

Abstract
Microglia are the primary immune cell of the brain and function to protect the central nervous system (CNS) from injury and invading pathogens. In the homeostatic brain, microglia serve to support neuronal health through synaptic pruning, promoting normal brain connectivity and development, and through release of neurotrophic factors, providing support for CNS integrity. However, recent evidence indicates that the homeostatic functioning of these cells is lost in neurodegenerative disease, including Alzheimer’s disease (AD), ultimately contributing to a chronic neuroinflammatory environment in the brain. Importantly, the development of compounds and genetic models to ablate the microglial compartment has emerged as effective tools to further our understanding of microglial function in AD. Use of these models has identified roles of microglia in several pathological facets of AD, including tau propagation, synaptic stripping, neuronal loss, and cognitive decline. Although culminating evidence utilizing these microglial ablation models reports an absence of CNS-endogenous and peripheral myeloid cell involvement in Aβ phagocytosis, recent data indicates that targeting microglia-evoked neuroinflammation in AD may be essential for potential therapeutics. Therefore, identifying altered signaling pathways in the microglia-devoid brain may assist with the development of effective inflammation-based therapies in AD.
Keywords: Microglia, Colony-stimulating factor 1 receptor, Inflammation, Alzheimer’s disease, Amyloid, Tau, Spines
I. Introduction
Microglia are the brain’s resident immune cells, comprising approximately 5–12% of all cells found in the brain. They function as the brain’s first line of defense to protect the CNS from injury and invading pathogens. Originally presumed to be “resting,” microglia in the healthy adult brain are highly dynamic, surveying the entire brain parenchyma every 24 hours (Nimmerjahn et al., 2005). In this “surveying” state, microglia exhibit a ramified morphology and serve to support neuronal function and health via physical and biochemical interactions. Upon detection of an insult, microglia respond by becoming activated. This process may involve the migration to and proliferation of these cells at the site of the insult, as well as dramatic transformation into an amoeboid morphology, depending on the type and extent of the insult. Activated microglia produce and secrete several proinflammatory mediators, including tumor necrosis factor-α (TNFα), interleukin (IL)-6, and nitric oxide (NO), all of which can confer neurotoxicity (Akiyama et al., 2000). Neuroimmune regulatory proteins (NiReg) modulate the microglia-mediated immune response to resolve the inflammatory process (Hoarau et al., 2011), which then promotes tissue repair through the secretion of several neurotrophic factors, including insulin-like growth factor 1 (IGF-1), brain-derived neurotrophoc factor (BDNF), transforming-growth factor-β (TGF-β), and nerve growth factor (NGF) (Polazzi and Monti, 2010). In acute inflammatory events, the pro-inflammatory response resolves and microglia continue their surveillance of the brain parenchyma. However, in neurodegenerative disease, the equilibrium between microglial surveillance and activation is disturbed, creating a feedforward loop that results in a chronic neuroinflammatory state, promoting inflammation and tissue atrophy. Although astrocytes also participate in and propagate the neuroinflammatory environment in AD, for the purposes of this review, the focus will remain on microglia-mediated inflammation in AD pathogenesis. Here, we briefly review the various implications of microglia and other myeloid cells in neurodegeneration and discuss current methods that allow for investigations into the biology of microglia in AD.
2. Homeostatic microglial functions
2.1. Phagocytosis
One of the more extensively studied functions of microglia in the brain is their role in clearance via phagocytosis, by which these cells both protect the brain from invading pathogens as well as remove cellular debris from the neural environment. Aside from the clearance of cellular debris, microglia may also phagocytose viable neurons in a process known as “phagoptosis,” which specifically targets senescent or damaged cells (Brown and Neher, 2014). For this reason, proper degradation of internalized components by microglia is essential for normal CNS function. Consequently, dysregulated or abnormal degradation of material can result in the intracellular accumulation of toxic molecules, including reactive oxygen species (Underhill and Goodridge, 2012). Importantly, studies have shown that microglia are involved in the phagocytosis of supranumerous apoptotic neuroblasts in the subgranular zone of young mice, implicating their involvement in neurogenesis (Sierra et al., 2010).
2.2 Synaptic sculpting and cognition
In addition to phagocytosis, microglia are also involved in the removal of synapses from neuronal cell bodies via synaptic stripping. This phenomenon was first observed in a model of facial nerve injury in rats (Blinzinger and Kreutzberg, 1968), in which microglial localization to the site of injury and interaction with facial motor neurons resulted in the removal of synaptic contacts. In the developing brain, microglia form brief, repetitive contacts with synapses, eliminating any weak or unnecessary synaptic structures, a process which is modulated by sensory experiences (Tremblay et al., 2010). Recently, it was shown that knockout of the microglial purigenic receptor P2Y12, which mediates process motility during injury response, also disrupts plasticity in the visual system (Sipe et al., 2016). While the exact mechanism behind microglia-mediated synaptic elimination (whether it be by phagocytosis or the secretion of various factors) has yet to be elucidated, it is clear that the interaction between microglia and synapses is crucial for activity-dependent plasticity in the developing brain. Accumulating evidence points to neuron-microglia crosstalk as an essential mechanism for proper synapse and network maintenance. One pathway implicated in this crosstalk involves the fractalkine receptor (CX3CR1) expressed on microglia and its ligand CX3CL1, released by neurons. For example, knockout of CX3CR1 during development produces deficits in synaptic pruning, characterized by an excess of dendritic spines and immature synapses, resulting in weakened synaptic transmission and decreased functional brain connectivity (Paolicelli et al., 2011). Behaviorally, loss of CX3CR1 in juvenile mice manifests in impaired social interactions reminiscent of autism spectrum disorder and other neuropsychiatric disorders (Zhan et al., 2014). Furthermore, disruption of signaling between complement 3 (C3), which is localized to synaptically-enriched regions, and its receptor, complement receptor 3 (CR3), in the mouse retinogeniculate system impairs microglial phagocytosis of synaptic inputs, leading to sustained deficits in brain wiring (Schafer et al., 2012). Collectively, these studies underscore the role of microglia as regulators of the synaptic landscape in the developing brain, implicating neuron-microglia crosstalk as a crucial process for proper brain development. In addition to complement and fractalkine signaling, paired immunoglobulin-like receptor B (PirB) is also involved in the regulation of synaptic plasticity. In the CNS, PirB is expressed by both neurons and glia and binds several ligands, including major histocompatibility complex-I (MHC-I) (Syken et al., 2006), which is believed to direct cellular recognition by immune cells. In adult mice, disruption of PirB signaling in the visual cortex increased dendritic spine density and induced the formation of functional synapses, as evidenced by increases in miniature excitatory postsynaptic currents (Bochner et al., 2014). Whether microglia are mediating the PirB-induced synaptic changes remains unresolved, although this cell type is a likely candidate.
While the role of microglia in synaptic sculpting during development is well-described, it remains unclear whether microglia contribute to the synaptic landscape in adulthood. We recently reported that the absence of microglia in the healthy adult mouse brain increases total dendritic spine density and intensity of immunolabeling for the synaptic surrogates PSD95 and synaptophysin (Rice et al., 2015), indicating that microglia continue to regulate the synaptic landscape in adulthood. Collectively, these studies point to microglia as critical mediators of synaptic sculpting in development and adulthood, providing an important role in shaping and modulating neuronal circuitry to maintain normal brain connectivity.
As microglia are heavily implicated in shaping the synaptic landscape of the brain during the incorporation of new memories and experiences into the neural network, the CNS immune system is also thought to be involved in cognitive function. Genetic ablation of microglia using CX3CR1CreER mice to drive diphtheria toxin receptor expression in these cells found that mice devoid of microglia, following administration of diphtheria toxin, exhibited impaired performance on cue-based fear condition and novel object recognition tasks, as well as impaired dendritic spine remodeling (Parkhurst et al., 2013). However, it is important to note that since behavioral analysis occurred no later than postnatal day 34, an age at which microglia actively refine synapses and eliminate weak and unnecessary neuronal connections, it is not surprising that microglia-deficient mice exhibit memory-related deficits. Additionally, death of microglia with this genetic approach may induce widespread inflammatory processes from surviving cells (Bruttger et al., 2015), including the production of pro-inflammatory cytokines, which can impair cognitive functioning (Bellinger et al., 1993; Dugan et al., 2009; Terrando et al., 2010). In contrast to this, the administration of the CSF1R inhibitor, PLX3397, to eliminate microglia in adult mice revealed no behavioral or cognitive impairments, indicating that microglia are not necessary in these tasks in adulthood (Elmore et al., 2014). Providing an additional explanation for the differences between these studies, one report indicated impairments in spatial memory with one week of PLX3397 treatment, which subsided with longer treatments (Torres et al., 2015). The transient impairment in cognitive function is presumably due to behavioral assessment occurring at a time point in which microglia are still in the process of dying. Importantly, multiple groups have utilized PLX3397, as well as the CSF1R-specific inhibitor PLX5622 to eliminate microglia (De et al., 2014; Valdearcos et al., 2014; Asai et al., 2015; Klein et al., 2015; Schreiner et al., 2015), without inducing adverse effects on cognitive function in adult mice (Dagher et al., 2015; Rice et al., 2015; Spangenberg et al., 2016).
3. AD pathophysiology
In contrast to the beneficial roles of microglia in the homeostatic brain, the activation of these cells is heavily implicated in the progression of several neurodegenerative diseases, including AD. AD is a progressive neurodegenerative disease characterized by the extracellular deposition of Aβ-associated plaques and intracellular tau-associated neurofibrillary tangles. It is also one of the most common forms of dementia, affecting roughly 10% of the population aged 65, and up to 50% of people aged 85 and over (Hebert et al., 2003), with the number of cases expected to triple by 2050 (Hebert et al., 2013). Therefore, understanding the mechanistic changes in the human brain leading to pathophysiology of the disease is essential to creating effective therapies. According to the amyloid cascade hypothesis, the triggering factor for the disease is the accumulation of amyloid aggregates composed of Aβ peptides (Hardy and Selkoe, 2002). The presence of aggregated Aβ initiates downstream effects, including sustained chronic neuroinflammation, tau hyperphosphorylation, and a loss of synapses and neurons that ultimately lead to considerable brain atrophy and cognitive decline. In human AD, evidence indicates that these amyloid deposits may present decades before the cognitive deficits associated with this disorder are evident (Mintun et al., 2006). Following amyloid deposition, the presence of intraneuronal neurofibrillary tangles comprised of hyperphosphorylated tau is observed in AD, and importantly, tau pathology more closely correlates with cognitive status, and neurodegeneration, than amyloid pathology (Nelson et al., 2009). Another critical facet of AD is chronic neuroinflammation, which is characterized by astro- and microgliosis (Akiyama et al., 2000). Together, these neuropathological alterations exert toxic effects on the brain, leading to brain degeneration. Even in the mild cognitive impairment (MCI) phase of the disease, which is the intermediate stage between the expected cognitive decline associated with normal aging and dementia, there appears to be substantial accumulation of amyloid and tau, as well as neuronal loss (Morris and Price, 2001). These findings highlight the importance of treating the disease at the earliest stages in order to effectively control and limit the progression of AD.
4. Microglial contribution to AD pathogenesis
4.1. Microglial dysfunction and inflammation in AD
Over one hundred years ago, Alois Alzheimer first identified the presence of plaque-associated microglia in post-mortem brains (Alzheimer et al., 1995) and since then, activated microglia have been considered a key feature of AD pathology. In the AD brain, the number and size of microglia directly increase proportionally to the size of the plaques (Wegiel et al., 2001) and proliferate in the vicinity of plaques leading to the accumulation of these cells at amyloid deposits (Frautschy et al., 1998; Bornemann et al., 2001). Plaque-associated microglia have been shown to encircle plaques and it is postulated that the close proximity of microglia regulates plaque dynamics in AD mice (Condello et al., 2015). Recently, the emergence of inflammation-associated positron emission tomography (PET) imaging radioligands have allowed researchers to track inflammatory processes in humans with various diseases, including AD. Prospective studies utilizing PET ligands in AD patients found that microglial activation occurs well before clinical symptoms of cognitive decline present (Yasuno et al., 2012; Hamelin et al., 2016), highlighting the potential for immune-specific PET ligands as tools to monitor the inflammatory progression in a clinical setting for AD.
The hypothesis of compromised microglial function as a contributor to AD pathogenesis gained momentum following the publication of recent genome wide association (GWAS) studies. These studies identified several single nucleotide polymorphisms (SNPs) that convey risk of developing AD, with many of these SNPs associated with or related to microglial function (Malik et al., 2015) including TREM2, CD33, CR1, CLU, CD2AP, EPHA1, ABCA7, and INPP5D (Lambert et al., 2009; Hollingworth et al., 2011; Naj et al., 2011; Guerreiro et al., 2013; Lambert et al., 2013), indicating that microglia play a critical role in the development of AD. The generation of knockout mice for various immune-associated GWAS genes have allowed for in vivo investigations into the contribution of these genes in AD pathogenesis. Inactivation of CD33 in APP/PS1 mice reduced Aβ accumulation and plaque burden (Griciuc et al., 2013), whereas ABCA7 deficiency in APP/PS1 and TgCRND8 mice exacerbated Aβ load (Satoh et al., 2015; Sakae et al., 2016), highlighting the importance of these genes in regulating Aβ pathology. In contrast to CD33 and ABCA7, attempts to define the exact function of TREM2 in AD have been less clear. In APP/PS1 mice, removal of TREM2 mitigated Aβ accumulation (Wang et al., 2015), whereas in 5xfAD mice, knockout of TREM2 worsened Aβ pathology (Jay et al., 2015). In both studies, however, removal of TREM2 signaling prevented the association of myeloid cells – whether it be microglia or monocytes – with plaques. A subsequent parabiosis study using both AD models found no indication of infiltrated monocytes surrounding Aβ plaques (Wang et al., 2016), providing evidence that endogenous microglia are the source of plaque-associated myeloid cells. Importantly, loss of one (Yuan et al., 2016) or two copies of TREM2 (Wang et al., 2016) impaired the ability of microglia to compact amyloid into dense deposits, thereby increasing damage to local neuritic structures. Collectively, these studies indicate that TREM2 signaling regulates the microglial response to Aβ in AD, although whether this alteration in microglial function is beneficial or detrimental requires further experimentation. Furthermore, the role of inflammation in AD progression, as well as facilitating Aβ deposition, neuronal loss, and cognitive deficits is well described (see Fig. 1). Exposing microglia to Aβ induces the production and release of pro-inflammatory cytokines, including IL-1β, IL-6, TNFα, TGF-β, as well as chemokines, such as macrophage inflammatory proteins (MIP)-1a, 1b, -2, and chemokine (C-C Motif) ligand 2 (CCL2) (Akiyama et al., 2000) in an attempt to facilitate the clearance of Aβ. However, microglia in AD are ineffective Aβ phagocytes, as evidenced by the continued presence of plaques surrounded by activated microglia in the AD brain, resulting in an enduring chronic neuroinflammatory environment for the duration of the disease. Furthermore, the sustained release of neurotoxic factors likely leads to neurodegeneration and many of these neurotoxic factors are evidenced to be microglia-derived, including TNF-α, NO, IL-1β, and ROS (Lull and Block, 2010). Knockout of NLRP3 – a critical component in the inflammasome pathway and major contributor to neuroinflammatory insult in the CNS – in APP/PS1 mice mitigated inflammatory signaling, decreased deposition of Aβ, restored synaptic plasticity, and improved cognitive function as assessed by Morris water maze (Heneka et al., 2013). Importantly, the formation and secretion of the NLRP3 inflammasome is restricted to the microglial compartment in the mouse brain (Gustin et al., 2015), which together with the NLRP3 knockout data indicate that dysfunctional regulation of microglial NLPR3 signaling may underlie AD pathogenesis.
Figure 1.
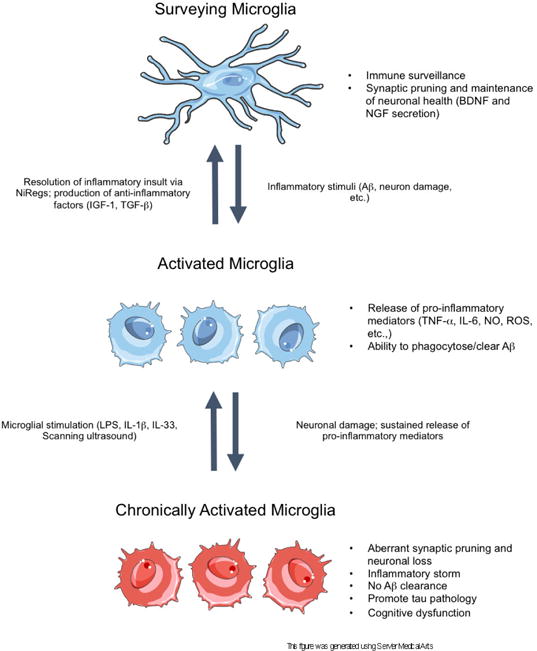
A schematic of acute and chronic microglial activation in the context of Alzheimer’s disease. In the surveillance state, microglia sample the brain parenchyma to detect anomalous materials, while concomitantly secreting anti-inflammatory factors and pruning unnecessary or weak synapses to support neuronal health. Disturbances in brain homeostasis, such as detection of Aβ, result in the activation of microglia. In the activated state, microglia release pro-inflammatory mediators to recruit other cells to the insult and to phagocytose the detected insult. When the insult is cleared, the inflammatory response resolves via NiReg (Neuroimmune regulatory proteins) activity, and microglia secrete anti-inflammatory factors to promote tissue repair and growth. However, in neurodegenerative diseases, the sustained release of pro-inflammatory mediators, as well as increasing neuronal death, drives the cells into a state of chronic activation. In this state, the homoeostatic functioning of microglia is lost, resulting in an accumulation of aggregated proteins, the stripping of synapses from neurons, as well as further neuronal loss. Importantly, in this reactive state, microglia can be stimulated with certain proteins or methods to induce microglial phagocytosis of Aβ, thereby restoring the phagocytic ability of microglia.
In addition to the general functions of these cells in AD, the timing of neuroinflammation in the progression of AD is also crucial to our understanding of the disease – recent evidence indicates that inflammatory changes occur before the appearance of amyloid plaques (Heneka et al., 2005; Ferretti et al., 2012; Wright et al., 2013). One study exploring early immune activation in AD revealed that inflammation alone can drive AD pathogenesis. Specifically, systemic immune stimulation in prenatal mice with a viral mimetic (i.e., double stranded RNA) followed by immune challenge later in adulthood, was sufficient to induce the development of sporadic-like AD, characterized by deposition of Aβ and tau proteins, impairments in working memory, as well as chronic microglial activation (Krstic et al., 2012). Interestingly, this study provides evidence for immune activation early in life as a sufficient factor to trigger the onset of AD. In addition to chronic microglial activation, the senescence of this cell type that normally ensures neuroprotection may underlie AD pathogenesis (Streit, 2002). Analysis of postmortem AD brains showed elevated expression of the microglial dystrophic marker, ferretin, as well as thinning and fragmentation of microglial processes (Lopes et al., 2008). Subsequent studies found that tau-positive neuronal structures were associated with dystrophic rather than chronically activated microglia, suggesting that microglial senescence, which results in diminished neuroprotection, may contribute to the onset of AD through neurofibrillary degeneration (Streit et al., 2009).
Although gray matter is predominantly affected in AD, reports indicate that white matter from AD patients also displays considerable damage (Rose et al., 2000). It is proposed that axonal degeneration may occur in early AD (Terry, 1998), although the contribution of microglia in white matter damage remains to be clarified. Assessment of cerebrospinal fluid (CSF) markers and regional microstructure from asymptomatic adults found that the neuroinflammatory markers were associated with markers of axonal damage and altered microstructure on diffusion tensor imaging (DTI) (Melah et al., 2016), suggesting that neuroinflammation worsens white matter damage in preclinical AD. However, PET imaging with the second generation TPSO ligand DPA-714 in early stage and prodromal AD patients showed that patients with increased ligand binding declined slower, suggesting that microglial activation may be protective in the early stages of the disease (Hamelin et al., 2016). Collectively, these studies highlight the importance of inflammation-targeted therapeutic approaches for treating this disease, but caution that timing may be critical.
4.2. Microglial regulation of Aβ dynamics
Traditionally, it was believed that microglia respond to the presence of Aβ deposits by clearing them from the brain via phagocytosis of Aβ-fibrils (Pan et al., 2011). In vitro studies have shown clear evidence that microglia are able to internalize and degrade Aβ aggregates (Paresce et al., 1997; Li et al., 2000; Koenigsknecht-Talboo and Landreth, 2005; Hellwig et al., 2015). However, there was no clear consensus in vivo as to whether microglia possess the capacity to clear Aβ, as some studies show internalization of Aβ to the lysosome of microglia (Bolmont et al., 2008), whereas others do not observe Aβ plaque clearance by these cells (Stalder et al., 2001; Meyer-Luehmann et al., 2008). In order to directly assess the roles of microglia in Aβ clearance and plaque dynamics, several methods for in vivo microglial ablation have been developed, which allows researchers to probe the roles of AD-associated microglia, and how they contribute to pathophysiology. For example, clodronate liposomes induce apoptosis in phagocytizing macrophages (Claassen et al., 1990). Using this method in P0 5xfAD mouse organotypic hippocampal slice cultures, microglia are depleted leading to the rapid accumulation of Aβ deposits, showing that microglia in development and/or in slice cultures actively clear Aβ (Hellwig et al., 2015). However, upon replenishment with juvenile microglia, Aβ clearance is restored resulting in fewer Aβ deposits. Notably, replenishment with adult 5xfAD microglia does not reduce plaque deposits, indicating that the phagocytic ability of microglia is quickly lost with age, somewhere between 1 and 6 months of age. In CD11b-HSVTK (TK) mice, the thymidine kinase of herpes simplex virus is expressed under the CD11b promoter. Thymidine kinase converts ganciclovir into cytotoxic kinases, leading to cell death. In the brain, microglia exclusively express CD11b, and are therefore inducibly ablated with this method, with current studies maintaining ablation for up to four weeks. Researchers crossed TK and AD mice to assess alterations in plaque dynamics with the elimination of microglia and found no changes in plaque load, Aβ levels, or dystrophic neuritic structures near plaques, in either young or aged animals (Grathwohl et al., 2009). As the absence of microglia had no impact on Aβ dynamics, this suggests that aged microglia may not be actively phagocytosing and clearing plaques in the AD brain.
In addition to toxin models, microglia can also be eliminated via inhibition of the CSF1R. Knocking out the CSF1R (Ginhoux et al., 2010) or either of its ligands, CSF1 (Wegiel et al., 1998) and IL34 (Wang et al., 2012), results in robust decreases in microglial number, emphasizing the importance of this signaling pathway in myeloid cell development. Our lab discovered that pharmacological inhibition of the CSF1R in adult mice led to the rapid elimination of microglia from the entire CNS. With seven days of treatment, >95% of microglia can be eliminated with the drug PLX3397, and microglia remain eliminated for as long as treatment is continued (Elmore et al., 2014). This provides a non-invasive approach to study microglial dynamics in any mouse model. Importantly, treatment with PLX3397 induces microglial cell death, as opposed to a downregulation of microglial-related genes (Spangenberg et al., 2016). CSF1R inhibitors can be formulated in chow and consumed by mice ad-libitum, while also maintaining blood-brain barrier (BBB) integrity. Thus, this provides a non-invasive context in which to study microglial dynamics in the brain, in both health and disease. Consistent with the prior data (Grathwohl et al., 2009), elimination of microglia from either aged or pre-pathological 5xfAD mice had no effects on Aβ accumulation or deposition (Spangenberg et al., 2016), confirming that microglia are not serving to remove amyloid from the brain. Notably, we found evidence of heterogeneity of myeloid cell populations associated with plaques, as ~50% of plaque-associated myeloid cells resistant to elimination with CSF1R inhibitor treatment. These surviving cells may be infiltrated monocytes, which are not dependent on CSF1R signaling for their survival (Jay et al., 2015), or may alternatively upregulate TREM2, which can act as a survival signal in place of CSF1R (Wang et al., 2015).
In accordance with these findings, recent evidence suggests that plaque-associated microglia in vivo are in a suppressed phagocytic state due to the overproduction of IL-10 (Chakrabarty et al., 2015), prostaglandin E2 (PGE2) (Johansson et al., 2015), and arginase-1 (Kan et al., 2015). Indeed, subsequent exposure of microglia to certain stimuli, including LPS (DiCarlo et al., 2001), IL-1β (Shaftel et al., 2007), IL-33 (Fu et al., 2016), as well as the retinoid X receptor agonist bexarotene (Cramer et al., 2012) and ultrasound (Leinenga and Götz, 2015) are sufficient to induce microglial phagocytosis of Aβ. Together, these studies indicate that microglia are fully capable of Aβ phagocytosis, given a favorable environment in which to do so. Additionally, in a transgenic model of Aβ arrest, switching off the APP transgene halted the progression of Aβ pathology, but did not induce the breakdown or clearance of plaques (Jankowsky et al., 2005), providing further evidence that microglia are not regulating Aβ dynamics in the AD brain. Although microglial association with plaques is pervasive in different murine models of AD, as well as AD patients, the function of this association was previously unknown. Using in vivo methods to examine the role of microglia in regulating plaque dynamics revealed that microglia constitute a barrier surrounding amyloid deposits, serving to limit their outward expansion. Moreover, plaque-associated microglia limited the toxic effects of Aβ42 hotspots on nearby neurons (Condello et al., 2015). As smaller (and presumably newer) plaques were most restricted in size, these data provide evidence for a neuroprotective role of the microglial barrier early in the disease to shield neurons from toxic species of Aβ, and perhaps with age, this method of plaque restriction loses its effectiveness. Recently, it was reported that TREM2-deficient (Wang et al., 2016) and –haplodeficient (Yuan et al., 2016) mice display greater plaque diffusion and damage to nearby neuronal structures by modulating the microglial response, indicating that TREM2 signaling is crucial for the compaction of amyloid plaques, as well as limiting their toxicity to nearby neurons. Although these studies may appear to contradict data published by our lab, it is important to note that thorough Aβ plaque analysis in microglia-devoid and -intact animals was performed only in aged 5xfAD mice, whereby the ability of microglia to compact amyloid into dense plaques is presumably lost. Moreover, and perhaps more importantly, microglia-association with plaques and their ability to restrict plaque growth is perturbed throughout the entire lifetime of TREM2-deficient mice. With PLX3397-treatment, plaque-associated microglia are reduced only during treatment with the CSF1R inhibitor; therefore, earlier and longer treatment paradigms may allow for the detection of alterations in plaque compaction with the absence of microglia in AD mice, and may be informative as to how aging modulates microglial function to facilitate AD pathologies.
Collectively, numerous studies implicate microglial reactivity in AD pathogenesis, but the current consensus is that these cells are not key regulators of Aβ/plaque levels in vivo. Thus, further experimentation is needed to determine what effect these cells have in the disease and how GWAS identified myeloid gene changes influence the risk for AD.
5. Impacts of chronically reactive microglia in the AD brain
5.1 Cognitive dysfunction mediated by synaptic and neuronal loss
Modulation of microglial function in AD has been repeatedly shown to improve cognitive function (i.e. (Parachikova et al., 2010; Yamanaka et al., 2012)), indicating that chronically reactive microglia are promoting the cognitive decline that occurs in the disease. In accordance with this, chronically eliminating microglia in 5xfAD mice improved hippocampal dependent-memory, as assessed by contextual fear conditioning (Spangenberg et al., 2016). Notably, synapse degeneration is the best pathological correlate of cognitive decline in AD (Scheff et al., 2006), raising the question of whether microglia continue to prune synapses in adulthood and if this process goes askew in AD, resulting in the pathological stripping of synapses in the brain and, potentially, impairments in cognitive function. We recently reported a 35% increase in number of synapse-bearing dendritic spines in healthy, adult mice devoid of microglia compared to microglia-intact mice (Rice et al., 2015), suggesting that these cells continue to sculpt the synaptic landscape into adulthood. Subsequent examination into the role of microglia in mediating pathological synaptic stripping in 5xfAD mice revealed a significant loss of total dendritic spine density, and in 5xfAD microglia-devoid mice dendritic spine analysis showed a significant increase in total dendritic spine densities, particularly in mushroom and thin spines (Spangenberg et al., 2016). These data indicate that microglia are stripping dendritic spines in the AD brain, and mechanistically, microglial phagocytosis of synaptic material in the AD brain was recently reported as being mediated by the complement system (Hong et al., 2016). As synapse degeneration is the best pathological correlate of cognitive deficits in AD, preventing the loss of dendritic spines or allowing lost spines to regenerate through modulating microglial function may be an important area to pursue for AD therapeutics. Importantly, the 5xfAD mouse model is the only model of AD to date that exhibits profound neuronal loss, specifically in the subiculum and layer V cortex (Eimer and Vassar, 2013), permitting inquiries into the role of chronically activated microglia in promoting the loss of neurons. We reported a 25% loss of subiculum neurons in these mice, and the neuronal loss was entirely prevented with the elimination of microglia (Spangenberg et al., 2016). Moreover, studies investigating the neurotoxic roles of microglia have revealed that knockout of CX3CR1, which is myeloid/microglia-expressed, prevents neuronal loss in 3xTg-AD mice (Fuhrmann et al., 2010), again suggesting that these cells are critically involved in the loss of neurons that occurs in AD. CX3CR1-deficient mice did not show any differences in soluble or insoluble Aβ levels, indicating that this signaling pathway is either not affected or not involved in the phagocytosis of Aβ, while also uncoupling Aβ/plaque pathology from neuronal loss. Collectively, these studies indicate that chronically activated microglia in the AD brain are major contributors to pathological synaptic stripping and neuronal loss, both of which likely underlie deficits in cognitive function.
5.2 Intervention in AD with CSF1R inhibitors that modulate microglial function
As prolonged treatment with high-doses of CSF1R inhibitors may not be a viable option for the treatment of AD, we have conducted studies to explore the effects of lower, clinically-relevant doses of CSF1R inhibitors on AD pathologies (Dagher et al., 2015). Importantly, lower doses modulate microglial function without eliminating all microglia from the brain. We identified a dose of PLX5622 that had minimal effects on microglial numbers in the brain (up to a 30% sustained overall reduction). This dose in 3xTg-AD mice, an AD transgenic model, administered just prior to the initial plaque forming period for 6 or 12 weeks, improved hippocampal dependent memory. Investigations into possible mechanisms underlying this improvement in cognition revealed no changes in amyloid pathology, but a complete prevention of the association of myeloid cells/microglia with plaques. These findings have been confirmed with a different CSF1R inhibitor, GW2580, in APP/PS1 mice, which reduced the number of plaque-associated microglia, normalized behavioral impairments, and recovered dendritic spine density (Olmos-Alonso et al., 2016). Collectively, modulating CSF1R signaling appears to affect both chemotaxis and proliferation, thus preventing the myeloid cells/microglia from migrating to and reacting to the plaques. Again, preventing microglia from associating with plaques did not affect the number of plaques or Aβ levels, supporting the argument that these cells do not clear plaque derived Aβ from the brain. Curiously, CSF1R inhibition results in a phenotype that recapitulates that seen in TREM2-deficient mice – a lack of plaque-associated myeloid cells (Jay et al., 2015; Wang et al., 2015; Wang et al., 2016), further suggesting a relationship between CSF1R and TREM2.
6. Contribution of peripheral myeloid cells in Aβ clearance
In certain murine models, such as repeated social defeat and irradiation conditioning in AD, peripheral myeloid cells are capable of crossing the BBB to mediate differential effects in the brain (Mildner et al., 2007; Wohleb et al., 2013). It is theorized that peripherally-derived myeloid cells possess a greater capacity to phagocytose amyloid than endogenous microglia, as reducing their association with plaques (Jay et al., 2015) or restricting the infiltration of myeloid cells into the brain (Mildner et al., 2011) promotes AD-like pathology, although the latter found reductions in cerebrovascular Aβ load while parenchymal Aβ levels remained unchanged. In line with this, increasing the recruitment of myeloid cells in the AD brain, through the specific overexpression of TGFβ in peripheral populations, enhances the removal of Aβ deposits (Town et al., 2008). Therefore, understanding the roles of peripheral myeloid cells in Aβ clearance is crucial, as these cells may provide some therapeutic benefit to AD. To that end, researchers depleted microglia from APPPS1/TK mice and allowed for repopulation to occur following the cessation of ganciclovir treatment. Analysis of CNS cells evidenced a substantial portion of these cells to be peripherally-derived myeloid cells, thereby allowing researchers to address the role of these cells in Aβ clearance. Assessment of the monocyte-repopulated brain revealed that the number of Aβ deposits was unchanged (Prokop et al., 2015). In an analogous study published at the same time, a different research group used the same APPPS1/TK mice, as well as APP23/TK mice, to repopulate the brain with peripheral myeloid cells and also found that amyloid burden in the brain was unchanged in both APP transgenic models (Varvel et al., 2015). In contrast to the previous study, repopulation was allowed to occur for up to six months in these mice, and even long-term exposure of amyloid to peripheral myeloid cells did not impact amyloid dynamics. Together, these studies indicate that the origin of myeloid cells does not necessarily govern their functionality in AD, and rather, the brain environment dictates the behavior of myeloid cells in the AD brain, as the repopulated peripheral myeloid cells inherited a similar phenotype to that of endogenous microglia. As it is generally believed that BBB breakdown in AD patients and animal models accelerates disease onset, and may also provide a route for potential therapies into the brain, researchers sought to assess BBB integrity in patients with AD and mouse models of AD. Analysis of BBB antibody permeability showed no differences between transgenic (e.g., hTau P301L, ApoEKO and ApoEKI, PS2-APP, etc.) and wildtype mice (Bien-Ly et al., 2015), and in humans, no differences in brain farct percentage or volume were observed between AD patients and controls. This indicates that in AD, BBB disruption is not overtly observed, diminishing the likelihood of infiltration of peripheral myeloid cells in AD. Altogether, these data suggest that peripheral myeloid populations are not largely contributing to the maintenance of amyloid pathology, even in circumstances in which these cells can cross the BBB.
7. Microglial maintenance of tau pathology
In the healthy brain, tau proteins are abundant in the CNS and function to stabilize microtubules in axons. However, in a disease state, such as AD, the binding of tau proteins to microtubules is interrupted, leading to high levels of free tau which is ultimately converted to aggregated and fibrillized tau (Kuret et al., 2005). Microglial activation is implicated as a driver of this pathological change, as it was shown to precede the accumulation of neurofibrillary tangles in a tauopoathy mouse model (PS301S) (Yoshiyama et al., 2007). In a study investigating the relationship between microglia-induced inflammation and neurofilbrillary tangles, researchers found that a deficiency in CX3CR1 in a humanized tau (hTau) mouse model altered microglial activation and increased levels of phosphorylated tau protein, resulting in behavioral impairments (Bhaskar et al., 2010). Importantly, this provides clear evidence that microglial activation plays a direct role in accelerating tau pathology. Further studies in CX3CR1-deficient hTau mice found that microglial activation precedes the spreading of tau pathology and is correlated with the propagation of tau protein from the CA1 to subiculum (Maphis et al., 2015). The transfer of microglia from CX3CR1-deficient hTau mice into non-transgenic recipient mice found that reactive microglia are sufficient to induce tau hyperphosphorylation (Maphis et al., 2015), providing perhaps the most compelling evidence for microglial activation in driving tau pathology. Interestingly, tau protein has been identified in exosomes from CSF samples of AD patients (Saman et al., 2012) and is elevated in AD (Fiandaca et al., 2015), suggesting that secretion of phosphorylated tau via exosomes may play a role in tau-associated neurodegeneration. As microglia have secretory properties, the focus shifted towards these cells in mediating the spreading of tau in the brain. Moreover, in human brains, the nonsynpatic spread of tau pathology is often observed, and the mechanism by which this happened remained elusive. In an AAV-based, rapid tau propagation model, it was demonstrated that the elimination of microglia, using the CSF1R inhibitor PLX3397 as described earlier, halted the propagation of tau. Furthermore, inhibiting exosome synthesis in primary microglia revealed a reduction in transmission of tau from microglia to neurons in culture (Asai et al., 2015), indicating a necessity for microglia in the propagation of tau protein. However, the method of microglial phagocytosis of tau from neurons remains undetermined, although the authors propose that it may occur a phagoptosis-related mechanism. Altogether, these studies point to the activation of microglia as a driver of tau deposition and implicate these cells in spreading pathological tau protein in the brain. Therefore, microglia-targeted therapies may prove beneficial to suppress tau pathologies in AD, and other tau-related neurodegenerative disorders.
8. Anti-inflammatories to alleviate AD
Increasing evidence indicates that inflammation is a major element in the promotion of progressive CNS damage. Approaches to mitigate microglia-mediated neuroinflammation in neurodegenerative disease have long been developed, with PPARy agonists, statins, flavonoids, COX2 inhibitors, minocycline, and glatiramer acetate showing positive effects on mouse models of AD or patients enrolled in clinical trials (Zipp and Aktas, 2006). Delivery of an IL-1 receptor antagonist via injection of neural precursor cells in AD mice rescued spatial and contextual learning impairments and restored neurogenesis deficits, suggesting that blocking pro-inflammatory signaling may ameliorate cognitive outcomes in AD (Ben-Menachem-Zidon et al., 2014). Additionally, the administration of bexarotene, an RXR agonist which exerts anti-inflammatory effects, to APP/PS1 mice revealed stark reductions in Aβ levels and plaque burden, as well as restoration of learning and memory function, as assessed by contextual fear conditioning (Cramer et al., 2012). This compound proceeded to clinical trials, in which four weeks of treatment of bexarotene did not reduce brain amyloid in AD patients, nor did it improve cognitive function (Cummings et al., 2016). However, in AD patients with the ApoE4 allele, bexarotene treatment slightly lowered amyloid levels, hinting that the drug may help clear amyloid in an APOE isoform-dependent manner. Moreover, findings from epidemiological studies indicate that chronic intake of non-steroidal anti-inflammatory drugs (NSAIDs) is associated with a reduced incidence of AD (Zandi et al., 2002), and additionally, the administration of certain NSAIDs to AD mouse models reduced Aβ plaque load (Prokop et al., 2013). Although treatment with NSAIDs has failed to slow cognitive decline in patients with mild to moderate AD (de Jong et al., 2008; Green et al., 2009) and has failed to prevent AD in prospective clinical trials of aged subjects (Group et al., 2007), there may still be value in pursuing this as a preventative AD therapeutic if intervention occurs well before disease onset.
9. Acute inflammation mediated by microglia
Although it is accepted that chronic neuroinflammation is detrimental to the brain, acute inflammation may serve an important function for tissue repair and angiogenesis (Varin and Gordon, 2009). Therefore, the timing of microglial modulation is critical, as these cells can have both harmful and beneficial effects in the brain during inflammatory events. In a genetic hippocampal lesion model, we sought to determine the effects of eliminating microglia during different phases of a neuronal lesion. Importantly, this model has relevance to TBI, stroke, hippocampal sclerosis, and AD, as these diseases feature extensive neuronal loss and chronic microglial reactivity. Notably, the elimination of microglia for 4 weeks immediately following the lesion promoted functional recovery, whilst the elimination of microglia during and following the lesion promoted further neuronal loss, suggesting that microglia are beneficial during the acute phase of an injury (Rice et al., 2015). Importantly, these findings highlight the diverse roles of microglia in the brain during different phases of an insult. In line with this, microglia have been reported to serve beneficial functions in acute damage to the BBB via a localized laser lesion (Nimmerjahn et al., 2005). Importantly, the severity of the lesion dictated the number of microglia responding to the injury, and the accumulation of microglial processes in the lesion vicinity served to shield the injured area, demonstrating a neuroprotective role of microglia that is dependent on injury severity. Collectively, these studies emphasize the complexity of microglia biology, and that careful consideration needs to be given when developing/employing microglia-targeted interventions to the phase and severity of the injury or disease.
10. Conclusions – Net impact of microglia in AD
Together, much remains to be determined about the role of microglia in AD. In development, these cells serve to shape neuronal connectivity through refinement of extranumerous synapses to establish a functional network, and importantly, microglia continue to sculpt the synaptic landscape throughout adulthood. However, the ways in which their function changes in AD and to what extent requires further experimentation. Whether microglia are exerting neuroprotective or neurodegenerative effects in the AD brain largely depends on the severity/stage of the disease, with some evidence indicating beneficial roles of microglial activation in early AD (Condello et al., 2015; Hamelin et al., 2016). Moreover, whether these cells are phagocytosing amyloid in the human AD brain remains uncertain. Is Aβ phagocytosis a microglial function that goes awry with age or are these cells simply not involved in the clearance of amyloid? If microglia are phagocytosing Aβ, do these cells contribute to the spreading of amyloid throughout the brain in an exosome-related manner, as has been observed with tau protein? Current studies implicate the spreading of amyloid to involve exosome trafficking (Nath et al., 2012), but whether microglia are propagating this spread is unclear. Furthermore, teasing out the effects of peripheral versus endogenous myeloid cells in the regulation of amyloid requires additional study. To date, distinguishing myeloid cell populations (resident versus infiltrating) in the brain is challenging due to shared expression of marker proteins, and so, identification of unique cell signatures will be necessary to determine if and/or which population of myeloid cells are involved in amyloid phagocytosis. Here, we argue that microglia are not involved in the phagocytosis of amyloid in the AD brain, unless provided with sufficient stimulation, and thus, identifying signaling molecules/pathways that revert microglia back into a phagocytic phenotype may prove beneficial in AD.
Several studies assessing cognitive function of AD patients with mini mental status scores found that the number of activated microglia is highly correlated with cognitive decline (Cagnin et al., 2001; Versijpt et al., 2003; Edison et al., 2008). Possibly, microglia become problematic in AD and other neurodegenerative diseases due to excessive proliferation, and therefore, the supranumerous microglia may be contributing to the pathophysiology of the disease. In this way, possible therapies that target the excess number of microglia could prove beneficial for patients with AD. Regardless of what effects these cells are exerting in the AD brain, one point is clear – therapeutic intervention at late stages of AD has little hope of being effective, as lost neurons are incapable of regeneration, and rather, successful therapies likely require intervention during preclinical stages of AD. Collectively, microglia appear to become dysfunctional in AD, whereby these cells exert detrimental effects in the brain, and thus, developing ways to harness and mitigate microglia-mediated effects in the brain may provide the key to creating effective therapies for AD.
Highlights.
Genetic and pharmacological manipulations of CSF1R signaling allow for investigations into microglial function in AD
Chronically activated microglia promote tau pathology, synaptic stripping, neuronal loss, and cognitive decline in AD, but do not regulate Aβ dynamics
Peripheral myeloid cell populations are not largely involved in the maintenance of amyloid pathology
Targeting microglia-mediated effects may highlight important therapeutic avenues for AD
Acknowledgments
This work was generously funded by the National Institutes of Health under awards 1R01NS083801 (NINDS) and P50 AG016573 (NIA) to K.N.G., in addition to the American Federation of Aging Research to K.N.G., the Alzheimer’s Association to K.N.G., and a Glenn Foundation Award to K.N.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors E.E.S and K.N.G have no conflicting financial interests.
References
- Akiyama H, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, Ikezu T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- Ben-Menachem-Zidon O, Ben-Menahem Y, Ben-Hur T, Yirmiya R. Intrahippocampal transplantation of neural precursor cells with transgenic overexpression of IL-1 receptor antagonist rescues memory and neurogenesis impairments in an Alzheimer’s disease model. Neuropsychopharmacology. 2014;39:401–414. doi: 10.1038/npp.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Boswell CA, Jeet S, Beach TG, Hoyte K, Luk W, Shihadeh V, Ulufatu S, Foreman O, Lu Y, DeVoss J, van der Brug M, Watts RJ. Lack of Widespread BBB Disruption in Alzheimer’s Disease Models: Focus on Therapeutic Antibodies. Neuron. 2015;88:289–297. doi: 10.1016/j.neuron.2015.09.036. [DOI] [PubMed] [Google Scholar]
- Blinzinger K, Kreutzberg G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat. 1968;85:145–157. doi: 10.1007/BF00325030. [DOI] [PubMed] [Google Scholar]
- Bochner DN, Sapp R, Adelson JD, Zhang S, Lee H, Djurisic M, Syken J, Dan Y, Shatz CJ. Blocking PirB up-regulates spines and functional synapses to unlock visual cortical plasticity and facilitate recovery from amblyopia. Sci Transl Med. 2014;6 doi: 10.1126/scitranslmed.3010157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Aβ-Induced Inflammatory Processes in Microglia Cells of APP23 Transgenic Mice. Am J Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15:209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N, Klein M, Blank T, Yona S, Wolf Y, Mack M, Pinteaux E, Muller W, Zipp F, Binder H, Bopp T, Prinz M, Jung S, Waisman A. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity. 2015;43:92–106. doi: 10.1016/j.immuni.2015.06.012. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. The Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, Sacino A, Nix S, Janus C, Price ND, Das P, Golde TE. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85:519–533. doi: 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen I, Van Rooijen N, Claassen E. A new method for removal of mononuclear phagocytes from heterogeneous cell populations in vitro, using the liposome-mediated macrophage ‘suicide’ technique. J Immunol Methods. 1990;134:153–161. doi: 10.1016/0022-1759(90)90376-7. [DOI] [PubMed] [Google Scholar]
- Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015;6:6176. doi: 10.1038/ncomms7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CYD, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A, Pontecorvo M, Devous M, Tang A, Bena J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer’s disease. Alzheimers Res Ther. 2016;8:4. doi: 10.1186/s13195-016-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, West BL, Green KN. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12:139. doi: 10.1186/s12974-015-0366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De I, Nikodemova M, Steffen MD, Sokn E, Maklakova VI, Watters JJ, Collier LS. CSF1 overexpression has pleiotropic effects on microglia in vivo. Glia. 2014;62:1955–1967. doi: 10.1002/glia.22717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong D, Jansen R, Hoefnagels W, Jellesma-Eggenkamp M, Verbeek M, Borm G, Kremer B. No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PLoS One. 2008;3:e1475. doi: 10.1371/journal.pone.0001475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Aβ load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Ali SS, Shekhtman G, Roberts AJ, Lucero J, Quick KL, Behrens MM. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS One. 2009;4:e5518. doi: 10.1371/journal.pone.0005518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Eimer WA, Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol Neurodegener. 2013;8 doi: 10.1186/1750-1326-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC. Intracellular Aβ-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol Aging. 2012;33:1329–1342. doi: 10.1016/j.neurobiolaging.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, Abner EL, Petersen RC, Federoff HJ, Miller BL, Goetzl EJ. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015;11:600–607.e601. doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- Fu AK, Hung KW, Yuen MY, Zhou X, Mak DS, Chan IC, Cheung TH, Zhang B, Fu WY, Liew FY, Ip NY. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1604032113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH, TPS Group Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. 2009;302:23. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AR Group. Lyketsos CG, Breitner JC, Green RC, Martin BK, Meinert C, Piantadosi S, Sabbagh M. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. 2007;68:1800–1808. doi: 10.1212/01.wnl.0000260269.93245.d2. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P, Dostert C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS One. 2015;10:e0130624. doi: 10.1371/journal.pone.0130624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelin L, Lagarde J, Dorothee G, Leroy C, Labit M, Comley RA, de Souza LC, Corne H, Dauphinot L, Bertoux M, Dubois B, Gervais P, Colliot O, Potier MC, Bottlaender M, Sarazin M, It Clinical Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain. 2016;139:1252–1264. doi: 10.1093/brain/aww017. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. 2013;80:1778–1783. doi: 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert SE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Hellwig S, Masuch A, Nestel S, Katzmarski N, Meyer-Luehmann M, Biber K. Forebrain microglia from wild-type but not adult 5xFAD mice prevent amyloid-beta plaque formation in organotypic hippocampal slice cultures. Sci Rep. 2015;5:14624. doi: 10.1038/srep14624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation. 2005;2:22. doi: 10.1186/1742-2094-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoarau JJ, Krejbich-Trotot P, Jaffar-Bandjee MC, Das T, Thon-Hon GV, Kumar S, Neal JW, Gasque P. Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg) CNS Neurol Disord Drug Targets. 2011;10:25–43. doi: 10.2174/187152711794488601. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016 doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, Borchelt DR. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212:287–295. doi: 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson JU, Woodling NS, Wang Q, Panchal M, Liang X, Trueba-Saiz A, Brown HD, Mhatre SD, Loui T, Andreasson KI. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J Clin Invest. 2015;125:350–364. doi: 10.1172/JCI77487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan MJ, Lee JE, Wilson JG, Everhart AL, Brown CM, Hoofnagle AN, Jansen M, Vitek MP, Gunn MD, Colton CA. Arginine deprivation and immune suppression in a mouse model of Alzheimer’s disease. J Neurosci. 2015;35:5969–5982. doi: 10.1523/JNEUROSCI.4668-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D, Patzko A, Schreiber D, van Hauwermeiren A, Baier M, Groh J, West BL, Martini R. Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot-Marie-Tooth disease in mice. Brain. 2015;138:3193–3205. doi: 10.1093/brain/awv240. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, Manalastas A, Hilfiker M, Pfister S, Schwerdel C, Riether C, Meyer U, Knuesel I. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. 2012;2:151. doi: 10.1186/1742-2094-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuret J, Congdon EE, Li G, Yin H, Yu X, Zhong Q. Evaluating triggers and enhancers of tau fibrillization. Microsc Res Tech. 2005;67:141–155. doi: 10.1002/jemt.20187. [DOI] [PubMed] [Google Scholar]
- Lambert JC, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lambert JC, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinenga G, Götz J. Scanning ultrasound removes amyloid-β and restores memory in an Alzheimer’s disease mouse model. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aaa2512. [DOI] [PubMed] [Google Scholar]
- Li R, Shen Y, Yang LB, Lue LF, Finch C, Rogers J. Estrogen enhances uptake of amyloid beta-protein by microglia derived from the human cortex. J Neurochem. 2000;75:1447–1454. doi: 10.1046/j.1471-4159.2000.0751447.x. [DOI] [PubMed] [Google Scholar]
- Lopes KO, Sparks DL, Streit WJ. Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia. 2008;56:1048–1060. doi: 10.1002/glia.20678. [DOI] [PubMed] [Google Scholar]
- Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, LaDu MJ, Fardo DW, Rebeck GW, Estus S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol Neurodegener. 2015;10:52. doi: 10.1186/s13024-015-0048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138:1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melah KE, Lu SY, Hoscheidt SM, Alexander AL, Adluru N, Destiche DJ, Carlsson CM, Zetterberg H, Blennow K, Okonkwo OC, Gleason CE, Dowling NM, Bratzke LC, Rowley HA, Sager MA, Asthana S, Johnson SC, Bendlin BB. Cerebrospinal Fluid Markers of Alzheimer’s Disease Pathology and Microglial Activation are Associated with Altered White Matter Microstructure in Asymptomatic Adults at Risk for Alzheimer’s Disease. J Alzheimers Dis. 2016;50:873–886. doi: 10.3233/JAD-150897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, Quinn M, Brück W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Morris JC, Price JL. Pathological Correlates of Nondemented Aging, Mild Cognitive Impairment, and Early-Stage Alzheimer’s Disease. J Mol Neurosci. 2001;17:101–118. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- Naj AC, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath S, Agholme L, Kurudenkandy FR, Granseth B, Marcusson J, Hallbeck M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J Neurosci. 2012;32:8767–8777. doi: 10.1523/JNEUROSCI.0615-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, F H. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas-Caballero M, Holscher C, Perry VH, Gomez-Nicola D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain. 2016;139:891–907. doi: 10.1093/brain/awv379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP, Chen XC. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: implications for Alzheimer’s disease. Mol Neurodegener. 2011;6 doi: 10.1186/1750-1326-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Parachikova A, Vasilevko V, Cribbs DH, LaFerla FM, Green KN. Reductions in amyloid-beta-derived neuroinflammation, with minocycline, restore cognition but do not significantly affect tau hyperphosphorylation. J Alzheimers Dis. 2010;21:527–542. doi: 10.3233/JAD-2010-100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Chung H, Maxfield FR. Slow Degradation of Aggregates of the Alzheimer’s Disease Amyloid b-Protein by Microglial Cells. J Biol Chem. 1997;272:29390–29397. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polazzi E, Monti B. Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol. 2010;92:293–315. doi: 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer’s disease. Acta Neuropathologica. 2013;126:461–477. doi: 10.1007/s00401-013-1182-x. [DOI] [PubMed] [Google Scholar]
- Prokop S, Miller KR, Drost N, Handrick S, Mathur V, Luo J, Wegner A, Wyss-Coray T, Heppner FL. Impact of peripheral myeloid cells on amyloid-beta pathology in Alzheimer’s disease-like mice. J Exp Med. 2015;212:1811–1818. doi: 10.1084/jem.20150479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RP, Hernandez MX, Tenner AJ, West BL, Green KN. Elimination of Microglia Improves Functional Outcomes Following Extensive Neuronal Loss in the Hippocampus. J Neurosci. 2015;35:9977–9989. doi: 10.1523/JNEUROSCI.0336-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose SE, Chen F, Chalk JB, Zelaya FO, Strugnell WE, Benson M, Semple J, Doddrell DM. Loss of connectivity in Alzheimer’s disease: an evaluation of white matter tract integrity with colour coded MR difusion tensor imaging. J Neurol Neurosurg Psychiatry. 2000;69:528–530. doi: 10.1136/jnnp.69.4.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakae N, et al. ABCA7 Deficiency Accelerates Amyloid-beta Generation and Alzheimer’s Neuronal Pathology. J Neurosci. 2016;36:3848–3859. doi: 10.1523/JNEUROSCI.3757-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Abe-Dohmae S, Yokoyama S, St George-Hyslop P, Fraser PE. ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J Biol Chem. 2015;290:24152–24165. doi: 10.1074/jbc.M115.655076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Schreiner B, Romanelli E, Liberski P, Ingold-Heppner B, Sobottka-Brillout B, Hartwig T, Chandrasekar V, Johannssen H, Zeilhofer HU, Aguzzi A, Heppner F, Kerschensteiner M, Becher B. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell Rep. 2015;12:1377–1384. doi: 10.1016/j.celrep.2015.07.051. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117:1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe GO, Lowery RL, Tremblay ME, Kelly EA, Lamantia CE, Majewska AK. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun. 2016;7:10905. doi: 10.1038/ncomms10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain. 2016;139:1265–1281. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–434. doi: 10.1016/s0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syken J, Grandpre T, Kanold PO, Shatz CJ. PirB Restricts Ocular-Dominance Plasticity in Visual Cortex. Science. 2006;313:1795–1800. doi: 10.1126/science.1128232. [DOI] [PubMed] [Google Scholar]
- Terrando N, Monaco C, Ma D, Foxwell BM, Feldmann M, Maze M. Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci U S A. 2010;107:20518–20522. doi: 10.1073/pnas.1014557107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD. The cytoskeleton in Alzheimer disease. J Neural Trasn Suppl. 1998;53:141–145. doi: 10.1007/978-3-7091-6467-9_12. [DOI] [PubMed] [Google Scholar]
- Torres L, Danver J, Ji K, Miyauchi JT, Chen D, Anderson ME, West BL, Robinson JK, Tsirka SE. Dynamic microglial modulation of spatial learning and social behavior. Brain Behav Immun. 2015 doi: 10.1016/j.bbi.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol. 2012;12:492–502. doi: 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014;9:2124–2138. doi: 10.1016/j.celrep.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009;214:630–641. doi: 10.1016/j.imbio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, Degenhardt K, Resch C, Bosch A, Jucker M, Neher JJ. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-beta deposition in mouse models of Alzheimer’s disease. J Exp Med. 2015;212:1803–1809. doi: 10.1084/jem.20150478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versijpt JJ, Dumont F, Van Laere KJ, Decoo D, Santens P, Audenaert K, Achten E, Slegers G, Dierckx RA, Korf J. Assessment of Neuroinflammation and Microglial Activation in Alzheimer’s Disease with Radiolabelled PK11195 and Single Photon Emission Computed Tomography. European Neurology. 2003;50:39–47. doi: 10.1159/000070857. [DOI] [PubMed] [Google Scholar]
- Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213:667–675. doi: 10.1084/jem.20151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Wiśniewski HM, Dziewiatkowski J, Tarnawski M, Kozielski R, Trenkner E, Wiktor-Jedrzejczak W. Reduced number and altered morphology of microglial cells in colony stimulating factor-1-deficient osteopetrotic op/op mice. Brain Res. 1998;804:135–139. doi: 10.1016/s0006-8993(98)00618-0. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Wang K, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APPSW mice. J Neurosci. 2001;31:11159–11171. doi: 10.1016/s0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013;33:13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, Clark IA, Abdipranoto A, Vissel B. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One. 2013;8:e59586. doi: 10.1371/journal.pone.0059586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuno F, Kosaka J, Ota M, Higuchi M, Ito H, Fujimura Y, Nozaki S, Takahashi S, Mizukami K, Asada T, Suhara T. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [(1)(1)C]DAA1106. Psychiatry Res. 2012;203:67–74. doi: 10.1016/j.pscychresns.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016;90:724–739. doi: 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC, CCS Investigators Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59:880–886. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17:400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- Zipp F, Aktas O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 2006;29:518–527. doi: 10.1016/j.tins.2006.07.006. [DOI] [PubMed] [Google Scholar]