

Abstract
Syntheses and enzymological characterization of fluorogenic substrate probes targeting secretory phospholipase A2 (sPLA2) for detection and quantitative assays are presented. Three fluorogenic phosphatidylcholine analogs PC-1, PC-2, and PC-3 each containing the duo of 7-mercapto-4-methyl-coumarin fluorophore and 2,4-dinitroanaline quencher on either tail were synthesized from (R)-3-amino-1,2-propanediol and R-(−)-2,2-dimethyl-1,3-dioxolane-4-methanol. These small reporter groups are advantageous in preserving natural membrane integrity. Phosphocholine was incorporated into the sn-3 position of the glycerol backbone. Acyl amino group at the sn-1 position in PC-1 and PC-2 is meant to block sPLA1. The sn-1 and sn-2 positions of the glycerol backbone in PC-1 have a quencher terminated 12-carbon chain and fluorophore terminated 11-carbon chain respectively. PC-2 has a quencher terminated 3-carbon chain at the sn-2 and chain terminating fluorescent reporter at the sn-1 positions. PC-3 resembles PC-1 except for an ester instead of amide at the sn-1 position, because of which it is more similar to natural phospholipids than PC-1. It was designed to elucidate the effect of replacing the ester group with amide by comparing its hydrolysis rate with that of PC-1. Design principles apply to synthesis of other labeled phospholipids. Enzymological characterization using bee-venom sPLA2 was performed by a fatty-acid-binding-protein fluorescence assay and by pH-Stat method in which the amount of fatty acid released by hydrolysis is given by the amount of base required to maintain a constant pH of 8.0. Hydrolytic activity toward PC-1 and PC-3 were each about 238 ± 25 μmol/mg/min and 537 μmol/mg/min on unmodified phospholipid. Ester to amide change did not affect hydrolysis rates. Activity toward PC-2 was about 45-μmol/mg/min. PC-1 and PC-3 show potential for targeted real-time spectrophotometric assay of sPLA2.
Keywords: Phospholipid syntheses, Fluorescent phospholipid substrates, Phospholipase A2 assay
1. Introduction
Phospholipids are the most abundant of lipids in biological membranes. They are composed of a glycerol backbone, substituted with two fatty acid side chains and a phosphate group. Phospholipids can be degraded through phospholipases into precursors of signaling molecules, such as arachidonic acids. Phospholipases A2 (PLA2) are a large family of enzymes that catalyze the hydrolysis of the sn-2-ester bond of glycerophospholipids (Dennis et al., 2011) releasing fatty acids and lysophospholipids (Eq.1) that can serve as precursors of anti-inflammatory lipid mediators (Murakami et al., 2011).
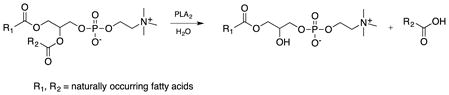 |
(1) |
The mammalian family of secreted phospholipases A2 enzymes (sPLA2s) are comprised of 10 enzymatically active isoforms and one inactive isoform (Lambeau and Gelb., 2008; Murakami et al., 2015). sPLA2 are small secreted enzymes of 14–18 kDa with the exception of group III (Dennis et al., 2011). These phospholipases have been found in a variety of physiological and pathophysiological conditions (Murakami et al., 2011). Secretory PLA2 are present in several mammalian tissues and extensively expressed at transcript level in some human tissues (Tribler et al., 2007). They may also play a role in tumorigenesis and tumor metastasis (Yamashita et al., 1994, Hu et al., 2011). In this context, the serum level of the enzyme is strongly dependent on factors such as the stage of cancer, and the degree of metastasis (Arouri et al., 2013). Concentration of the enzyme in serum has been found to correlate with the severity of pancreatitis. (Nevalainen et al., 2008). sPLA2 has been reported to be an inflammatory factor in Alzheimer’s disease (Chalbot et al., 2009), predicating its potential as a biomarker in screening neuroinflammatory disorders. Furthermore, sPLA2-IIA was shown to have a critical role in the development of cardiovascular diseases (Koenig, 2009). This enzyme is found at elevated levels in patients with coronary heart disease, and has thus been used as a biomarker to predict recurrent coronary events, independent of other risk factors (Kujiyama et al., 1999; Mallat et al., 2007; Zakynthinos and Pappa, 2009). The implications of sPLA2 in diverse pathologies has been a springboard for the development of sensitive real-time sPLA2 detection assays, the quantitation of sPLA2 catalysis, and for the kinetic characterization and mechanistic elucidation of the action of these enzymes. Delineation of the physiological roles of sPLA2, yet unclear, continues to be a vital area of research.
Phospholipase A2 activity has previously been assayed using titrimetric, colorimetric, fluorometric, and radiometric techniques (Reynolds et al, 1991; Kethineedi et al., 2013). These techniques are time consuming, suffer from limited sensitivity, and present significant safety concerns (Kethineedi et al., 2013). For example, early efforts that focused on development of chromogenic PLA2 assays using sn-2-thioester phospholipids led to PLA2-catalyzed formation of sn-2-thiolate to be trapped by reaction with DTNB (5,5’-dithio-bis-2-nitrobenzoic acid). However, thioester hydrolysis by the enzyme occurred at a substantially lower rate than the catalytic hydrolysis of natural substrate (Balet et al., 1988; Reynolds et al., 1991). In addition, subsequent research indicated that the PLA2-catalyzed thioester hydrolysis is likely to proceed via a different reaction mechanism (Linderoth et al., 2009).
There is a recognized need for fluorogenic substrate design to enable real-time in situ assays for diagnostic, functional, and mechanistic studies of PLA2 in various cells and tissues (Cho, 2006). More recently, fluorescence displacement (Wilton, 1990) and fluorescence resonance energy transfer (FRET) based methods, were developed to enable simpler and more direct measurements of PLA2 activity (Rose and Prestwich, 2006; Wichmann et al., 2007). The fluorogenic phospholipids in these assays were used mostly for qualitative to semi-quantitative activity measurements.
The photophysical stability of the fluorophores (Kethineedi et al., 2013) and the effect of the size of the reporter groups on lipid membranes are recognized as important issues in the design of fluorogenic probes and assays (Skaug et al., 2011). We have been engaged in developing new fluorogenic probes and assays for the detection and kinetic characterization of secretory phospholipase A2 enzymatic activity (Wang, et al., 2013). Small reporter groups employed in the present design should help minimize any impact on the structure of the lipid membrane system (Wang, et al., 2013). The FABP-assisted fluorescence assay described below enables a quantitative assessment of these considerations.
In this communication, we describe the design and enzymological characterization of PLA2-directed fluorogenic phosphatidylcholine analogs PC-1, PC-2 and PC-3 (Fig. 1). An sn-1-acylamino group is introduced to block the action of PLA1 and thus achieve selectivity toward PLA2. (R)-3-amino-1,2-propanediol was used to provide a chiral glycerol backbone. Phosphocholine was incorporated into the sn-3 position of the glycerol. PC-1 has a 12 carbon chain at the sn-1 position of the glycerol, terminated by the quencher and a long chain-terminal fluorescent reporter group at the sn-2 position of the glycerol. PC-2 has a 3 carbon chain at the sn-2 position of the glycerol with the terminal quencher and a chain-terminal fluorescent reporter group at the sn-1 position of the glycerol. The protecting group p-methoxy trityl placed in sn-3 position could be removed without acyl migration. This strategy reduced the number of steps in the synthesis. PC-3 resembles PC-1 except for an ester functional group placed in the sn-1 position rather than an acyl amino group. PC-3 bears more similarity, than PC-1, to natural phospholipids since almost all of which have an ester functional group in the sn-1 position. PC-1 and PC-2 were designed to block PLA1. PC-3 on the other hand can be hydrolyzed by PLA2 as well as PLA1. It was designed to test the effect of replacing the amide by ester on the rate of hydrolysis by sPLA2. Furthermore, the synthetic strategy to make PC-3 is different from the first two analogs and would give access to a variety of labeled phospholipids that contain ester group in the sn-1 position.
Figure 1.

Structures of fluorescently labeled phosphatidylcholines PC-1, PC-2 and PC-3.
For enzymological characterization of the probes, a fatty acid binding protein was added to the assay mixture to bind and sequester the fatty acid produced by the catalytic reaction. Before hydrolysis, the fluorescence is quenched because of the fluorophore-quencher proximity. The sequestering action of the water soluble FABP in the aqueous phase, separates the fluorophore and quencher, thus preventing intermolecular quenching. Presence of the hydrolysis reaction thus manifests as an increase in fluorescence emission intensity of the fluorophore. The method should be useful for development of real-time spectrophotofluorimetric assay of phospholipases to detect their biological activity. Furthermore, the synthesis described in this communication provides an efficient route for the preparation of a variety of phospholipid derivatives with chain-terminal reporter groups.
The rationale for the difference in the design of PC-1 and PC-2 was that the shorter 3 carbon chain in PC-2 being less hydrophobic than the corresponding longer chain in PC-1 would release into the aqueous phase upon hydrolysis without assistance from FABP, making the use of FABP unnecessary for PC-2 whereas PC-1 with a longer chain would require FABP assistance to detect hydrolysis.
2. Results and Discussion
2.2. Design and Syntheses
The synthetic strategy to make PC-1, PC-2 and PC-3 are shown in Scheme 1, Scheme 2 and scheme 3 respectively. 12-aminolauric acid 1 provided the desired carbon chain length to make long chain quencher 3. Pentafluorophenol 4 in the presence of DCC-DMAP was used to turn carboxylic acid 3 into an active ester 5. (R)-3-Amino-1,2-propanediol 6 provided the chiral glycerol backbone. Reaction of diol 6 with the active ester gives the amide 7. Primary alcohol of the amide 7 was protected by treating it with para-methoxytriylchloride and DMAP for 90 minutes to afford 8. Formation of a sulfide linkage by base catalyze alkylation of 7-mercapto-4-methylcoumarin using 11-bromoundecanoic acid in acetone gave chemically stable thioether 9 (Wang, et al 2013). sn-2 position of the glycerol 8 was acylated using DMAP as a catalyst to give 10 in quantitative yield. Methoxytrityl of 10 was successfully removed using 0.1 M HCl to yield alcohol 11. sn-3 phosphocholine head group was installed on alcohol 11 in two consecutive steps to yield PC-1. Similar reagents and reaction conditions were applied to synthesize PC-2.
Scheme 1.

Reagents and conditions: (a) KOH, 1 M HCl, dioxane, 24h; (b) DCC-DMAP, CH2Cl2, 35min; (c) (R)-3-Amino-1,2-propanediol, DIPEA, CHCl3/CH3OH, 45min; (d) 4-methoxytriphenylmethyl chloride, DMAP, CH2Cl2, 90min; (R)-3-Amino-1,2-propanediol (R)-3-Amino-1,2-propanediol (e) DCC-DMAP, CH2Cl2, 24h; (f) 0.1 M HCl, CHCl3/CH3OH,0°C 90min; (g) (i) ethylene chlorophosphate, (C2H5)3N, C6H6, rt., 24h, (ii) (CH3)3N, CH3CN, 67°C, 48h.
Scheme 2.

Reagents and conditions: (a) (i) Pentafluorophenol 4, DCC-DMAP, CH2Cl2, 35min (ii) (R)-3-Amino-1,2-propanediol, DIPEA, CHCl3/CH3OH, 45min; (b) TEA, DMAP, CH2Cl2, 1h; (c) DMAP, CH2Cl2, 1h; (d) 0.1 M HCl, CHCl3/CH3OH, 0°C 1h; (e) (i) ethylene chlorophosphate, (CH2CH3)3N, benzene, rt., 24h, (ii) (CH3)3N, CH3CN, 67°C, 48h.
Scheme 3.

Reagents and conditions: (a) DCC-DMAP, CHCl3, 30min; (b) 0.5 M HCl in 1,4-dioxane, 1M KOH, 2h; (c) 2,3,5 collidine, CH2Cl2, 2h; (d) DHP, PPTs, CH2Cl2, 1h; (e) 10% piperidine in CH2Cl2, 1h; (f) (i) N(CH2CH3)3, ethylene chlorophosphate, C6H6, r.t, 24 h; (ii) N(CH3)3, CH3CN, 67 °C, 18h; (g) 0.15 M HCl in 1,4-dioxane, r.t., 1h; h) #9, DCC-DMAP, ultrasound, 37°C, 12hrs.
A different synthetic approach had to be designed for PC-3 since it contained an ester functional group at the sn-1 position of glycerol. Acylation of chiral dioxolane 18 by quencher carboxylic acid afforded the ester 19. An acid catalyzed ring opening of the acetonide of 19 was carried out with 0.5 M HCl in dioxane to afford the diol 20 in 86% yield. Reaction conditions needed to be optimized in order to achieve a regioselective protection of the diol. We envisioned that the best approach in protecting the sn-2 and sn-3 of glycerol would be using an acid-labile and base-labile protecting group, respectively, allowing selective cleavage of one protecting group. To our delight, fluorenylmethyloxycarbonyl chloride (FMOC-Cl) in the presence of 2,3,5 collidine afforded the protected sn-3 hydroxyl group with decent yield (58%). Tetrahydropyranilation of sn-2 glycerol of 21 was achieved by using excess DHP and PPTS as catalyst. Removal of FMOC in 10% piperidine afforded the alcohol 23 in quantitative yield. Phosphorylation of 23 was performed in two consecutive steps to afford sn-3 phosphocholine 24 in 48% yield. Acid catalyzed cleavage of tetrahydropyranyl was subsequently carried out with 0.15 M HCl in aqueous dioxane to give 25. Finally, the sn-2-hydoxyl of 25 was acylated by a fluorescent carboxylic acid using ultrasound to yield the fluorescently labeled phosphatidylcholine analog, PC-3.
2.3. Enzymological Characterization of the Substrate Properties of the Synthetic Phospholipids
Rates of bee-venom PLA2 (BV-PLA2) catalyzed hydrolysis of PC-1 dispersed in DPS micelles were measured by an FABP based fluorescence assay. Hydrolysis of PC-1 by PLA2 breaks down the phospholipid sequestered in the mixed micelle into lysophospholipid with the quencher (PC-1) or fluorophore (PC-2) and fatty acid with the quencher (PC-2) or fluorophore (PC-1). The water soluble FABP present in the aqueous phase of the reaction mixture binds the fatty acid carrying the quencher (PC-2) or fluorophore (PC-1) and thus separates it from the lysophospholipid carrying the quencher (PC-1) or fluorophore (PC-2) with the result that the fluorescence intensity increases as the proximity of the quencher to the fluorescence moiety decreases. Fluorescence spectra, following addition of PLA2 to the PC-1 substrate/dodecyldimethylammonio propane sulfonate (DPS) mixed micelle solution, was measured. DPS was chosen as the host medium because DPS/lipid mixed micelles are stable, spherical, and do not change in shape with concentration as opposed to bile salts that have been the choice historically (Singh, et al., 2008). This property of DPS is important when comparing activities toward different lipids and concentration dependence studies. pH-Stat assays were conducted to compare the activities of BV-PLA2 toward each of the lipids PC-1, PC-2, and DPPC (dipalmitoylphosphatidylcholine)) dispersed in DPS micelles.
2.3.1. Preparation of Phospholipid/DPS Mixed Micelles and Reaction Mixture
The mixed micelle composition was [DPS] = 2 mM, [PC-1] = 100 μM for the fluorescence assay of PC-1 hydrolysis and [DPS]= 5 mM and [Phospholipid: PC-1, PC-2, PC-3, or DPPC] = 1 mM for the pH-Stat assay. Appropriate amount of the phospholipid to be investigated was mixed with a few drops of ethanol. The resulting ethanol solution mixture was vortexed thoroughly to produce a clear solution which was then dried under dry N2 flux to produce a film of the lipid. Thereafter, the required amounts of the DPS surfactant and phosphate buffer (for the fluorescence assay) or water (for the pH-Stat assay) were added to the dry film to achieve the final concentrations. The solution was stirred overnight to ensure the complete solubilization of phospholipid in surfactant micelles (Singh and Ranganathan, 2014). Reaction mixture consisted of mixed micelle solution with 10 mM CaCl2 and BV-PLA2.
Phospholipase A2 (PLA2) from honey bee-venom was obtained from Sigma as a lyophilized powder. The enzyme was dialyzed against 0.05 M sodium phosphate buffer at pH 8.0 for three days, changing the buffer every 8 hours (Singh and Ranganathan, 2014). Protein concentration was determined by Lowry’s method (Lowry et al,. 1951) using bovine serum albumin (BSA) as standard and also by the extinction coefficient method (Scopes 1974, Stoscheck 1990). Both of these methods gave results consistent to within ±5%. The dialyzed enzyme was stored at pH 7.5 in 0.05 M sodium phosphate buffer at 4°C.
2.3.2. Fluorescence Assay of Hydrolysis of PC-1
Experiments and Results
Fluorescence emission spectra were measured using a Fluoromax-4 Spectrometer (Horiba Scientific), before and at various times after addition of calculated amounts of the enzyme stock (0.11 mg/mL) to achieve the required final enzyme concentration. The excitation wavelength was 335 nm and the emission spectra were recorded from 350 nm to 500 nm. Sample temperature was maintained at 37°C with a thermostated water bath.
Fig. 2 shows the time evolution of fluorescence spectra following addition of PLA2. The peak of the emission is at 440 nm. The increase in the fluorescence intensity reflects the decrease in quenching due to hydrolysis and increase in separation between quencher and fluorophore.
Figure 2.

Fluorescence spectra of the Coumarin label of PC-1 following addition of BV-PLA2 to PC-1/DPS mixed micelles, at the times indicated.
The continuous time course of hydrolysis was determined by recording the fluorescence emission intensities at 440 nm at 6-second intervals, following addition of PLA2, using the Multi option mode of the Fluoromax-4. In this mode of operation, the spectrometer grating stays at one position and facilitates temporal measurements of the intensity at a user selected wavelength. Experiments were conducted for four different enzyme concentrations. The measured intensity variations with time are shown in Fig. 3.
Figure 3.

Time course of hydrolysis of PC-1 dispersed in DPS micelles for different enzyme concentrations, measured by continuously monitoring the peak emission intensity as a function of time lapsed after addition of BV- PLA2.
Linear regression analysis of the observed variation yielded the slope. The rate of formation of hydrolysis products is represented by the slope. Fig. 4 displays the linear dependence of the rate of hydrolysis on enzyme concentration. The linearity of the data in Fig. 3 and 4 demonstrate the potential of the present synthetic phospholipid PC-1 as a real-time photometric probe for detecting presence of PLA2.
Figure 4.

Dependence of the rate of BV-PLA2 catalyzed hydrolysis of PC-1 in DPS micelles on the concentration of the enzyme. The rate of hydrolysis is the slope of the linear fit to the data in Figure 3.
2.3.3. Comparison of PC-1, PC-2, PC-3 and DPPC as substrates for BV-PLA2 using pH-Stat assay
An underlying principle motivating our syntheses strategies has been the minimization of the effect of the labels on substrate hydrolysis rates. (Wang, et al., 2013) While the fluorescence data demonstrate the intended use of the flouorescence/quencher labeled phospholipid, the impact of attaching reporter groups on the rate of substrate hydrolysis can be assessed only by comparing the rates of hydrolysis of the labeled lipids with that of unmodified natural phospholipids.
Rates of hydrolysis of PC-1, PC-2, PC-3 and DPPC by BV-PLA2 were measured using a pH-Stat assay. This method makes comparisons between substrates meaningful because theis assay yields the enzyme activity directly in standard units of μmol of fatty acid released, due to lipid hydrolysis, per minute per milligram of enzyme (μmol/min/mg). The principle behind this method is that the fatty acid released by the hydrolysis reaction causes the pH to drop and the amount of NaOH needed to bring the pH back to a preset value is a direct measure of enzyme activity. (Singh, et al., 2008, Singh, et al., 2010)
The activity of BV-PLA2 in mixed micelles of phospholipid (PC-1, PC-2, PC-3 or DPPC)/DPS was measured by monitoring the amount of 0.01M NaOH required per minute to maintain a constant pH of 8.00 following addition of 5 μL of enzyme stock (concentration of 0.2-mg/mL) to 5 mL of the mixed micelles solution, The pH-Stat equipment was a Radiometer assembly of a titrator, an auto burette, Model TIM 854 electrode and a pH meter interfaced to a computer for recording data. The reaction was followed for 8-min. The initial rate of activity was determined from the first 2 to 4-min of data. The yield increases linearly with time and the slope of the line divided by the mass of the enzyme in mg gives the activity, in units of μmol/min/mg. The error in the fitted slopes was < 1%. All activity measurements were conducted at 37 °C.
BV-PLA2 activities toward the three lipids are compared in Fig. 5. The amount of fatty acid released per mg of enzyme increases linearly with time. Activity is the slope of this line. The slope is greatest for DPPC with a value of 537 μmol/mg/min. The activity toward PC-1 and PC-3 was about two-fold less at 238 μmol/mg/min. The activity toward the short chain phospholipid, PC-2, was about 45 μmol/mg/min (about twelve times less than that on DPPC). PC-2 was designed for investigating the ability to detect hydrolysis without using FABP. However results show that the shorter chain in PC-2 impairs hydrolysis.
Figure 5.

Amount of fatty acid released upon hydrolysis per mg of BV-PLA2 for each of PC-1, PC-2, PC-3, and DPPC in DPS micelles vs. time following addition of the enzyme. The slopes are the rates of hydrolysis.
In conclusion, the synthetic approach to make PC-1 and PC-2 described here is superior to our previous published work (Wang, et al., 2013) since one less protecting group is required; thereby, cutting the number of steps in the synthesis. Furthermore, methoxytrityl could be removed without intramolecular acyl migration. Principles are applicable to design of various other labeled phospholipids. The acyl amino group at the sn-1 position in PC-1 was designed to target sPLA2 and block sPLA1. Explicit experiments were not conducted to show the blocking of sPLA1; but it is reasonable to expect that the amide group should achieve its purpose. PC-1 and PC-3 exhibit identical rates of hydrolysis by BV sPLA2. Replacement of the acyl amino group in PC-1 by ester does not affect the rate of hydrolysis. The labeling moieties are small compared to those that have been used and reported in literature (Wichmann et al,. 2007). The present synthetic phospholipid substrates are thus an improvement over lipids with bulky labels. The doubly labeled phospholipids PC-1 and PC-3 show only a two-fold reduction in the rate of hydrolysis from that of unmodified phospholipids. Assays of BV-PLA2 activity show that the PC-1 and PC-3 phospholipids are sensitive spectrophotometric probes for in-vitro detection of PLA2.
3. Experimental Procedures for the Syntheses
3.1. Syntheses
3.1.1. N-(2,4-dinitrophenyl)aminododecanoic acid (3)
1.72 g (8 mmol) of 12-aminododecanoic acid 1 was dissolved in 16 mL of 1,4-dioxane followed by addition of 16 mL of 1 M KOH. The solution was stirred for 5 minutes using a magnetic stir bar. 1.49 g (8 mmol) of 1-fluoro-2,4-dinitrobenzene 2 (DNFB) was added dropwise to the reaction mixture. The solution turned yellow upon the addition of DNFB. After 18 hours, 1 M HCl was added slowly to the reaction mixture to bring the pH down to 2. Yellow precipitate was suction filtered. The bright yellow solid product 3 was air dried overnight (2.60 g, 86.0%). IR (CHCl3): 2938, 1623, 1523, 1342 cm−1. 1H NMR (CDCl3, 400MHz): δ 9.10 (s, 1H), 8.78 (s, 1H), 8.40 (dd, J = 2.8, 9.6 Hz, 1H), 7.60 (d, J = 10 Hz, 1H), 3.55 (q, J = 6.5 Hz, 2H), 2.50 (t, J =7.4 Hz, 2H), 1.94-1.34 (m, 18H); 13C NMR (CDCl3, 100 MHz) δ 179.7, 148.4, 135.9, 130.3, 124.3, 113.9, 67.0, 43.6, 34.0, 29.4, 29.3, 29.2, 29.1, 29.0, 28.7, 26.9, 24.6; Rf= (CHCl3/MeOH 98:2) = 0.40
3.1.2. Perfluorophenyl 12-((2,4-dinitrophenyl)amino)dodecanoate (5)
Pentafluorophenol 4 (0.58 g, 3.1 mmol) was added to a solution of compound 3 (1.0 g, 2.6 mmol) in 20 mL dichloromethane. A catalytic amount (50 mg) of 4-Dimethylaminopyridine (DMAP) and (DCC) (0.65 g, 2.6 mmol) were added to the reaction mixture. The reaction was stopped after 35 minutes and the white precipitate (DCC-urea) was removed by vaccum filtration and rinsed with cold dichloromethane. The liquid was collected and the solvent was removed on a rotary evaporator at 37°C. The product 5 was purified by column chromatography on silica gel using as eluent afforded a yellow solid. (1.10 g, 74.0%) Rf (CHCl3) = 0.92.
3.1.3. N-(2,3-dihydroxypropyl)-12-((2,4-dinitrophenyl)amino)dodecanamide (7)
Compound 5 (1.0 g, 1.8 mmol) was dissolved in 15 mL of freshly distilled chloroform and to it was added, 6 (R)-3-Amino-1,2-propanediol (0.32 g, 3.6mmol) in 15 mL methanol. Diisopropylethylamine (DIPEA) (0.23 g, 1.8 mmol) was added to the reaction mixture. The reaction was stopped after 35 minutes and the solvent was removed on a rotary evaporator at 37°C. Purification was performed on silica gel column chromatography twice using (CHCl3/MeOH 9:1) to afford a yellow solid 7 (1.30g, 84.0%). IR (CHCl3): 3362, 2925, 2854,1589 cm−1; 1H NMR (CDCl3, 400 MHz): δ 9.16 (s, 1H), 8.57 (s, 1H), 8.27 (d, J = 10 Hz, 1H), 6.93 (d, J = 10 Hz, 1H), 5.97 (s, 1H), 3.80-3.72 (m, 1H), 3.60-3.48 (m, 2H), 3.47-3.36 (m, 4H), 3.14-2.97 (m, 2H), 2.22 (t, 7.6 Hz, 2H), 1.83-1.72 (m, 2H), 1.68-1.58 (m, 3H), 1.52-1.41 (m, 2H), 1.29 (br s, 12H); 13C (NMR) (CDCl3, 100 MHz); δ 175.3, 148.4, 130.4, 124.4, 113.9, 71.2, 63.5, 43.6, 42.2, 36.5, 29.3, 29.2, 29.1, 29.0, 28.6, 26.9, 25.6; MS MH+ (C21H34NO7H+) Calcd: 455.2500, Found: 455.2515; Rf (CHCl3/MeOH 9:1) = 0.35.
3.1.4. 12-((2,4-dinitrophenyl)amino)-N-(2-hydroxy-3-((4methoxyphenyl)diphenylmethoxy) propyl) dodecanamide (8)
Compound 7 (1.0 g, 2.2 mmol) was dissolved in 15 mL dichloromethane and to it, was added DMAP (0.32 g, 2.6 mmol) followed by 4-methoxytrityl chloride (0.82 g, 2.6 mmol). The reaction was followed by TLC and stopped after 90 minutes and the solvent was evaporated using a rotary evaporator at 37°C. Purification was performed on silica gel column chromatography using CHCl3/MeOH (9.7:0.3) as eluent to give a yellow product 8 (0.70 g, 87.0%). IR (CHCl3): 3363, 2853, 1524, cm−1; 1H NMR (CDCl3, 400 MHz): δ 9.09 (s, 1H), 8.51 (s, 1H), 8.20 (d, J = 9.2, 1H), 7.50-7.15 (m, 13H), 6.86 (d, J = 9.2 Hz, 1H), 6.78 (d, J= 7.6 Hz, 2H), 5.68 (t, 1H), 3.85 (s, 1H), 3.74 (s, 3H), 3.55-3.45 (m, 1H), 3.40-3.30 (m, 2H), 3.22-3.05 (m, 4H), 2.03 (t, J = 8.0 Hz, 2H), 1.75-1.65 (m, 2H), 1.55-1.35 (m, 4H), 1.22 (br s, 11H); 13C (NMR) (CDCl3, 100 MHz); δ 174.5, 158.6, 148.4, 144.2, 135.9, 135.2, 130.3, 130.2, 128.3, 127.9, 127.0, 124.4, 113.9, 113.2, 86.5, 70.4, 64.7, 55.2, 43.6, 42.9, 36.5, 29.4, 29.3, 29.2, 29.1, 28.7, 26.9, 25.6); MS MNa+ (C41H50N4O8Na+) Calcd: 749.3521, Found: 749.3538; Rf (CHCl3/CH3OH 9.7:0.3) = 0.34; [α]D25: 8.2° (c 1.00, CHCl3-MeOH 4:1).
3.1.5. 11-(7′-mercapto-4′-methyl-coumarin)undecanoicacid (9)
To a suspension of K2CO3 (1.0g, 7.2mmol) in 20mL acetone that has been bubbled by nitrogen for 20 minutes, 7-mercapto-4-methyl coumarin (0.72 g, 3.8 mmol) was added. Reaction mixture was stirred at room temperature for 30 minutes and a cloudy orange solution formed. To this solution was added 11-bromoundecanoicacid (1.2 g, 4.5 mmol) in 15 mL acetone, yielding a pale yellow precipitate. The reaction was stirred for 2 hours at room temperature and solvent was evaporated with a rotary evaporator to form a pale yellow product. 100 mL deionized water was added to this pale yellow residue, followed by addition of 1 M HCl until the pH was 4. The resulting solution was extracted with 100 mL CHCl3. The organic layer was washed with 100 mL deionized water and extracted again. The organic layer was dried over anhydrous sodium sulfate and the solvent was removed with a rotary evaporator to give a light yellow residue. This residue was chromatographed twice on silica gel using CHCl3-EtOAc (9:1) as eluent. The fractions of the product were collected and solvents were evaporated to give a very light yellow powder 10 (1.30 g, 87%); Rf (CHCl3-EtOAc 9:1) = 0.20. Our experimental data matched that of a previous report (Wang, et al., 2013).
3.1.6. 12-((2,4-dinitrophenyl)amino)-N-(3-((4-methoxyphenyl)diphenylmethoxy)-2-((11-((4-methyl-2-oxo-2H-chromen-7-yl)thio)-1-oxo-1λ3,2λ5-undecan-2-yl) oxy)propyl) dodecanamide (10)
To a solution of compound 8 (1.0 g, 1.4 mmol) in 15 mL dichloromethane was added fluorescent carboxylic acid 9 (0.78 g, 3.8 mmol), DCC (0.43 g, 3.8 mmol) and 50 mg of DMAP. The reaction was stopped after 18h and solvent was removed on a rotary evaporator at 37°C. Purification was performed on silica gel column chromatography using (CHCl3/EtOAc 9:1) to give a yellow oil product 10 (1.42 g, 95.0%). 1H NMR (CDCl3, 400MHz): δ 9.08 (s, 1H), 8.48 (s, 1H), 8.19 (d, J = 8.8, 1H), 7.38-7.05 (m, 14H) 6.84 (d, J = 9.2 Hz, 1H), 6.76 (d, J = 6.8Hz, 2H) 7.60-7.20 (m, 13H), 6.17 (s, 1H), 5.62 (app, br s, 1H), 4.97 (app, br t, 1H), 3.75 (s, 3H), 3.60-3.15 (m, 6H), 2.90 (t, J = 6.4 Hz, 2H), 2.39 (s, 3H), 2.32 (t, J = 7.2Hz, 2H), 1.98 (app. t, J = 7.2Hz, 2H), 1.70-1.50 (m, 2H), 1.30 (br s, 26H); MS MNa+C62H76N4O11SCalcd: 1107.5124, Found: 1107.5113; Rf (CHCl3/EtOAc 9:1)= 0.55.
3.1.7. 12-((2,4-dinitrophenyl)amino)-N-(3-hydroxy-2-((11-((4-methyl-2-oxo-2H-chromen-7-yl)thio)-1-oxo-1λ3,2λ5-undecan-2-yl)oxy)propyl)dodecanamide (11)
Compound 10 (1.0 g, 0.9 mmol) was dissolved in 10 mL CHCl3:MeOH (1:1). 5 mL HCl (1 mL 12 M HCl in 125 mL CHCl3: MeOH (1:1) at 0°C was added dropwise to the solution. The reaction mixture was stirred for 90 min. The reaction was quenched with 10 mL of NaHCO3 solution (1.5 g in 50 mL H2O) stirred for 15 min at 0°C. After neutralization, the solution was extracted with 2 × 10 mL of chloroform. The organic layer was recombined and re-extracted with 5 mL brine dried over anhydrous sodium sulfate. The solvent was evaporated on a rotary evaporator and the residue purified with a silica gel chromatography CHCl3/EtOAc (9:1) was used to give a yellow solid product 11 (0.68g, 91.0%). IR (CHCl3): 3364, 2925, 1601, 1524, 1312 cm−1; 1H NMR (CDCl3, 400 MHz): δ 9.11 (s, 1H), 8.55 (s, 1H), 8.25 (d, J = 6.8 Hz, 1H), 7.45 (d, J = 8.8 Hz, 1H), 7.16-7.10 (m, 2H), 6.91 (d, J = 9.6 Hz, 1H), 6.18 (s, 1H), 6.0 (app. br t, 1H), 4.83 (app, pen, J = 4.8 Hz, 1H), 3.86 (t, J = 7.2 Hz, 1H), 3.60-3.25 (m, 6H), 2.97 (t, J = 8 Hz, 2H), 2.37 (s, 3H), 2.31 (t, J = 7.6 Hz, 2H), 2.22 (t, J = 7.2 Hz, 2H), 1.75-1.30 (m, 13H), 1.32 (br s, 20H); 13C (NMR) (CDCl3, 100 MHz) δ (175.1, 173.3, 160.8, 153.9, 152.4, 148.4, 143.8, 135.9, 130.3, 130.2, 124.5, 124.3, 123.0, 117.0, 113.9, 113.7, 113.6, 72.8, 60.4, 43.6, 38.8, 36.5, 34.3, 32.1, 31.2, 29.3, 29.2, 29.1, 29.0, 28.9, 28.8, 26.9, 25.6, 24.9, 18.6; MS MNa+ (C42H60N4O10SNa+) Calcd: 835.3922, Found: 835.3925; Rf (CHCl3/EtOAc 9:1) = 0.17.
3.1.8. 3-(12-((2,4-dinitrophenyl)amino)dodecanamido)-2-((11-((4-methyl-2-oxo-2H-chromen-7-yl)thio)undecanoyl)oxy)propyl (2-(trimethylammonio)ethyl) phosphate (PC-1)
To a solution of 11 (0.5 g, 0.62 mmol) in 35 mL dried acetonitrile was added 2-chloro-2-oxo-1,3,2-dioxaphospholane (0.13 g, 0.93 mmol). After mixing, the solution was placed on ice bath for a few minutes, then triethylamine (0.1 g, 0.99 mmol) was added to the solution. The reaction was stirred overnight. After 24 h, a white precipitate formed that was filtered and the solvent was evaporated on a rotary evaporator to give cyclic phosphate intermediate as an oil. This oily residue was placed in a pressure bottle and dissolved in 35 mL of dried acetonitrile. Reaction flask was cooled to −10°C and 3 mL of trimethylamine was added to it. Then the reaction was heated to 67°C for two days. After 48 hours, reaction was stopped and cooled to room temperature. The solid was filtered and the filtrate was evaporated using a rotary evaporator. Two silica gel chromatography columns (CHCl3/CH3OH/H2O 65:25:4) were used to purify the solid that was formed and the oily residue was treated the same way separately. The pure fractions were collected and the solvent was evaporated. The sample was freeze-dried from benzene to give a yellow oily product PC-1 (0.29 g, 48%); IR (CHCl3): 3368, 2926, 1621, 1524, 1313cm−1; 1H NMR (CDCl3/CD3OD 1:1, 400 MHz); δ 8.96 (s, 1H), 8.54 (s, 1H), 8.16 (dd, J = 2.4, 9.6 Hz, 1H), 7.70 (t, J = 6.4 Hz, 1H), 7.48-7.42 (m, 1H), 7.09 (dd, J = 2.0, 8.0 Hz, 1H), 7.06-7.02 (m, 1H), 6.92 (d, J = 9.6 Hz, 1H), 6.12 (s, 1H), 4.95 (pen, J = 5.2 Hz, 1H), 4.2 (br s, 2H), 3.86 (t, J = 6.8 Hz, 2H), 3.58-3.22 (m, 8H), 3.14 (br s, 9H), 2.91 (t, J = 7.6 Hz, 2H), 2.35 (s, 3H), 2.22 (t, J = 7.6 Hz, 2H), 2.08 (t, J = 6.8 Hz, 2H), 1.75-1.30 (m, 14H), 1.21 (br s, 22H). 13C (NMR) (CDCl3/CD3OD 1:1, 100 MHz) δ 175.9, 174.4, 162.4, 154.4, 149.3, 145.0, 136.4, 131.0, 125.6, 124.9, 123.8, 117.7, 115.1, 114.4, 113.8, 106.9, 71.7, 70.2, 67.1, 64.8, 59.8, 54.7, 44.2, 39.7, 37.0, 34.8, 32.7, 30.2, 29.3, 27.6, 26.6, 25.5 19.0; MS MH+ (C47H72N5O13PSH+) Calcd: 978.4658, Found: 978.4638 Rf (CHCl3/CH3OH/H2O 65:25:4)= 0.66; [α]D25: 1.2° (c 1.00, CHCl3-MeOH 4:1).
3.1.9. (R)-N-(2,3-dihydroxypropyl)-11-((4-methyl-2-oxo-2H-chromen-7-yl) thio)undecanamide (13)
Pentafluorophenol 4 (0.53 g, 3.2 mmol) was added to a solution of fluorescent carboxylic acid 9 (1.0 g, 2.7 mmol) in 20 mL dichloromethane followed by addition of DCC (0.66 g) and 50 mg of DMAP. The reaction was stirred at room temperature for 35 min and DCC-urea was filtered off and the filtrate was evaporated at 37°C to give a pale yellow product (1.3 g). Rf(CHCl3/CH3OH 9.7:0.3) = 0.80. To the crude product (1.3 g) in 15 mL of spectrograde chloroform was added (R)-3-Amino-1,2-propanediol 6 (0.40 g, 4.3 mmol) in 15mL methanol. The mixture was stirred for 10 minutes at room temperature. At last, DIPEA (0.31g, 2.4 mmol) was added to the reaction mixture. The reaction was stopped after 35 minutes and the solvent was evaporated on a rotary evaporator at 37°C and the residue purified by silica gel chromatography (CHCl3/CH3OH 9:1) to give a colorless solid product 13 (1.2 g). IR (CHCl3): 3310, 2915, 1725, 1635, 1605 cm−1; 1H NMR (CDCl3/CD3OD 1:1, 400 MHz) δ7.48 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 7.08 (s, 1H), 6.14 (s, 1H), 3.61 (pen, J = 5.2 Hz, 2H), 3.36-3.15 (m, 4H), 2.93 (t, J = 7.6Hz, 2H), 2.34 (s, 1H), 2.12 (t, J = 7.2 Hz, 2H), 1.62 (pen, J = 7.2 Hz, 2H), 1.52 (pen, J = 7.2 Hz, 2H), 1.38 (pen, J = 7.6 Hz, 2H), 1.21 (br s, 12H). 13C (NMR) (CDCl3/CD3OD 1:1, 100 MHz) (δ179.7, 165.7, 157.7, 148.2, 132.0, 128.7, 127.0, 120.8, 117.5, 116.8, 114.6, 74.6, 67.4, 45.9, 40.0, 35.8, 33.2, 33.1, 33.0, 32.9, 32.5, 32.4, 29.6, 21.9); MS MH+ (C24H35NO5SH+) Calcd: 450.2309, Found: 450.2305; Rf (CHCl3/CH3OH 9:1) = 0.27.
3.1.10. (R)-N-(2-hydroxy-3-((4-methoxyphenyl)diphenylmethoxy)propyl)-11-((4-methyl-2-oxo-2H-chromen-7-yl)thio)undecanamide (14)
To 13 (1.1 g, 2.4 mmol) in 15 mL CH2Cl2 was added 2 mL triethylamine to obtain a basic pH. DMAP (0.45 g, 3.6 mmol) was added to the flask followed by the addition of 4-methoxytrityl chloride (1.14g, 3.6 mmol). After 30 min, another 2 mL triethylamine was added to the reaction mixture followed by the addition of DMAP (0.45 g, 3.6 mmol) and 4-methoxytrityl chloride (1.14 g, 3.6 mmol) in that order. The reaction was stopped after 30 min. Solvent was removed on a rotary evaporator at 37°C. Purification was done by silica gel column chromatography using (CHCl3/Et3N 9.5:0.5) to give an oily pale yellow product 14 (1.43 g, 81.0%). 1H NMR (CDCl3, 400 MHz) δ 7.55-7.22 (m, 17H), 6.91 (d, J = 6.8 Hz, 1H), 6.28 (s, 1H), 5.81 (app. br t, 1H), 3.93 (br s, 1H), 3.87 (s, 1H), 3.55-3.42 (m, 1H), 3.19-3.05 (m, 4H), 3.01 (t, J = 7.2 Hz, 2H), 2.48 (s, 3H), 2.17 (t, J = 7.6 Hz, 2H), 1.77 (pen, J = 7.6 Hz, 2H), 1.63 (pen, J = 6.8 Hz, 2H), 1.53 (pen, J = 7.6 Hz, 2H), 1.35 (br s, 12H). 13C (NMR) (CDCl3, 100 MHz) δ ( 174.5, 160.8, 158.6, 153.9, 152.3, 144.1, 135.3, 130.3, 128.3, 127.9, 127.0, 124.5, 123.0, 116.9, 113.8, 113.7, 113.2, 86.5, 70.5, 64.7, 55.2, 42.9, 36.5, 32.1, 29.4, 29.3, 29.2, 29.0, 28.8, 28.7, 25.7, 18.6); MS MNa+ (C44H51NO6SNa+) Calcd: 744.3329, Found: 744.3329; Rf (CHCl3/CH3OH 9:1) = 0.75; [α]D25: 3.2° (c 1.00, CHCl3-MeOH 4:1).
3.1.11. 3-((2,4-dinitrophenyl)amino)propanoic acid (15)
15 was synthesized according to an existing procedure (Sharko and Kisel, 2011).
3.1.12. (R)-1-((4-methoxyphenyl)diphenylmethoxy)-3-(11-((4-methyl-2-oxo-2H-chromen-7-yl)thio)undecanamido)propan-2-yl 3-((2,4-dinitrophenyl)amino)propanoate (16)
To a solution of 14 (0.70 g, 0.97 mmol) in 15 mL dichloromethane was added the short chain quencher 15 (0.38 g, 1.46 mmol). The reaction mixture turned yellow upon mixing them. DCC (0.30 g, 1.46 mmol) and catalytic amount of DMAP (50 mg) was added to the reaction mixture. The reaction was stopped after 1hr then the DCC-urea precipitate was removed by filtration and the solvent was evaporated at 37°C using a rotary evaporator. Purification was done by silica gel column chromatography using (CHCl3/EtOAc 9:1) to give a yellow product 16 (0.89 g, 92.0%). IR (CHCl3): 2926, 1728, 1618, 1605, 1334cm−1; 1H NMR (CDCl3, 400 MHz) δ 9.0 (d, J = 2.8 Hz, 1H), 8.63 (app, br t, 1H), 8.14 (dd, J = 2.4, 9.2 Hz, 1H), 7.35-7.01 (m, 10H), 6.82 (d, J = 9.6Hz, 1H), 6.70 (d, J = 8.8 Hz, 1H), 6.10 (s, 1H), 5.36 (t, J = 5.6 Hz, 1 H), 5.03 (pen, J = 5.6 Hz, 1H), 3.67 (s, 3H), 3.64-3.54 (m, 4H), 3.4-3.10 (m, 4H), 2.85 (t, J = 7.6 Hz, 2H), 2.68-2.57 (m, 2H), 2.28 (s, 1H), 1.88 (t, J = 7.2 Hz, 2H), 1.57 (pen, J = 7.6 Hz, 3H), 1.48-1.35 (m, 9H), 1.28 (br s, 14H); MS MNa+ (C53H58N4O11SNa+) Calcd: 981.3715, Found: 981.3694; Rf (CHCl3/EtOAc 9:1) = 0.80; [α]D25: 3.5° (c 1.00, CHCl3/MeOH 4:1).
3.1.13. (R)-1-hydroxy-3-(11-((4-methyl-2-oxo-2H-chromen-7-yl)thio) undecanamido) propan-2-yl 3-((2,4-dinitrophenyl)amino)propanoate (17)
Compound 16 (0.80 g, 0.83 mmol) was dissolved in 10 mL CHCl3: MeOH (1:1) and to it was added dropwise a solution of 5 mL HCl (1 mL 12 M HCl in 125 mL CHCl3:MeOH at 0°C. The reaction mixture was stirred for 60 min. After that, 10 mL NaHCO3 solution (1.5 g in 50 mL H2O) was added and stirred for 15 minutes at 0°C. The solution was extracted with 2 X 10 mL CHCl3 and the organic layer was combined and re-extracted with 5 mL brine. The organic layer was then dried over anhydrous sodium sulfate and the solvent was evaporated on a rotary evaporator. The yellow product 17 was isolated on a silica gel column chromatography (CHCl3/EtOAc 9:1) to yield (0.53 g, 92.0%). IR (CHCl3): 3372, 2926, 1728, 1615, 1087 cm−1; 1H NMR (CDCl3, CD3OD 400 MHz) δ 9.09 (d, J = 2.4 Hz, 1H), 8.76 (app. br t, 1H), 8.26 (dd, J = 2.8, 8.8 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.14 (s, 1H), 6.94 (d, J = 9.6 Hz, 2H), 6.18 (s, 1H), 4.90 (pent, J = 4.8 Hz, 1H), 3.79-3.43 (m, 6H), 2.95 (t, J = 7.2 Hz, 2H), 2.78 (t, J = 6.4 Hz, 2H), 2.38 (s, 3H), 2.22 (t, 3H J = 7.6 Hz, 3H), 1.67 (pen, J = 8.0 Hz, 2H), 1.60 (pen, J = 7.6 Hz, 2H), 1.42 (pen, J = 7.6 Hz, 2H), 1.24 (br s, 12H); 13C (NMR) (CDCl3, 100 MHz) δ (175.4, 170.9, 160.9, 160.0, 153.9, 152.6, 147.9, 143.9, 138.2, 136.4, 130.7, 130.5, 124.6, 124.4, 124.3, 123.2, 116.9, 113.7, 113.6, 74.0, 60.1, 39.1, 38.5, 36.4, 33.6, 32.1, 29.2, 28.9, 28.6, 28.3, 25.6, 21.7, 18.5); MS MH+ (C33H42N4O10SH+) Calcd: 686.2624, Found: 686.2690; Rf (CHCl3/CH3OH 9:1) = 0.75.
3.1.14. (R)-2-((3-((2,4-dinitrophenyl)amino)propanoyl)oxy)-3-(11-((4-methyl-2-oxo-2H- chromen-7-yl)thio)undecanamido)propyl (2-(trimethylammonio)ethyl) phosphate (PC-2)
To a solution of 17 (0.5 g, 1.45 mmol) in 35 mL dried acetonitrile in a pressure bottle, was added 2-chloro-2-oxo-1,3,2-dioxaphospholane (0.31 g, 2.2 mmol). After mixing, the solution was placed in a dry-ice acetone bath for a few minutes, and then 2 mL of anhydrous trimethylamine was added to the solution. The reaction went for 24h to give a cyclic phosphate intermediate. After 24 h, 3 mL of anhydrous trimethylamine was added to the reaction mixture at −10°C. The reaction was heated to 67°C for two days. After two days, reaction was stopped and cooled to room temperature. The solid was filtered and the solvent was evaporated using a rotary evaporator. Two silica gel chromatography columns (CHCl3/CH3OH/H2O 65:25:4) were done on the solid that was formed and the oily residue was treated the same way separately. After chromatography, the pure fractions were collected and the solvent was evaporated. The sample was freeze-dried from benzene to give a yellow oily product PC-2 (0.27 g, 44%). IR (CHCl3): 3372, 2927, 1718, 1620, 1336 cm−1; 1H NMR (CDCl3/CD3OD 1:1, 400 MHz) δ 9.97 (s, 1H), 8.17 (d, J = 9.6 Hz, 1H), 7.43-6.92 (m, 6H), 6.09 (s, 1H), 4.94 (app. br s 1H), 3.67-3.20 (m, 7H), 3.12 (br s, 9H), 2.87 (m, 2H), 2.31 (s, 3H), 2.01 (m, 2H), 1.63-1.25 (m, 6H), 1.15 (br s, 13H); 13C (NMR) (CDCl3/CD3OD 1:1, 100 MHz) δ (179.5, 175.1, 165.7, 157.5, 152.0, 148.2, 139.9, 134.1, 132.0, 128.7, 128.6, 128.1, 127.8, 127.0, 120.8, 118.0, 117.5, 116.8, 77.9, 64.7, 42.5, 40.0, 37.1, 35.8, 33.2, 32.5, 29.6, 22.0); MS MNa+ (C33H42N4O10SNa+) Calcd: 852.3249, Found: 852.3243; Rf (CHCl3/CH3OH/H2O 65/25/4)= 0.36; [α]D25: 3.6° (c 1.00, CHCl3/MeOH 4:1).
3.1.15. Perfluorophenyl 12-((2,4-dinitrophenyl)amino)dodecanoate (19)
To a solution of 3 (2.0 g, 5.2 mmol) in 15 mL of spectograde chloroform, R-(2,2-dimethyl-1,3-dioxalan-4-yl) (0.69 g, 5.2 mmol) 18 was added drop-wise at room temperature, followed by DCC (1.08 g, 5.2 mmol). The reaction mixture turned to a turbid yellow suspension. Then 4-dimethylaminopyridine (DMAP) (0.64 g, 5.2 mmol) was added to the reaction mixture, followed by stirring at room temperature for 30 minutes. The DCC-urea precipitate was filtered with suction, and the solvent was removed by rotatory evaporation. Purification was done using column chromatography (CHCl3/EtOAc 9.3:0.7) to obtain yellow solid 19 (2.1 g, 81%). IR (CH2Cl2): 3367, 2928, 2855, 1735, 1334; 1H NMR: (400 MHz, CDCl3) δ: 9.16 (d, J = 2.7 Hz, 1H), 8.57 (br s, 1H), 8.28 (dd, J = 2.5, 9.6 Hz, 1H), 6.92 (d, J = 9.8 Hz, 1H), 4.32 (m, 1H), 4.17 (m, 1H), 4.09 (m, 2H), 3.73 (dd, J = 6.3, 8.2 Hz, 1H), 3.41 (m, 2H), 2.35 (t, J = 7.4 Hz, 2H), 1.78 (pent, J = 7.3 Hz, 2H), 1.63 (pent, J = 8.0 Hz, 2H), 1.42 (m, 8H), 1.30 (br s, 12H); 13C NMR: (100 MHz, CDCl3) δ: 173.3, 148.1, 135.6, 130.0, 129.9, 124.1, 113.5, 109.5, 73.3, 66.0, 64.2, 43.3, 33.8, 33.6, 29.1, 29.0, 28.9, 28.8, 28.7, 28.4, 26.6, 26.4, 25.1; MS MNa+(C24H40N3O8Na+) Calcd: 518.2479 Found: 518.2147; Rf (CHCl3/EtOAc 9.7:0.3) = 0.60.
3.1.16. (R)-2,3-dihydroxypropyl 12-((2,4-dinitrophenyl)amino)dodecanoate (20)
To a solution of 19 (1.0 g, 2.0 mmol) in 8 ml 1,4-dioxane, was added a solution of HCl (0.4 mL of 12 M HCl diluted in 1.6 mL of 1,4-dioxane) at room temperature. The reaction was stopped after 2h. The mixture was purified directly on Brockman I, neutral alumina gel column using chloroform, to remove any unreacted starting material. CHCl3/MeOH (9.8:0.2) was used to elute the product 20 (0.79 g, 1.73 mmol, 86%). IR (CH2Cl2): 3365, 2938, 2832,1732, 1266; 1H NMR: (400 MHz, CDCl3) δ: 9.11 (d, J = 2.8 Hz, 1H), 8.50 (br s, 1H), 8.42 (dd, J = 2.8, 8.8 Hz 1H), 6.89 (d, J = 9.6 Hz, 1H), 4.21-4.09 (m, 2H), 3.93-3.79 (m, 1H), 3.68-3.60 (m, 1H) 3.58-3.50 (m, 1H), 3.34 (m, 2H), 2.29 (t, J = 7.4 Hz, 2H), 1.72 (pen, J = 7.3 Hz, 2H), 1.59-1.53 (m, 2H), 1.44-1.35 (m, 2H), 1.29 (br s, 12H); 13C NMR: (100 MHz, CDCl3) δ: 173.9, 148.1, 135.6, 130.0, 129.9, 124.1, 113.6, 69.9, 64.8, 63.0, 43.3, 33.8, 29.0, 29.0, 28.8, 28.7, 28.3, 26.6, 24.5; MS MNa+ (C21H33N3O8Na+) Calcd: 478.2198 Found: 478.2210; Rf (CHCl3/CH3OH 9.8:0.2) = 0.35.
3.1.17. 3-((((9H-fluoren-9-yl)methoxy)carbonyl)oxy)-2-hydroxypropyl 12-((2,4-dinitrophenyl)amino)dodecanoate (21)
To a solution of 20 (1.0 g, 2.19 mmol) and 9-Fluorenylmethyl chloroformate (1.13 g, 4.36 mmol), 4-(Dimethylamino) pyridine DMAP (0.05 g, 0.4 mmol) in 10 mL of dichloromethane kept at 0° C, was added a solution of 2,3,5-Collidine (0.8 g, 6.6 mmol) in 5 mL of dichloromethane. Collidine solution was added very slowly over a period of 20 minutes. After 90 minutes of stirring, the reaction was stopped. The mixture was purified directly on silica column loaded with chloroform and eluted with CHCl3/EtOAc (6:1). The fractions corresponding to the mono-acylated compound were combined and evaporated by rotovapor to give yellow oil 21 (0.86 g, 58%). IR (CH2Cl2): 3339, 2942, 2832, 1737, 1266; 1H NMR: (400 MHz CDCl3) δ: 9.15 (d, J = 2.7 Hz, 1H), 8.55 (br s, 1H), 8.30-8.23 (dd, J = 2.5, 9.2 Hz, 1H), 7.43 (d, J = 7.4 Hz, 2H) 7.62 (d, J = 7.4 Hz, 2H), 7.42 (m, 2H), 7.47 (s, 2H), 7.34 (m, 2H), 6.91 (d, J = 9.4 Hz, 2H), 4.45 (d, J = 7.4 Hz, 2H), 4.18 (m, 6H), 3.39 (m, 2H), 2.37 (t, J = 7.5 Hz, 2H), 1.77 (pent, J = 7.2 Hz, 2H), 1.42 (m, 4H), 1.31 (br s, 10H); 13C NMR: (100 MHz, CDCl3) δ: 173.7, 155.0, 148.2, 144.3, 143.1, 141.3, 141.1, 135.7, 130.1, 127.8, 127.4, 127.1, 126.9, 124.9, 124.6, 124.1, 119.9, 119.8, 113.7, 70.0, 68.4, 67.9, 64.6, 46.5, 43.4, 33.9, 29.3, 29.2, 29.1, 29.0, 28.9, 28.5, 26.7, 24.7, 14.0; MS MNa+ (C36H43N3O10H+) Calcd: 679.3028 Found: 679.4166; Rf (CHCl3/EtOAc 6:1) = 0.5
3.1.18. 3-((((9H-fluoren-9-yl)methoxy)carbonyl)oxy)-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl 12-((2,4-dinitrophenyl)amino)dodecanoate (22)
To a solution of 21 (1.0 g, 1.56 mmol) in 10 mL dichloromethane was added 3,4-dihydro-2H-pyran 10 (0.63 g, 7.5 mmol) and pyridinium toluene-4-sulfonate (1.13 g, 4.5 mmol). The reaction mixture was stirred at room temperature for 1hr. The mixture was purified on silica gel with dichloromethane and chromatographed with cyclohexane/ethyl acetate (2:1). The fractions corresponding to the product were combined and evaporated to give 22 (0.97 g, 86%). IR (CH2Cl2): 3370, 2929, 2857, 1739, 1263; 1H NMR: (400 MHz, CDCl3) δ: 9.11 (d, J = 2.8 Hz, 1H), 8.50 (br s, 1H), 8.25-8.30 (dd, J = 2.8, 9.2 Hz, 1H), 7.80 (d, J = 7.2 Hz, 2H), 7.61 (d, J = 7.1 Hz, 2H), 7.40 (t, J = 7.6 Hz, 2H), 7.30 (m, 2H), 6.99 (d, J = 9.2 Hz, 1H), 5.17 (br, s, 1H), 4.40-4.09 (m, 8H), 3.92-3.89 (m, 1H), 3.58-3.47 (m, 1H), 3.49 (q, J = 7.2 Hz, 2H), 2.33 (td, J = 4.9, 7.5 Hz, 2H), 1.85-1.67 (m, 4H), 1.65-1.46 (m, 8H), 1.41 (br s, 12H); 13C NMR: (100 MHz, CDCl3) δ: 173.6, 155.2, 148.5, 143.5, 143.4, 141.4, 136.0, 130.4, 128.1, 128.0, 127.3, 125.3, 125.2, 124.5, 120.2, 114.0, 98.4, 97.7, 94.8, 72.1, 71.6, 70.1, 62.5, 62.2, 46.9, 43.7, 34.3, 31.0, 29.5, 29.3, 28.8, 27.1, 25.5, 25.0, 19.2; MS MNa+ (C41H51N3O11Na +) Calcd: 784.3422 Found: 784.3647; Rf (Hex:EtOAc 2:1) = 0.6; [α]D20: +1.02° (c 0.062, CH2Cl2).
3.1.19. 3-hydroxy-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl 12-((2,4-initrophenyl)amino) dodecanoate (23)
To a solution of 22 (1.0 g, 1.31 mmol) in 9 mL of dichloromethane kept at room temperature was added piperidine (1 mL, 10 mmol). The reaction was followed by TLC with Hex/EtOAc (1:1) and found to be complete in 30 minutes. The solvent was removed on a rotary evaporator at 25°C. Purification was done on silica gel column using (Hex/EtOAc 1:1) to give yellow oil 23 (0.4 g, 56%). IR (CH2Cl2): 3363, 2928, 2854, 1732, 1335; 1H NMR: (400 MHz, CDCl3) δ: 9.16 (d, J = 2.7 Hz, 1H), 8.57 (br, s, 1H), 8.28 (dd, J = 2.5, 9.6 Hz, 1H), 6.92 (d, J = 9.8 Hz, 1H), 4.76 (t, J = 3.2 Hz, 1H), 4.34 (dd, J = 5.3, 11.5 Hz, 1H), 3.94 (m, 2H), 3.64 (m, 4H), 3.45 (m, 2H), 2.41 (td, J = 5.4, 7.0 Hz, 2H), 1.78 (pent, J = 1.78 Hz, 4H), 1.57 (m, 10H), 1.30 (m, 10H); 13C NMR: (100 MHz, CDCl3) δ: 173.3, 148.2, 135.6, 130.1, 100.3, 98.2, 78.3, 64.2, 63.4, 63.0, 61.4, 43.4, 34.0, 31.0, 30.7, 30.5, 29.2, 29.1, 29.0, 28.8, 28.5, 26.7, 24.9, 24.7, 20.4; MS MNa+ (C26H41N3O9Na +) Calcd: 562.2741 Found: 562.2754; R f (C6H14/EtOAc 1:1) = 0.5.
3.3.20. 3-((12-((2,4-dinitrophenyl)amino)dodecanoyl)oxy)-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl (2-(trimethylammonio)ethyl) phosphate (24)
To a solution of 23 (1.1 g, 2.0 mmol) in 35 mL dried benzene, was added 2-chloro-2-oxo-1,3,2-dioxaphospholane (0.58 g, 4 mmol). After mixing, the solution was placed on ice bath for a few minutes, at which point triethylamine (1 mL, 7.1 mmol) was added to the solution. The reaction mixture was kept for 24h at room temperature and followed by TLC in Hex/EtOAc (1:1). The phospholipid formation was detected as a blue spot upon treating the TLC plate with molybdenum spray. After 24 h, a precipitate formed that was filtered and the solvent was evaporated on a rotary evaporator to give an oil as intermediate. The compound was suspended in dry cold acetonitrile (35 mL) and transferred to a pressured bottle and cooled until the mixture was frozen. Cold trimethylamine (3 mL, 34 mmol) was then added and the reaction was heated to 67°C for 18 hours, at which point a precipitate was formed. The reaction was stopped and the pressure tube was cooled to room temperature. The solid was filtered and the solvent was evaporated using a rotary evaporator. The mixture was purified using silica gel chromatography narrow column (CHCl3/CH3OH/H2O 65:25:4) to obtain a yellow sticky solid product 24. (0.67 g, 48%). IR (CH2Cl2): 3309, 2942, 2842, 1725, 1337; 1H NMR: (400 MHz, CDCl3/CD3OD 1:1) δ: 9.22 (d, J = 2.7 Hz, 1H), 8.39 (dd, J = 2.7, 9.8 Hz, 1H), 7.54 (s, 1H) 7.08 (d, J = 9.4 Hz, 1H), 4.40 (m, 2H), 4.30 (m, 2H), 4.13 (m, 1H), 3.88 (m, 1H), 3.74 (m, 1H), 3.69 (m, 2H), 3.55 (t, J = 8.0 Hz, 2H), 3.31 (br s, 9H), 3.25 (m, 2H), 2.41 (m, 2H), 1.87 (pent, J = 8.0 Hz, 2H), 1.68 (m, 2H), 1.64-1.53 (m, 4H) 1.18 (m, 16H).); 13C NMR: (100 MHz, CDCl3/CD3OD 1:1) δ: (ppm) 174.4, 148.4, 135.8, 130.4, 124.4, 114.1, 66.3, 64.9, 64.2, 63.1, 62.1, 59.5, 46.1, 43.6, 43.5, 34.2, 34.1, 30.8, 30.5, 29.4, 29.2, 29.1, 28.7, 26.9, 25.4, 24.8, 19.8; MS MNa+ (C31H53N4O12PNa+) Calcd: 727.3296 Found: 727.3572; Rf (CHCl3/CH3OH/H2O 65:25:4) = 0.24; [α]D20: +3.05° (c 0.022, CH2Cl2).
3.3.21. 3-hydroxy-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl 12-((2,4-dinitrophenyl) amino) dodecanoate (25)
To a solution of 24 (0.5 g, 0.71 mmol) in 24.69 mL 1,4-dioxane, 0.31 mL of 12 M HCl was added. The reaction was followed by TLC with a solvent mixture of CHCl3/CH3OH/H2O (65:25:4) and found to be complete in one hour. Additional 25 mL of 1,4-dioxane was added to the reaction mixture, which was then freeze-dried to form yellow sticky oil. The product was then chromatographed on silica gel on a narrow column using CHCl3/CH3OH/H2O (65:25:4) to obtain the yellow sticky solid product 25 (0.25 g, 57%). IR (CH2Cl2): 3362, 2926, 2853, 1731, 1621,1270 cm−1; 1H NMR: (400 MHz, CDCl3) δ: 8.97 (d, J = 2.7 Hz, 1H), 8.46 (br s, 1H), 8.11(dd, J = 2.5, 9.6 Hz, 1H), 6.85 (d, J = 9.8 Hz, 1H), 5.52 (br s, 1H), 4.41 (m, 2H), 4.13 (m, 1H), 3.96 (m, 4H), 4.74 (m, 2H), 3.31 (br t, J = 6.8 Hz, 2H), 3.15 (br s, 9H), 3.05 (m, 2H) 2.19 (m, 2H), 1.67 (pent, J = 8.2 Hz, 2H), 1.53-1.38 (m, 6H), 1.16 (m, 10H); 13C NMR: (100 MHz, CDCl3/CD3OD 1:1) δ: (ppm) 173.8, 148.1, 135.1, 129.7, 129.6, 123.5, 114.0, 67.6, 65.4, 64.1, 59.8, 53.4, 46.1, 42.8, 33.4, 29.0, 28.7, 28.6, 28.5, 28.1, 26.3, 24.3, 24.2. MS MNa+ (C26H45N4O11PNa+) Calcd: 643.2720 Found: 643.2810; Rf (CHCl3/CH3OH/H2O 65:25:4) = 0.20; [α]D20: +3.5° (c 0.064, CH2Cl2).
3.1.22. 3-((12-((2,4-dinitrophenyl)amino)dodecanoyl)oxy)-2-((11-((4-methyl-2-oxo-2H- chromen-7-yl)thio)undecanoyl)oxy)propyl (2-(trimethylammonio)ethyl) phosphate (PC-3)
To a solution of phosphocholine 25 (0.40 g, 0.64 mmol) in 10 mL dichloromethane, was added fluorescent carboxylic acid 9 (1.21 g, 3.21 mmol), DCC (0.66 g, 3.20 mmol) and DMAP (0.39 g, 3.20 mmol) in a long necked flask containing 2 g of glass beads. The flask was placed in a sonicator at 37°C such that the flask was close to the bottom without touching it. The reaction reached completion in 18 hr as shown by TLC CHCl3/CH3OH/H2O (65:25:4). The product was purified with silica gel chromatography using CHCl3/MeOH/H2O (65:25:4). The pure product was obtained as a yellow sticky solid PC-3 (0.38 g, 60%). IR (CH2Cl2): 2928, 2854, 1721, 1336 cm−1; 1H NMR: (400 MHz, CDCl3/CD3OD 1:1) δ: 8.95 (d, J = 2.7 Hz, 1H), 8.11 (dd, J = 2.8, 9.6 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.01 (m, 2H), 6.81 (m, 1H), 6.12 (s, 1H), 5.06 (m, 1H), 4.23 (dd, J = 3.6, 12.4 Hz, 1H), 4.13 (br s, 2H), 4.01 (dd, J = 6.8, 12.4 Hz, 1H), 3.49 (m, 2H), 3.27 (t, J = 6.8 Hz, 2H), 3.22 (m, 2H), 3.08 (s, 9H), 2.84 (t, J = 7.4 Hz, 2H), 2.27 (s, 3H), 2.20 (m, 4H), 1.62 (m, 4H), 1.55 (m, 4H), 1.50 (m, 4H), 1.12 (br s, 22H); 13C NMR: (100 MHz, CDCl3/CD3OD 1:1) δ: 173.7, 173.3, 161.2, 153.7, 152.9, 148.3, 143.9, 139.0, 130.2, 124.5, 124.2,123.0, 116.9, 113.9, 113.7, 113.3, 106.8, 70.0, 66.1, 64.1, 62.3, 59.6, 54.2, 43.4, 39.9, 34.0, 33.9, 32.0, 29.3, 29.1, 29.0, 28.9, 28.7, 28.5, 26.8, 24.7, 18.4; MS MNa+ (C47H71N4O14PS Na+) Calcd: 1001.4323 Found: 1001.4377; Rf (CHCl3/CH3OH/H2O (65:25:4) = 0.3. [α]D20 +5.0° (c 0.001, CH2Cl2).
Highlights.
Syntheses of fluorogenic substrates targeting phospholipase A2 for quantitative detection
Acyl amino group at the sn-1 position blocks phospholipase A1
Small reporter groups to preserve membrane and lipid integrity
Fatty-acid-binding-protein fluorescence assay for enzymological characterization of probes
Activities of phospholipase A2 toward the analogs and unmodified lipids are similar
Acknowledgments
This project was supported by NIH grant awards GM SC3096878 and GM SC3096876. One of the authors (RR) expresses gratitude to the Office of Research and Sponsored Projects, CSU Northridge for their support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arouri A, Hansen AH, Rasmussen TE, Mouritsen OG. Lipases, liposomes, and lipid-prodrugs. Curr Opin Coll Int Sci. 2013;18:419–431. [Google Scholar]
- Balet C, Clingman KA, Hajdu J. 1-Palmitoyl-2-thiopalmitoyl phosphatidylcholine, a highly specific chromogenic substrate of phospholipase A2. Biochemical and Biophysical Research Communications. 1988;150:561–567. doi: 10.1016/0006-291x(88)90430-5. [DOI] [PubMed] [Google Scholar]
- Brogan AP, Widger WR, Kohn H. Bicyclomycin fluorescent probes: synthesis and biochemical and biophysical properties. Journal of Organic Chemistry. 2003;68:5575–5587. doi: 10.1021/jo030020u. [DOI] [PubMed] [Google Scholar]
- Chalbot S, Zetterberg H, Blennow K, Fladby T, Grundke-Iqbal I, Iqbal K. Cerebrospinal fluid secretory Ca2+-Ddependent phospholipase A2 activity is increased in Alzheimer disease. Clinical Chemistry. 2009;12:2171–2179. doi: 10.1373/clinchem.2009.130286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W. Seeing Is Believing: Real-time cellular activity assay for phospholipase A2. ACS Chem Biol. 2006;1(2):65–66. doi: 10.1021/cb600079t. [DOI] [PubMed] [Google Scholar]
- Dennis EA, Cao J, Hsu HY, Magrioti V, Kotokos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutics intervention. Chemical Reviews. 2011;111:6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JT, Zhang N. Cytosolic phospholipase A2 and its role in cancer. Clin Oncol Cancer Res. 2011;8:71–76. [Google Scholar]
- Kethineedi VR, Crivat G, Tarr MA, Rosenzweig Z. Quantum dot–NBD–liposome luminescent probes for monitoring phospholipase A2 activity. Anal Bioanal Chem. 2013;405:9729–9737. doi: 10.1007/s00216-013-7422-z. [DOI] [PubMed] [Google Scholar]
- Koenig W, Vossen CY, Mallat Z, Brenner H, Benessiano J, Rothenbacher D. Association between type II secretory phospholipase A2 plasma concentrations and activity and cardiovascular events in patients with coronary heart disease. European Heart journal. 2009;30:2742–2748. doi: 10.1093/eurheartj/ehp302. [DOI] [PubMed] [Google Scholar]
- Kugiyama K, Doi H, Takazoe K, Kawano H, Soejima H, Mizuno Y, et al. Remnant lipoprotein levels in fasting serum predict coronary events in patients with coronary artery disease. Circulation. 1999;99:2858–2860. doi: 10.1161/01.cir.99.22.2858. [DOI] [PubMed] [Google Scholar]
- Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annual Review of Biochemistry. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- Linderoth L, Peters GH, Madsen R, Andresen TL. Drug delivery by an enzyme-mediated cyclization of a lipid prodrug with Unique bilayer-formation properties. Angew Chem Int Ed. 2009;48:1823–1826. doi: 10.1002/anie.200805241. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Lewis Farr A, Randall RJ. Protein Measurement with the Folin Phenol Reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mallat Z, Benessiano J, Simon T, Ederhy S, Sebella-Arguelles C, Cohen A, Huart V, et al. Circulating secretory phospholipase A2 activity and risk of incident coronary events in healthy men and women. Arteriosclerosis, Thermbosis, and Vascular Biology. 2007;27:1177–1183. doi: 10.1161/ATVBAHA.107.139352. [DOI] [PubMed] [Google Scholar]
- Nevalainen TJ, Graham GG, Scott KF. Antibacterial actions of secreted phospholipases A2. Review Biochimica et Biophysica Acta. 2008;1781:1–9. doi: 10.1016/j.bbalip.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Wichmann O, Gelb MH, Schultz C. Probing phospholipase A2 with fluorescent phospholipid substrates. Chembiochem. 2007;8(13):1555–1569. doi: 10.1002/cbic.200600462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: From cells to animals to humans. Progress in Lipid Research. 2011;50:152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Murakami M, Taketomi Y, Sato H, Yamamoto K. Secreted phospholipases A2 revisited. J Biochem. 2011;150:233–255. doi: 10.1093/jb/mvr088. [DOI] [PubMed] [Google Scholar]
- Murakami M, Sato H, Miki Y, Yamamoto K, Taketomi Y. A new era of secreted phospholipase A2. The journal of lipid research. 2015;56:1248–1261. doi: 10.1194/jlr.R058123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds LJ, Washburn WN, Deems RA, Dennis EA. Assay strategies and methods for phospholipases. Methods in Enzymology. 1991;197:3–23. doi: 10.1016/0076-6879(91)97129-m. [DOI] [PubMed] [Google Scholar]
- Rose TM, Prestwich GD. Fluorogenic phospholipids as head group-selective Reporters of Phospholipase A Activity. ACS Chem Biol. 2006;1(2):83–92. doi: 10.1021/cb5000014. [DOI] [PubMed] [Google Scholar]
- Scopes RK. Measurement of protein by spectrophotometry at 205 nm. Analytical Biochemistry. 1974;59:277–282. doi: 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- Scott KF, Sajinovich M, Hein J, Nixdorf S, Galettis P, De Souza WP, Dong Q, Graham GG, Russell PJ. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie. 2010;92:601–610. doi: 10.1016/j.biochi.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Sharko O, Kisel M. 1-Acyl-2-[N-(2,4-dinitrophenyl)aminopropionyl]-sn-glycero-3-phosphocholine as a chromogenic substrate for phospholipase A2 substrate. Analytical Biochemistry. 2011;413:69–71. doi: 10.1016/j.ab.2011.02.018. [DOI] [PubMed] [Google Scholar]
- Singh J, Ranganathan R. Surface Dilution Kinetics of Phospholipase A2 Catalyzed Lipid-Bilayer Hydrolysis. J Phys Chem B. 2014;118:2077–2083. doi: 10.1021/jp411512c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Ranganathan R, Hajdu J. Kinetics of Bacterial Phospholipase C Activity at Micellar Interfaces: Effect of Substrate Aggregate Microstructure and a Model for the Kinetic Parameters. J Phys Chem B. 2008;112(51):16741–16751. doi: 10.1021/jp807067g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Ranganathan R, Hajdu J. Surface dilution kinetics using substrate analog enantiomers as diluents: Enzymatic lipolysis by bee-venom phospholipase A2. Analytical Biochemistry. 2010;407:253–260. doi: 10.1016/j.ab.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug MJ, Longo ML, Faller R. The impact of Texas Red on lipid bilayer. J Phys Chem B. 2011;115:8500–8505. doi: 10.1021/jp203738m. [DOI] [PubMed] [Google Scholar]
- Stoscheck CM. Quantitation of Protein. Methods in Enzymology. 1990;182:50–69. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- Tribler L, Jensen LT, Jørgensen K, Brünner N, Gelb MH, Nielsen HJ, Jensen SS. Increased expression and activity of group IIA and X secretory phospholipase A2 in peritumoral versus central colon carcinoma tissue. Anticancer. 2007;27:3179–3185. [PubMed] [Google Scholar]
- Wang M, Pinnamaraju S, Ranganathan R, Hajdu J. Chemistry and Physics of Lipids. 2013;172–173:78–85. doi: 10.1016/j.chemphyslip.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann O, Gelb MH, Schultz C. Probing phospholipase A2 with fluorescent phospholipid substrates. ChemBioChem. 2007;8:1555–1569. doi: 10.1002/cbic.200600462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton DC. A continuous fluorescence displacement assay for the measurement of phospholipase A2 and other lipases that release long-chain fatty acids. Biochem J. 1990;266(2):435–439. doi: 10.1042/bj2660435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakynthinos E, Pappa N. Inflammatory biomarkers in coronary artery disease Epaminondas. Journal of Cardiology. 2009;53:317–333. doi: 10.1016/j.jjcc.2008.12.007. [DOI] [PubMed] [Google Scholar]