

Abstract
Background
Chronic rhinosinusitis (CRS) is a multifactorial inflammatory airway disorder in which bacteria are implicated in the initiation and/or sustenance of disease in some patients. The sinuses are colonized by bacteria even in health, and the potential for sinus-specific niches harboring unique microbial consortia raises questions for clinical and research investigation.
Objective
To determine the degree to which resident upper airways microbiota differ between individuals and anatomic sites, in order to determine the optimal site of microbial sampling for study in CRS.
Methods
8 CRS patients undergoing primary surgery were sampled bilaterally at the anterior nares, middle meatus, nasopharynx, maxillary, frontal, and sphenoid sinuses, for investigation using broad-range bacterial 16S rRNA sequencing.
Results
Between-subject variability in bacterial microbiota was substantially greater than within-subject variability. The middle meatus was fairly representative of the underlying sinuses, although corynebacteria were detected at higher abundances in the middle meatus, relative to the maxillary (P<0.1), frontal (P<0.05), or sphenoid (P<0.1) sinuses.
Conclusion
Interpersonal variation of the upper airway microbiome greatly outweighs niche-specific differences. The middle meatus is a fair representation of the underlying sinuses and may be considered for use as a simple single site for sampling in longitudinal studies or in subjects who have not undergone sinus surgery.
Keywords: sinus, microbiome, sinusitis, CRS, chronic rhinosinusitis, microbiota, 16S
INTRODUCTION
Chronic rhinosinusitis (CRS) is a prevalent disorder in which microbes, particularly bacteria, are presumed to play a role in disease pathogenesis [1]. Study and clinical management of this disorder and other infectious sinonasal diseases has traditionally relied upon culture-based methods that may be biased and possibly inadequate to fully document microbial diversity in the sinonasal cavities [2, 3]. At this time, we are amid a revolution in the study and understanding of such disorders with the incorporation of modern microbiome concepts and techniques [4-10]. The paranasal sinuses comprise intricate, paired cavities within the craniofacial skeleton that are likely to contain unique microenvironments. As such, each niche could select for predominance of different bacterial consortia that are adapted to the unique properties of each microenvironment (e.g., hypoxia, pH, humidity). Currently, however, it is unknown if each individual sinus has a unique microenvironment and associated unique microbiota, or if they are fairly similar throughout.
In current clinical management of CRS, cultures of the middle meatus are utilized to represent the underlying sinuses, given the invasive nature of deeper sampling [1]. Prior culture-based study has demonstrated that the middle meatus is about 80-90% representative of underlying sinuses in detection of colonizing bacteria [11-13]. Although not perfect, these results imply that microbial similarities exist between the middle meatus and underlying ethmoid and maxillary sinuses. However, it seems plausible that “microgeography” of individual sinuses within a subject could vary, as local changes may influence microbial communities. Modern molecular detection methods have allowed for more sensitive detection of fastidious organisms and have described the large community of resident microbes within healthy and diseased sinuses, noting in particular the previously unrecognized abundance of anaerobic organisms [4,5,8,10]. It has been suggested that occluded sinuses may generate a hypoxic environment [14-15], or that disease may not be uniformly and evenly spread among tissues in CRS, and therefore a given sinus niche may represent a distinct environment suitable for different microbes.
Microbiome study has blossomed recently with a wealth of comparative cohort studies, but for proper understanding of its role in human health and disease, long-term prospective observational cohorts are necessary [16]. The sinonasal microbiome appears to be unique and potentially stable in health [7,17]. However, the sinuses themselves were not sampled in these studies, either the anterior nares or meati emanating from the sinuses were used as representations. Establishing a single site to sample longitudinally in the clinic setting is strategic, and the ability to study healthy and/or unoperated subjects would be ideal. Additionally, it would be of interest to re-examine the question of whether sinus microbiota are different among the sinuses utilizing these modern techniques. In this study we aimed to evalutte the degree of microbiome community differences between sinuses within and between individuals with chronic rhinosinusitis (CRS). A secondary aim was to determine if there is an ideal single site of sampling that is most representative of the sinuses as a whole.
METHODS
Study design
The diagnosis of CRS was made according to the 2007 Adult Sinusitis Guidelines, and accordingly CRS patients were initially managed medically with a minimum of three weeks of antibiotics and topical intranasal steroids prior to the decision for surgery [18]. Patients were included who underwent primary functional endoscopic sinus surgery (FESS) for CRS with bilateral inflammatory disease documented on CT scan. Patients less than age 18 years, those with cystic fibrosis, known or suspected immunodeficiency, or autoimmune disease, or those with a narrow nasal cavity precluding precise swab sampling, were excluded from the study. Patients were excluded if they had been treated with systemic antibiotic or corticosteroid therapy within one month prior to sampling, or if disease was unilateral or asymmetric. This study was approved by the Institutional Review Board of the University of Colorado (COMIRB protocol number 11-1442).
Specimen Collection
Swab samples were collected from adults undergoing primary FESS at a tertiary care rhinology practice at the University of Colorado between February and April, 2013. CultureSwabs™ (BD, Franklin Lakes, NJ) for DNA extraction were obtained from six sites on each side by applying the swab to the site surface, rotating at least 5 full turns until visibly saturated, and placing them on ice for immediate transport to the laboratory and storage at −80°C until DNA extraction. Careful sequential sampling under endoscopic guidance was used to limit the degree of potential contamination from other subsites. The anterior nares were swabbed first and then the nasal vibrissae trimmed. The anterior ethmoid region was sampled next in a similar fashion to prior study by placing the swab deep in the middle meatus, sampling the uncinate, ethmoid bulla and basal lamella [4]. The inferior turbinate was outfractured, and the nasopharynx was sampled with care taken to avoid contamination of the swab. FESS was then performed in the traditional manner [19], starting with maxillary antrostomy, then complete sphenoethmoidectomy, and finishing with Draf 2a frontal sinusotomy; as soon as each sinus was opened, an aggressive surface swab of that sinus was obtained. Externally-applied irrigation was not used, and microdebrider and/or endoscrub use was limited during the dissection so as to avoid irrigant contamination of a sinus prior to sampling.
Microbiome Profiling
DNA extraction and generation of bacterial 16S rRNA sequence libraries followed our previously described protocols [5,20]. In brief, a phenol:chloroform bead-beating method was used to extract total genomic DNA from culture swab heads, as previously described [8]. 16S rRNA gene templates were quantified in samples by quantitative PCR (QPCR) and the bacterial DNA load normalized to approximately 104 16S gene copies per μl [8,21]. Broad-range bacterial 16S rRNA gene amplicons were generated using primers that target approximately 300 base pairs of the V1V2 variable region of the 16S rRNA gene using barcoded [22] primers 27F-YM [23] (5’ AGAGTTTGATYMTGGCTCAG) and 338R [24] (5’ TGCTGCCTCCCGTAGGAGT). For barcoding PCR, 104 16S template copies were added to each reaction and amplified using the following program: first 95C for 7 minutes, then 94C for 30 seconds followed by 54.3C for 30 seconds and 72C for 1 minute × 34 cycles, then 72C for 10 minutes. Amplification was confirmed by running a 1.5% TAE agarose gel. Illumina paired-end sequencing was performed on the Illumina MiSeq platform using a 600-cycle version 3 reagent kit.
Analysis of Illumina Paired-end Reads
Illumina Miseq paired-end sequences were quality filtered and analyzed as previously described [20,25,26]. In brief, the sorted paired reads were assembled using phrap [27,28] and potential chimeras identified with Uchime (usearch6.0.203_i86linux32) [29] using the Schloss [30] Silva reference sequences. Assembled sequences were aligned and classified with SINA (1.2.11) [31] using the 418,497 bacterial sequences in Silva 115NR99 [32] as reference configured to yield the Silva taxonomy. Operational taxonomic units (OTUs) were generated by clustering sequences with identical taxonomic assignments. Relative abundances of OTUs were calculated for each subject by dividing the sequence counts observed for each OTU by the total number of high-quality 16S rRNA sequences generated for the subject. The Good's coverage index of each sequence library was >99.5% (estimated through 1000 replicate resamplings of rarefied sequence data [33]), indicating that most of the biodiversity in a sample was captured in its corresponding 16S sequence library.
Statistical Analysis
Within-patient similarity in microbiome composition between anatomical sites (i.e. beta-diversity) was measured using the abundance-based Morisita-Horn index. This abundance-based measurement of community structure reports the ecologic dissimilarities between pairs of samples, and is relatively less dependent on sample size and diversity than alternative measurements; here, a value of “0” means there is no overlap and a value of “1” means perfect overlap (i.e., two samples are completely identical). Comparison of percent relative abundances of OTUs between anatomical sites and subjects – such as the comparison of corynebacterial relative abundances -- was performed using the paired Wilcoxon Signed Rank Test, which was applied to all OTUs with mean relative abundances >1%. Because of the exploratory nature of this study, P-values were not adjusted for multiple comparisons. Principal coordinates analysis (PCoA) was utilized to explore and visualize data similarities in two-dimensional space; distance matrices were calculated using the Morisita-Horn index (vegdist function of vegan R package) and applied to the cmdscale function in R. Associations between principal coordinate scores and clinical variables were assessed by analysis of variance tests. Overall differences in microbiome composition between groups were quantified by non-parametric multivariate analysis of variance (PERMANOVA) tests using the adonis function of the vegan R package (v2.2-1), with dissimilarity measured by Morisita-Horn index. P-values were assessed through 50,000 replicate re-samplings and corrected for false discovery rate using the R p.adjust function [33]. All statistical analyses were performed using Explicet (www.explicet.org) [34], the R statistical package (v.2.14.0, Institute for Statistics and Mathematics, Wien, Austria), or Microsoft Excel.
RESULTS
The primary goal of this study was to examine the variability of the upper airways microbiome between individuals and between anatomic sites within each individual. To this end, eight subjects with diffuse bilateral CRS were consented and enrolled in the study, and bilateral sampling was performed. Swabs were collected from both the left and right side of the anterior nares, nasopharynx, middle meatus, frontal, maxillary, and sphenoid sinus. Demographics of subjects are shown in Table 1. High-throughput 16S rRNA gene sequencing demonstrated a rich consortium of bacteria among sampled sites, consistent with prior upper airway microbiome studies [5-9] (Figure 1). Nearly all of the 16S sequences (>99%) were classified to one of five bacterial phyla: Firmicutes (33% abundance, mainly streptococci and staphylococci), Proteobacteria (21% abundance, mainly pseudomonads and xanthomonads), Bacteroidetes (21% abundance, mainly prevotellas), Actinobacteria (15% abundance, mainly propionibacteria and corynebacteria), and Fusobacteria (10% abundance). Nonetheless, each subject displayed a characteristic assemblage of bacterial genera (Figure 1, lower panels).
Table 1.
Patient demographic and biodiversity characteristics (n=8).
| Mean age (years)1 | 62 (42, 73) |
| Male (%) | 4 (50%) |
| Allergic rhinitis (%) | 6 (75%) |
| Asthma (%) | 6 (75%) |
| Polyposis (%) | 4 (50%) |
| Purulence (%) | 5 (62.5%) |
| Never-smoker (%)2 | 5 (71.4%) |
| Diabetes (%) | 0 (0%) |
| Saline rinses (%) | 7 (87.5%) |
| Nasal steroid spray (%) | 7 (87.5%) |
| CT score1 | 14 (7.25, 14.5) |
| Endoscopy score1 | 6 (4.75, 11.25) |
| 16S Sequences per Subject1 | 139,000 (76,23, 244,211) |
| Good's Coverage (%)1 | 99.9% (99.8, 99.9) |
Data reported as median (interquartile range)
Data were available from 7 of 8 patients.
Figure 1.

Distribution of bacterial taxa across patients and anatomical subsites. Each stacked barchart summarizes the percent relative abundances (displayed on y-axes of plots) of bacterial taxa based on 16S rRNA sequencing. The top panels present average values across all 8 patients for phylum- and genus-level classifications. The bottom panels present genus-level data for each individual patient. To simplify data presentation, only taxa with relative abundances greater than 1% are shown; the remainder of low-abundance taxa are presented as “Other”. In the lower panels, subjects with polyps are labeled CRSwNP and those without polyps are labeled CRSsNP.
Similarities in microbiome composition between pairs of samples were quantified using the Morisita-Horn beta-diversity index. PERMANOVA tests revealed that interpersonal variability (P<0.0001) was a significant source of differences in microbiota, but not anatomic site (P=0.1) or side (P=0.94). Moreover, when interpersonal variability was accounted for in PERMANOVA tests, no significant differences between pairs of anatomic sites were noted following correction for multiple comparisons. These results were corroborated by principal coordinates analysis, which demonstrated that variability in microbial community composition was significantly associated with person-to-person differences (P < 0.0001 for PC1, P = 0.02 for PC2), but not differences between sampling sites (P = 0.89 for PC1, P = 0.99 for PC2 after adjusting for subjects) within the sinonasal tract (Figure 2). Morisita-Horn values displayed by heat map more clearly show within-subject similarity among the various sites sampled, and dissimilarity compared with most other subjects and sites (Figure 3).
Figure 2.

Principal coordinates analysis (PCoA) of microbial communities was performed on genus-level microbiome data using the Morisita-Horn similarity score calculated on each pair of samples. These plots indicate that interpersonal variability outweighs the differences noted among the sampled subsites within a subject. The top two plots show all samples plotted by subject (top left) and by site (top right), and the lower panels show samples from each individual. Subjects with polyps are denoted by asterisks.
Figure 3.

Heat map illustrating the Morisita-Horn beta diversity values by subject and site (1=identical, 0=complete dissimilarity).
To address the question of intra-subject differences in sinus microbiota, and identify a potential single site for sampling, Morisita-Horn similarity scores were plotted for within-subject sinus comparisons (Figure 4). Although a few outliers were noted, the microbial diversity of the middle meatus specimens exhibited general agreement (i.e., high Morisita-Horn similarity) with the other anatomical subsites. In searching for differences among the representation of bacterial OTUs in the sinuses and middle meatus, we consistently observed (Figure 5 and Supplemental Figure 1) higher relative abundances of corynebacterial species in the middle meatus (median relative abundance of 2.66% [IQR 1.1%-7.4%]) compared with the maxillary (0.22% [IQR 0.005%-1.2%]), frontal (0.41% [IQR 0.23%-1.1%]), and sphenoid sinuses (0.7% [IQR 0.05%-1.9%]). Corynebacterial abundances were highest in the anterior nares (4.1% [IQR 1.7%-9.1%]). Other taxa, such as Pseudomonales and Stenotrophomonas, were enriched in deeper sinuses compared with the middle meatus in some, but not all, statistical comparisons. For example, Pseudomonadales p-values ranged from 0.008 to 0.10, depending on the anatomical site and side of sampling. No other taxa were found to be reproducibly enriched or depleted in these analyses.
Figure 4.

Morisita-Horn comparison among sample sites demonstrates similarity of the middle meatus to other sites with a few outliers (1=identical, 0=complete dissimilarity). “Deeper sinuses” (maxillary, sphenoid, frontal) exhibit closest similarity, whereas the middle meatus demonstrates fairly good similarity to all sites.
Figure 5.
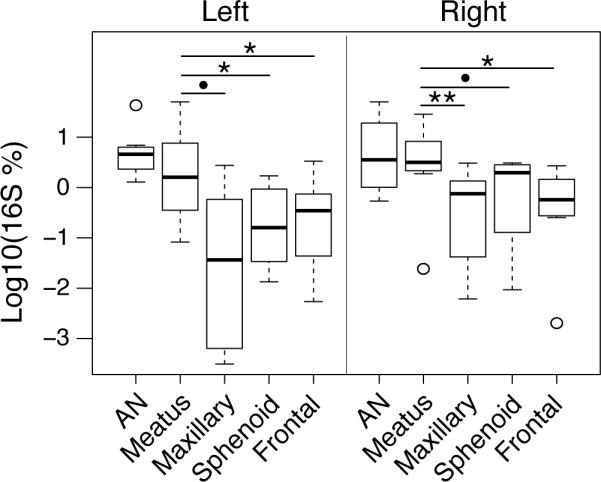
Corynebacterium spp. are more abundant in the middle meatus and anterior nares (AN) samples, possibly owing to a contribution from the anterior nares as seen on the heatmap. The results of paired Wilcoxon Signed Rank tests comparing middle meatus to AN, Maxillary, Sphenoid, and Frontal sinus corynebacterial relative abundances are noted [**=p<0.01, *=p<0.05, •=p<0.1].
DISCUSSION
Although we initially hypothesized that sinonasal anatomic subsites would harbor unique bacterial signatures resulting from distinctly altered microenvironments, we noted that interpersonal variability greatly outweighed the smaller differences apparent between anatomic sites. This finding is in concordance with prior microbiome studies that demonstrated that body habitat is a primary driver of community differences, but that have also noted that interpersonal variability is quite high for any given body site [35-41]. A recent study took this concept one step further, again reporting significant interpersonal variability, but also illustrating that within-subject community types at different body subsites are even predictive of each other [17]. Findings from these studies thus suggest that many different types of microbial consortia may contribute to health, and that although sites may differ in microbial community structure, they may still have some relationship with each other or result from specific host factors. Relating this to our current nascent understanding of microbiome study in CRS, interpersonal variation may very well outweigh subtler inter-sinus variability due to relatively mild alterations in the microenvironment of any given sinus, as seen in a recent study comparing nasal cavity sites [42]. This may be of importance in identifying subjects at risk for development of upper airway chronic inflammatory disease as well as predicting overall disease course. But, intra-individual, site-specific differences may still be significant in the consideration of localized sinus disease, which was not examined in the current study.
Although the primary goal of this study was not to conduct another survey of the sinus microbiome, the identified taxa aligned with those commonly found in prior studies [5,6]. As culture-independent study of sinus microbes continues to highlight the preponderance of anaerobes, it may be possible that effectors of tissue hypoxia previously identified in CRS [43-46] can drive changes in the local microbiome. In CRS, the significance of anaerobic organisms, as well as the nature of the hypoxic environment, are yet to be determined. But, it has been shown that hypoxia inhibits the activity of oxygen dependent enzymes required for clearance of organisms such as S..aureus [47-49], suggesting it may contribute to the persistence of replicating bacterial pathogens through modulation of host responses as well through selection of those preferring low oxygen tension.
A major implication of the current study is the potential moving forward for a single-site sampling method, an identified need to facilitate future longitudinal studies [50]. At this point, analyses of sinus microbiota have been challenging and difficult to replicate due to concerns about inclusion of control subjects, optimal sampling technique, sampling site, laboratory protocols, and data analysis pipelines [51]. One major advantage in CRS over microbiome study of other diseases is that patients are seen regularly over time and providers have ready access to the site of disease with nasal endoscopy. This potentially facilitates the study of the natural history of the disease process, patient-specific contributions to disease, and resilience of the sinus microbiome to perturbation, among many other possibilities. A recent investigation comparing surface swab to tissue biopsy demonstrated no difference in the microbial diversity revealed by the two methods, suggesting that swabs may serve as an adequate sampling method [52], a finding that has since been confirmed by a number of other groups. A recent publication also investigated inter-sinus differences, although only one subject studied had all sinuses from both sides sampled [53]. The authors found that compared sinuses were identical using unweighted UniFrac comparisons, suggesting no difference in presence or absence of taxa between specimens; using weight UniFrac distances that account for relative abundance, they concluded that when taking inter-individual variation into account, differences between sample sites were quite low. In the current study, we noted that interpersonal microbiome variability greatly outweighs inter-sinus, within-subject variability, and that the middle meatus is fairly representative of the underlying sinuses. However, we did note that the abundances of sinus-associated corynebacteria may be overestimated by middle meatus sampling. This result may reflect truly higher colonization density of corynebacteria within the middle meatus, or indicate potential contamination or migration arising from the anterior nares, which also harbors higher corynebacterial densities than the sinuses (Figures 1 and 5).
Overall, our results are analogous to the initial step in study of the gut microbiome, in which a single site of sampling (stool) was required to serve as a proxy for direct (i.e. invasive) sampling of each distinct segment of the alimentary tract. We selected patients with bilateral and generalized CRS (mean Lund-Mackay score =14) for study to evaluate the baseline degree of difference in each individual sinonasal niche. Findings may very well be different if one were to compare a healthy site to an infected sinus in patients with localized or unilateral CRS. Our data suggest that the middle meatus may be considered a representative sampling site for study of generalized host and environmental determinants of the sinonasal and upper airway microbiome, as well as in studies requiring healthy subjects who have not undergone sinus treatments, whereas direct sampling from within the sinus (and ideally from the same area of tissue harvest) may be preferred in studies of the direct interaction of the microbiome with host tissues.
Potential limitations of the current study include a relatively small sample size, although at least preliminary comparisons that warrant consideration were documented. The current study includes eight patients (16 sides) fitting strict criteria, and 96 total samples compares favorably to existing literature [6-8, 41]. The sample size serves as an initial inquiry and provides a basis for continued investigation utilizing the middle meatus as a single sample site in patients who have undergone prior surgery, and allows for the recruitment of healthy control subjects in which sinus openings have not been surgically created. In our study, we carefully placed swabs under endoscopic guidance to precise anatomic areas in sequence to limit potential contamination. The order of sampling was utilized to theoretically remove the easily sampled surface and limit potential contamination with subsequent deeper passes. It is possible, however, that anterior nares contaminants would be present on subsequent swab samples, but this concern appears to be limited as most subjects (2,3,4,6,7,8) showed a notable difference from the anterior nares to deeper subsites (Figure 1B). Despite these findings mitigating such concerns, efforts should be made to limit contamination from surrounding areas in future study, ideally with the use of a protected swab in future studies.
CONCLUSION
Microbiome study of the sinuses in chronic rhinosinusitis is in its early stages, and here we examine the possible use of the middle meatus as a representative proxy for the sinuses for sampling in future, prospective, longitudinal studies of health and disease state. Although small, idiosyncratic microbiome differences were observed among sinonasal anatomic subsites, interpersonal variability largely outweighed these differences. Examination of direct interactions of the microbiome on local tissues is still best served with sampling of the airway surface and matched, directly adjacent epithelium.
Supplementary Material
Acknowledgments
Funding Sources: Research reported in this publication was supported by the National Institute On Deafness And Other Communication Disorders of the National Institutes of Health under Award Number K23DC014747 (VRR), and by Flight Attendants Medical Research Institute grant CIA13006 (DNF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of interest: no conflicts of interest or financial disclosures for any author
Contributions: VRR conceived the study, analyzed the data, and wrote the manuscript; SG designed and performed the experiments; JMK performed the experiments and analyzed the data; CER performed the experiments and analyzed the data; DNF conceived the study, supervised the performed experiments, analyzed the data, and contributed to the manuscript. All authors discussed the results and implications and contributed to the final manuscript.
References
- 1.Fokkens W, Lund VJ, Mullol J, et al. European position paper on rhinosinusitis and nasal polyps 2012. Rhinology. 2012;(Suppl 23):1–298. [PubMed] [Google Scholar]
- 2.Feazel L, Frank DN, Ramakrishnan VR. Update on Bacterial Detection Methods: Implications for clinicians and researchers. Int Forum Allergy Rhinol. 2011;1(6):451–9. doi: 10.1002/alr.20071. [DOI] [PubMed] [Google Scholar]
- 3.Hauser LJ, Feazel LM, Ir D, et al. Sinus culture poorly predicts resident microbiota. Int Forum Allergy Rhinol. 5(1):3–9. doi: 10.1002/alr.21428. 20145. [DOI] [PubMed] [Google Scholar]
- 4.Ramakrishnan VR, Feazel LM, Gitomer SA, et al. The microbiome of the middle meatus in healthy adults. PLoS One. 2013 Dec 30;8(12):e85507. doi: 10.1371/journal.pone.0085507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramakrishnan VR, Hauser LJ, Feazel LM, et al. Sinus microbiota varies among chronic rhinosinusitis phenotypes and predicts surgical outcome. J Allergy Clin Immunol. 2015;136(2):334–42. doi: 10.1016/j.jaci.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Abreu NA, Nagalingam NA, Song Y, et al. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med. 2012;4(151):151ra124. doi: 10.1126/scitranslmed.3003783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan M, Pamp SJ, Fukuyama J, et al. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host Microbe. 2013;14(6):631–40. doi: 10.1016/j.chom.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feazel L, Robertson C, Ramakrishnan VR, Frank DN. Staphylococcus aureus and microbiome diversity in chronic rhinosinusitis. Laryngoscope. 2012;122(2):467–72. doi: 10.1002/lary.22398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank DN, Feazel LM, Bessesen MT, et al. The Human Nasal Microbiota and Staphylococcus aureus carriage. PLos ONE. 2010;(5):e10598. doi: 10.1371/journal.pone.0010598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephenson MF, Mfuna L, Dowd SE, et al. Molecular characterization of the polymicrobial flora in chronic rhinosinusitis. J Otolaryngol Head Neck Surg. 2010;(39):182–7. [PubMed] [Google Scholar]
- 11.Gold SM, Tami TA. Role of middle meatus aspiration culture in the diagnosis of chronic sinusitis. Laryngoscope. 1997;107(12 Pt 1):1586–9. doi: 10.1097/00005537-199712000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Oczan M, Unal A, Aksaray S, et al. Correlation of middle meatus and ethmoid sinus microbiology in patients with chronic sinusitis. Rhinology. 2002;40(1):24–7. [PubMed] [Google Scholar]
- 13.Dubin MG, Ebert CS, Coffey CS, et al. Concordance of middle meatal swab and maxillary sinus aspirate in acute and chronic sinusitis; a meta-analysis. Am J Rhinol. 2005;19(5):462–70. [PubMed] [Google Scholar]
- 14.Aust R, Drettner B. Oxygen tensions in the human maxillary sinus during normal and pathological conditions. Acta Otolaryngol (Stockh) 1974;78:264–9. doi: 10.3109/00016487409126354. [DOI] [PubMed] [Google Scholar]
- 15.Matsune S, Kono M, Sun D, et al. Hypoxia in paranasal sinuses of patients with chronic sinusitis with or without the complication of nasal allergy. Acta Otolaryngol. 2003;123(4):519–23. doi: 10.1080/0036554021000028113. [DOI] [PubMed] [Google Scholar]
- 16.Blaser M, Bork P, Fraser C, et al. The microbiome explored: recent insights and future challenges. Nat Rev Microbiol. 2013;11:213–217. doi: 10.1038/nrmicro2973. [DOI] [PubMed] [Google Scholar]
- 17.Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014;509(7500):357–60. doi: 10.1038/nature13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenfeld RM, Andes D, Bhattacharyya N, et al. Clinical practice guideline: adult sinusitis. Otolaryngol Head Neck Surg. 2007;137(3 Suppl):S1–31. doi: 10.1016/j.otohns.2007.06.726. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy DW, Ramakrishnan VR. Functional Endoscopic Sinus Surgery: Concepts, Surgical Indications, and Techniques. Chapter. In: Kennedy, Hwang, editors. Rhinology: Diseases of the Nose, Sinuses and Skull Base. Thieme; 2012. pp. 306–335. [Google Scholar]
- 20.Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339(6123):1084–8. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- 21.Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(1):257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 22.Frank DN. BARCRAWL and BARTAB: software tools for the design and implementation of barcoded primers for highly multiplexed DNA sequencing. BMC Bioinformatics. 2009;10:362. doi: 10.1186/1471-2105-10-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank JA, Reich CI, Sharma S, et al. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol. 2008;74:2461–2470. doi: 10.1128/AEM.02272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lane D. 16S/23S DNA sequencing. In: Stacklebrandt E, Goodfellow M, editors. Nucleic Acid Techniques in Bacterial Systematics. Hohn Wiley and Sons; New York, NY: 1991. pp. 115–175. [Google Scholar]
- 25.Dillon SM, Le EJ, Kotter CV, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014;7(4):983–994. doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frank DN, Bales ES, Monks J, et al. Perilipin-2 Modulates Lipid Absorption and Microbiome Responses in the Mouse Intestine. PLoS One. 2015;10(7):e0131944. doi: 10.1371/journal.pone.0131944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome research. 1998;8:186–94. [PubMed] [Google Scholar]
- 28.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–85. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 29.Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;(6):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson CE, Harris JK, Wagner BD, et al. Explicet: graphical user interface software for metadata-driven management, analysis and visualization of microbiome data. Bioinformatics. 2013;29:3100–3101. doi: 10.1093/bioinformatics/btt526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costello EK, Lauber CL, Hamady M, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–7. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarzberg K, Le R, Bharti B, et al. The personal human oral microbiome obscures the effects of treatment on periodontal disease. PLoS One. 2014;9(29)(1):e86708. doi: 10.1371/journal.pone.0086708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lavelle A, Lennon G, O'Sullivan O, et al. Spatial variation of the colonic microbiota in patients with ulcerative colitis and control volunteers. Gut. 2015 Jan 16; doi: 10.1136/gutjnl-2014-307873. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frank DN, Feazel LM, Bessesen MT, et al. The human nasal microbiota and Staphyloccous aureus carriage. PLoS ONE. 2010;5:e10598. doi: 10.1371/journal.pone.0010598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomcs era. Curr Opin Gastroenterol. 2008;24:4–10. doi: 10.1097/MOG.0b013e3282f2b0e8. [DOI] [PubMed] [Google Scholar]
- 40.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008;3:417–27. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biswas K, Hoggard M, Jain R, et al. The nasal microbiota in health and disease: variation within and between subjects. Front Microbiol. 2015;9:134. doi: 10.3389/fmicb.2018.00134. doi: 10.3389/fmicb.2015.00134. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Airley R, Loncaster J, Davidson S, et al. Glucose transporter GLUT-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix. Clin Cancer Res. 2001;7:928–34. [PubMed] [Google Scholar]
- 44.Zhou X, Tu J, Li Q, et al. Hypoxia induces mucin expression and secretion in human bronchial epithelial cells. Transl Res. 2012;160(6):419–27. doi: 10.1016/j.trsl.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Kim YJ, Cho HJ, Shin WC, et al. Hypoxia-mediated mechanism for MUC5AC production in human nasal epithelia and its implication for rhinosinusitis. PLoS One. 2014;9:e98136. doi: 10.1371/journal.pone.0098136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelly CJ, Glover LE, Campbell EL, et al. Fundamenal role for HIF-1alpha in constitutive expression of human beta defensin-1. Mucosal Immunol. 2013;6(6):1110–8. doi: 10.1038/mi.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGovern NN1, Cowburn AS, Porter L, et al. Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J Immunol. 2011;186(1):453–63. doi: 10.4049/jimmunol.1002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daniliuc S, Bitterman H, Rahat MA, et al. Hypoxia inactivates inducible nitric oxide synthase in mouse macrophages by disrupting its interaction with alpha-actinin 4. J Immunol. 2003;171:3225–3232. doi: 10.4049/jimmunol.171.6.3225. [DOI] [PubMed] [Google Scholar]
- 49.Wiese M, Gerlach RG, Popp I, et al. Hypoxia-mediated impairment of the mitochondrial respiratory chain inhibits the bactericidal activity of macrophages. Infect Immun. 2012;80(4):1455–66. doi: 10.1128/IAI.05972-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramakrishnan VR, Hauser LJ, Frank DN. The sinonasal bacterial microbiome in health and disease. Curr Opin Otolaryngol Head Neck Surg. 2016;24(1):20–5. doi: 10.1097/MOO.0000000000000221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JT, Frank DN, Ramakrishnan VR. Microbiome of the paranasal sinuses: update and literature review. Am J Rhinol Allergy. doi: 10.2500/ajra.2016.30.4255. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bassiouni A, Cleland EJ, Psaltis AJ, et al. Sinonasal microbiome sampling: a comparison of techniques. PLoS One. 2015;10(4):e0123216. doi: 10.1371/journal.pone.0123216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joss TV, Burke CM, Hudson BJ, et al. Bacterial communities vary between sinuses in chronic rhinosinusitis patients. Front Microbiol. 2016;6:1532. doi: 10.3389/fmicb.2015.01532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.