mce4 loci, that of RHA1 is not proximal to a gene encoding an Mlk family ATPase. However, the RHA1 genome includes two genes, ro01974 and ro02744, encoding ATPases orthologous to MceG, the Mlk ATPase gene of M. tuberculosis. Both of these RHA1 genes are associated with other mce loci of RHA1. One or both of these ATPases may function in the RHA1 Mce4 system.
Growth Phenotypes of Mutants
We made an unmarked, in-frame deletion mutant of the mce4HI-ro04706 genes (Δmce4HI) in RHA1 (Fig. 1). Like the previous Δsup and Δmce4AF deletion mutants (20), the Δmce4HI mutant completely lost the ability to grow on cholesterol (Fig. 2A). We constructed two complementation strains to confirm that deleted genes were the cause of the mutant phenotypes.
FIGURE 2.
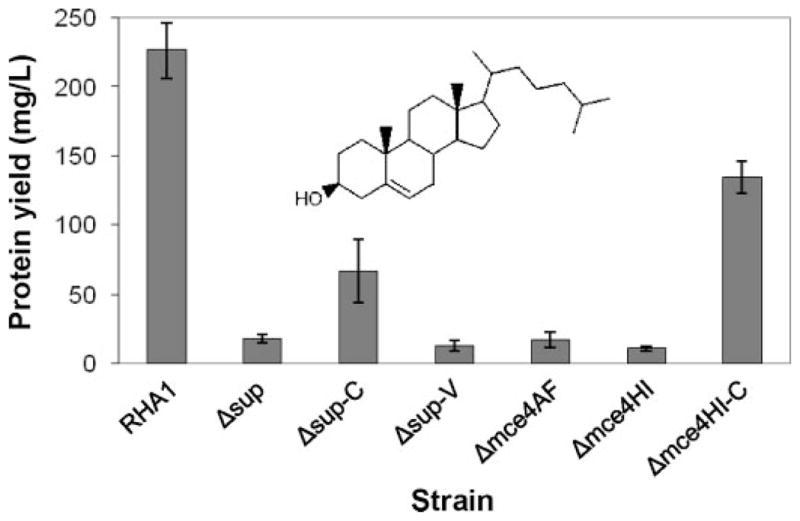
Growth of RHA1 and mutant strains on cholesterol (n ≥ three independent cultures, bars indicate standard deviation).
The Δsup-C complementation strain consists of the Δsup mutant with plasmid pTip (23) containing supAB. The Δsup-C strain partially regained the ability to grow on cholesterol. An “empty vector” control strain (Δsup-V), consisting of the Δsup mutant with plasmid pTip, could not grow on cholesterol. Given this complementation and the complete inability of the Δmce4AF and Δmce4HI deletion mutants to grow on cholesterol, it is clear that the Δsup deletion did not have a polar effect preventing expression of the downstream genes. The unmarked, in-frame Δmce4AF and Δmce4HI deletion mutants are also very unlikely to have polar effects.
The Δmce4HI-C complementation strain consists of the Δmce4HI mutant with plasmid pTip (23) containing mce4HI, but not ro04706. The Δmce4HI-C strain also partially regained the ability to grow on cholesterol. This indicates that the last gene of the mce4 operon, ro04706, is not essential for growth on cholesterol. Overall, the above results demonstrate that each group of genes, supAB, mce4ABCDEF, and mce4HI, encode one or more proteins essential for growth on cholesterol.
Cholesterol Uptake
The role of the Mce4 system in cholesterol uptake was directly examined by measuring cholesterol uptake by wild-type RHA1 and the mutant strains. We assayed uptake of 14C-labeled cholesterol by resting cell suspensions. Attempts to use cholesterol-induced cells were unsuccessful as cholesterol used for induction could not be washed from the cells, making it impossible to control the concentration and specific activity of cholesterol in the assay. However, uninduced RHA1 cells grown on pyruvate had measurable cholesterol uptake activity. Uptake followed Michaelis-Menten kinetics and exhibited a low Km (0.6 ± 0.1 μM) for cholesterol (Fig. 3A). Uptake activity was abolished by preincubation of cells with the respiratory inhibitor, sodium azide, or either of two ATPase inhibitors, DCCD and vanadate, indicating that uptake is energy-dependent, and more specifically, ATP-dependent (Fig. 3B). The intended effect of DCCD in RHA1 is supported by previous studies (26, 27) that have shown that DCCD inhibits proton-driven ATPases of mycobacteria that are phylogenetically related, and physiologically similar, to RHA1. Further, the consistent effect of these two, mechanistically distinct ATPase inhibitors strongly indicates that they indeed inhibited the ATPase. The maximum specific uptake activity for the uninduced cells was 15 ± 2 μm/min/mg of protein, but we predict a higher rate for induced cells.
FIGURE 3. Cholesterol uptake by RHA1 and mutant strains.

A, kinetics of uptake by RHA1 (n = three independent experiments using different cultures). The curve represents the fitted Michaelis-Menten equation. B, specific uptake rates of RHA1 and mutants as well as effects of inhibitors (n ≥ three suspensions of cells from independent cultures, except n = two for DCCD treatment, bars indicate range).
The Δsup, Δmce4AF, and Δmce4HI strains had no measurable cholesterol uptake activity (Fig. 3B). These uptake phenotypes are entirely consistent with the growth phenotypes of the mutants. Moreover, the Δsup-C and Δmce4HI-C complementation strains had wild-type cholesterol uptake activity. These phenotypes are consistent with the growth phenotypes, but it is interesting that complementation is more complete in the uptake assay. The explanation for this difference in degree of complementation may be the use of uninduced cells for the uptake assay. Thus, expression of the supAB and mce4HI genes from pTip may be sufficient for uninduced uptake activity but may not be sufficient for maximal growth on cholesterol. These results clearly indicate that products of the mce4 genes are part of a cholesterol uptake system.
Uptake of Other Steroids
Wild-type RHA1 and the three deletion mutants were additionally tested for growth on a range of steroids and related compounds. RHA1 was able to grow on 4-androstene-3,17-dione, 5-α-cholestanol, 5-α-cholestanone, cholic acid, progesterone, and β-sitosterol, but not on cholcalciferol, cholestane, ergocalciferol, ergosterol, estradiol, or hydrocortisone. The Δsup, Δmce4AF, and Δmce4HI mutants also lost the ability to grow on β-sitosterol (Fig. 4), 5-α-choles-tanol, and 5-α-cholestanone (not shown). Cholesterol, 5-α-cholestanol, 5-α-cholestanone, and β-sitosterol have very similar structures. The deletion mutants were unaffected in growth on 4-androstene-3,17-dione, cholic acid, and progesterone, which differ notably from the above four compounds in having shorter, polar side chains. These results indicate that the Mce4 system takes up cholesterol, 5-α-cholestanol, 5-α-choles-tanone, and β-sitosterol. One or more other systems likely transport 4-androstene-3,17-dione, cholic acid, and progesterone, but we cannot exclude the possibility that the Mce4 system also transports the latter three compounds.
FIGURE 4.

Growth of RHA1 and mutant strains on various steroids (n ≥2 independent cultures, bars indicate range).
Association of mce4 Genes with Steroid Metabolism Genes
To obtain evidence that Mce4 systems are involved in steroid uptake in other bacteria, we searched available genome sequences with mce4 loci for genetically linked steroid metabolism genes. We searched for homologs of the RHA1 kshA gene, hsaADCB, and hsaEGF gene clusters. These genes encode most of the steps involved in degradation of rings A and B of the steroid nucleus to citric acid cycle intermediates. We previously found that, although the gene order varied in the genomes of RHA1 versus H37Rv, these genes were all in the region upstream of the mce4 loci of both genomes, with the hsaADCB and hsaFGE putative operons conserved in both organisms (20). For the current analysis, we examined the genomes of M. avium (104), M. bovis BCG, M. bovis (AF2122/97), M. marinum (M), M. smegmatis (MC2 155), M. ulcerans (Agy99), M. paratuberculosis (k-10), and N. farcinica (IFM 10152). Orthologs of all of the above steroid metabolism genes were found linked to (within 150 kbp of) the mce4 locus in each genome. The encoded proteins had greater than 70% sequence identity to their RHA1 orthologs, with the sole exceptions of KshA_k-10 (63%) and HsaB_IFM 10152 (67%). Interestingly, the Mce proteins are much less conserved than these catabolic enzymes, with only 26–46% identity between orthologous Mce proteins of RHA1 and H37Rv. The hsaADCB and hsaFGE putative operons were conserved in all the genomes. Thus, in all available genomes with mce4 loci, those loci are genetically linked to steroid metabolism genes.
DISCUSSION
This study provides the first direct experimental evidence that an mce locus encodes an uptake system (Fig. 3), which substantiates previous suggestions that mce loci encode a novel type of ABC transporter (9, 15, 25). This study further demonstrates that the mce4 locus of RHA1 encodes a system for steroid uptake. Like other ABC transporters (28), the Mce4 system of RHA1 is an active transporter that requires ATP. Minimally, this system transports cholesterol, 5-α-cholestanol, 5-α-cholestanone, and β-sitosterol (Fig. 4).
Several lines of evidence strongly indicate that all Mce4 systems function in steroid uptake. First, mce4 loci are invariably associated with putative steroid metabolism genes in the currently available genome sequences. Second, of the species known to have mce4 loci, four have been tested and consistently found to grow on cholesterol, M. smegmatis (29) R. jostii (20), M. bovis BCG (20), and M. tuberculosis (10). Third, the mce4 genes and putative cholesterol metabolism genes are co-regulated in M. tuberculosis and M. smegmatis (25) as well as R. jostii (20). Fourth, additional Mycobacterium spp. can grow on steroids (29). Thus, steroid metabolism and the Mce4 uptake system may be nearly ubiquitous in the genus, Mycobacterium, and may be widespread in others such as Rhodococcus and Nocardia. Importantly, steroids are abundant in nature, being present in nearly all phyla, making steroids valuable growth substrates for saprophytic microorganisms, which include most known actinomycetes and the ancestors of contemporary pathogenic actinomycetes. It appears that the Mce4 system diverged from other Mce systems when it evolved the capacity to transport steroids and became associated with steroid metabolism genes. It further appears that the Mce4 system was subsequently recruited for a role in pathogenesis, accounting for its conservation in many pathogenic actinomycetes with reduced genomes.
In RHA1, the sup, mce4, and ro04706 genes appear to be co-transcribed and regulated as a very large 11-gene operon (Fig. 1), consistent with the prediction that the mce1 operon of M. tuberculosis is transcribed as one large polycistronic message (30). Minimally, supA plus co-transcribed genes are induced during growth on cholesterol, which provides further evidence for the role of the mce4 genes in steroid uptake. Analysis of M. tuberculosis and M. smegmatis (25) suggests that the mce4 operon of RHA1 may be part of a much larger regulon that includes steroid metabolism genes and possibly additional genes and that is negatively regulated by KstR. Despite the inducibility of the mce4 operon, there appears to be substantial constitutive expression in RHA1 because uptake activity was measurable in uninduced cells. This is consistent with the relatively modest, 4-fold, up-regulation of sup4A that we measured. Constitutive activity would presumably be adaptive for substrate uptake systems because cells with no uptake activity might be unable to detect the presence of available substrates in their environment.
The available evidence implicates at least 11 proteins in a functional Mce4 steroid uptake system. These include an ATPase, two permease subunits (YrbE or Sup), and eight additional proteins of unknown function (Mce4ABCDEFHI). The absolute requirement for supAB, mce4ABCDEF, and mce4HI genes for uptake activity and the identical growth phenotypes of all three RHA1 deletion mutant strains (Figs. 2–4) indicate that a partially active system does not exist in the absence of any of the corresponding groups of proteins. In particular, although some Mce systems appear to lack the MceHI (Mas) proteins, those proteins are essential for a functional Mce4 system. Our results do not prove that all 10 of the Sup and Mce4 proteins are essential for steroid uptake activity but are consistent with this hypothesis. The essentiality of all 10 Sup and Mce4 proteins is also strongly suggested by the conservation of all 10 genes in the mce4 loci of diverse actinobacteria (15).
It is unclear why Mce systems require so many more proteins than do classical ABC transporters. It seems likely that the Mce proteins form a complex that fulfills the role of substrate-binding proteins in classical bacterial ABC uptake transporters. Additionally, such a complex may mediate movement of the substrate across thick cell walls. This function is consistent with the prediction that these proteins are extracytoplasmic, and in some cases, tethered to the membrane (15). This role might resemble that proposed for Isd proteins, relaying heme iron through Gram-positive bacterial cell walls to which Isd proteins are tethered (31). Notably, mce loci are almost exclusively found in mycolic acid bacteria, such as members of Mycobacterium, Rhodococcus, and Nocardia, which have thick, hydrophobic cell walls (32). The only exception is that mce loci are also present in members of Streptomyces. Interestingly, genes with similarity to mce genes are also present in some Gram-negative bacteria (15) and plants (33). Thus, this broader group of genes may encode a corresponding group of proteins functioning in transport across impermeable cell walls and outer membranes.
Understanding the biological function of the Mce4 system provides new insight to the apparent role of Mce4 proteins in pathogenesis of M. tuberculosis, suggesting many possible mechanisms. The published studies of mutations within the M. tuberculosis mce4 locus report attenuation of virulence in mice (8, 9) or impairment of macrophage infection (11). A straightforward explanation for this effect is that steroid metabolism by M. tuberculosis is an important part of pathogenesis. One possibility is that host cell cholesterol, which is relatively abundant, is an important growth substrate for the bacterium at some time during infection, intracellular growth, or intracellular persistence. Another possibility is that metabolic transformation of a steroid might permit the pathogen to manipulate its host. Alternatively, uptake of cholesterol may be an important signal allowing the pathogen to recognize and respond to the host environment. It is also possible that the ability of Mce4 proteins to bind cholesterol might play an important role in pathogenesis by localizing the bacterium, modifying the host membrane or eliciting a signal response from the host. Consistent with these possibilities, it was proposed that a possible lipid-binding motif of Mce4 proteins may be involved in binding or other bacterial interactions with host cells (4). Finally, previous suggestions that Mce proteins elicit an important immune response from the host (14) or facilitate host cell entry (1, 2) may be correct, with induction of the Mce4 proteins by host cell cholesterol possibly stimulating the processes. All of the above possibilities are consistent with reports that the presence of cholesterol is essential for infection of host cells by various mycobacteria (34–36). Given the complex and variable virulence phenotypes of M. tuberculosis mce mutants, effects of Mce proteins are likely complex and, possibly, multiple. Knowing that Mce systems function in uptake and that the Mce4 system functions specifically in steroid uptake suggests numerous testable hypotheses that may ultimately lead to elucidation of roles for the Mce proteins in pathogenesis of diverse actinobacteria, foremost of which is M. tuberculosis.
Supplementary Material
Oligonucleotides used in this study.
Acknowledgments
We thank Gerda Hessels (University of Groningen) for excellent technical assistance in the construction of RHA1 gene deletion mutants.
Footnotes
This work was supported by Discovery Grants from the Natural Sciences and Engineering Research Council of Canada (to W. W. M. and L. D. E.) and an Operating Grant from the Canadian Institutes of Health Research (to W. W. M.).
The on-line version of this article (available at http://www.jbc.org) contains a supplemental table.
The abbreviations used are: ABC, ATP-binding cassette; RT-PCR, reverse transcription-PCR; DCCD, dicyclohexylcarbodiimide; BCG, Bacille de Calmette et Guèrin.
References
- 1.Arruda S, Bomfim G, Knights R, Huimabyron T, Riley LW. Science. 1993;261:1454–1457. doi: 10.1126/science.8367727. [DOI] [PubMed] [Google Scholar]
- 2.Chitale S, Ehrt S, Kawamura I, Fujimura T, Shimono N, Anand N, Lu S, Cohen-Gould L, Riley LW. Cell Microbiol. 2001;3:247–254. doi: 10.1046/j.1462-5822.2001.00110.x. [DOI] [PubMed] [Google Scholar]
- 3.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Nature. 1998;396:190–198. [Google Scholar]
- 4.Mitra D, Saha B, Das D, Wiker HG, Das AK. Tuberculosis (Edinb) 2005;85:337–345. doi: 10.1016/j.tube.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Casali N, Konieczny M, Schmidt MA, Riley LW. Infect Immunol. 2002;70:6846–6852. doi: 10.1128/IAI.70.12.6846-6852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu S, Tager LA, Chitale S, Riley LW. Anal Biochem. 2006;353:7–14. doi: 10.1016/j.ab.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 7.El-Shazly S, Ahmad S, Mustafa AS, Raja Al-Attiyah R, Krajci D. J Med Microbiol. 2007;56:1145–1151. doi: 10.1099/jmm.0.47095-0. [DOI] [PubMed] [Google Scholar]
- 8.Sassetti CM, Rubin EJ. Proc Natl Acad Sci U S A. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. Proc Natl Acad Sci U S A. 2006;103:11760–11765. doi: 10.1073/pnas.0603179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandey AK, Sassetti CM. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosas-Magallanes V, Stadthagen-Gomez G, Rauzier J, Barreiro LB, Tailleux L, Boudou F, Griffin R, Nigou J, Jackson M, Gicquel B, Neyrolles O. Infect Immun. 2007;75:504–507. doi: 10.1128/IAI.00058-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gioffre A, Infante E, Aguilar D, de la Paz Santangelo M, Klepp L, Amadio A, Meikle V, Etchechoury I, Romano MI, Cataldi A, Hernandez RP, Bigi F. Microbes Infect. 2005;7:325–334. doi: 10.1016/j.micinf.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Senaratne RH, Sidders B, Sequeira P, Saunders G, Dunphy K, Marjanovic O, Reader JR, Lima P, Chan S, Kendall S, McFadden J, Riley LW. J Med Microbiol. 2008;57:164. doi: 10.1099/jmm.0.47454-0. [DOI] [PubMed] [Google Scholar]
- 14.Shimono N, Morici L, Casali N, Cantrell S, Sidders B, Ehrt S, Riley LW. Proc Natl Acad Sci U S A. 2003;100:15918–15923. doi: 10.1073/pnas.2433882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casali N, Riley LW. BMC Genomics. 2007;8:60. doi: 10.1186/1471-2164-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLeod MM, Warren RL, Hsiao WWL, Araki N, Myhre M, Fernandes C, Miyazawa D, Wong W, Lillquist AL, Wang D, Dosanjh M, Hara H, Petrescu A, Morin RD, Yang G, Stott JM, Schein JE, Shin H, Smailus D, Siddiqui AS, Marra MA, Jones SJM, Holt R, Brinkman FSL, Miyauchi K, Fukuda M, Davies JE, Mohn WW, Eltis LD. Proc Natl Acad Sci U S A. 2006;103:15582–15587. doi: 10.1073/pnas.0607048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haile Y, Caugant DA, Bjune G, Wiker HG. FEMS Immunol Med Microbiol. 2002;33:125–132. doi: 10.1111/j.1574-695X.2002.tb00581.x. [DOI] [PubMed] [Google Scholar]
- 18.Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST. Tuber Lung Dis. 1999;79:329–342. doi: 10.1054/tuld.1999.0220. [DOI] [PubMed] [Google Scholar]
- 19.Mathur M, Kolattukudy PE. J Biol Chem. 1992;267:19388–19395. [PubMed] [Google Scholar]
- 20.van der Geize R, Heuser T, Hara H, Wilbrink MH, Yam K, Anderton MC, Sim E, Davies JE, Dijkhuizen L, Mohn WW, Eltis LD. Proc Natl Acad Sci U S A. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauchop T, Elsden R. J Gen Microbiol. 1960;23:457–469. doi: 10.1099/00221287-23-3-457. [DOI] [PubMed] [Google Scholar]
- 22.van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L. FEMS Microbiol Lett. 2001;205:197–202. doi: 10.1016/s0378-1097(01)00464-5. [DOI] [PubMed] [Google Scholar]
- 23.Nakashima N, Tamura T. Biotechnol Bioeng. 2004;86:136–148. doi: 10.1002/bit.20024. [DOI] [PubMed] [Google Scholar]
- 24.Cornish-Bowden A. Analysis of Enzyme Kinetic Data. Oxford University Press; New York: 1995. [Google Scholar]
- 25.Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B, Frita R, ten Bokum A, Besra GS, Lott JS, Stoker NG. Mol Microbiol. 2007;65:684–699. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agarwal N, Kalra VK. Biochim Biophys Acta. 1983;723:150–159. doi: 10.1016/0005-2728(83)90114-7. [DOI] [PubMed] [Google Scholar]
- 27.Parrish NM, Ko CG, Hughes MA, Townsend CA, Dick JD. J Antimicrob Chemother. 2004;54:722–729. doi: 10.1093/jac/dkh408. [DOI] [PubMed] [Google Scholar]
- 28.Davidson AL, Chen J. Annu Rev Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 29.Av-Gay Y, Sobouti R. Can J Microbiol. 2000;46:826–831. [PubMed] [Google Scholar]
- 30.Kumar A, Bose M, Brahmachari V. Infect Immun. 2003;71:6083–6087. doi: 10.1128/IAI.71.10.6083-6087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Science. 2003;299:906–909. doi: 10.1126/science.1081147. [DOI] [PubMed] [Google Scholar]
- 32.Sutcliffe I. Antonie Leeuwenhoek. 1998;74:49–58. doi: 10.1023/a:1001747726820. [DOI] [PubMed] [Google Scholar]
- 33.Garcia O, Bouige P, Forestier C, Dassa E. J Mol Biol. 2004;343:249–265. doi: 10.1016/j.jmb.2004.07.093. [DOI] [PubMed] [Google Scholar]
- 34.de Chastellier C, Thilo L. Cell Microbiol. 2006;8:242–256. doi: 10.1111/j.1462-5822.2005.00617.x. [DOI] [PubMed] [Google Scholar]
- 35.Gatfield J, Pieters J. Science. 2000;288:1647–1650. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- 36.Peyron P, Bordier C, N’Diaye EN, Maridonneau-Parini I. J Immunol. 2000;165:5186–5191. doi: 10.4049/jimmunol.165.9.5186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotides used in this study.