

Abstract
The Aβ1-42 peptide that is overproduced in Alzheimer's disease (AD) from a large precursor protein has a normal amino acid sequence but, when liberated, misfolds at neutral pH to form “protofibrils” and fibrils that are rich in β-sheets. We find that these protofibrils or fibrils are toxic to certain neuronal cells that carry Ca-permeant α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. Disrupting the structure of the Aβ1-42 fibrils and protofibrils might lead to the discovery of molecules that would be very useful in the treatment of AD. A high-throughput screen of a library of >3,000 small molecules with known “biological activity” was set up to find compounds that efficiently decrease the β-sheet content of aggregating Aβ1-42. Lead compounds were characterized by using thioflavin T (ThT) as a β-sheet assay. The most effective of six compounds found was 4,5-dianilinophthalimide (DAPH) under the following conditions: DAPH at low micromolar concentrations abolishes or greatly reduces previously existing fully formed Aβ1-42 fibrils, producing instead amorphous materials without fibrils but apparently containing some protofibrils and smaller forms. Coincubation of the Aβ1-42 peptide with DAPH produces either amorphous materials or empty fields. Coincubation of DAPH and Aβ1-42 greatly reduces the β-sheet content, as measured with ThT fluorescence, and produces a novel fluorescent complex with ThT. When the Aβ1-42 peptide was coincubated with DAPH at very low micromolar concentrations, the neuronal toxicity mentioned above (Ca2+ influx) was eliminated. Clearly, DAPH is a promising candidate for AD therapy.
Keywords: Ca ions, amyloid peptide, aggregation, toxicity, aggregation
The pathology of Alzheimer's disease (AD) was first defined by Alois Alzheimer in 1907. He saw “senile plaques” and also “tangles” in stained sections from the postmortem brain of his first diagnosed patient. It turned out (1, 2) that the plaques were composed of fibrils formed by the aggregation of short peptides, which were mostly 40 and 42 aa long. These peptides had a perfectly normal amino acid sequence (common to both peptides) and represented a part of a much larger common transmembrane “amyloid precursor protein” (APP) of unknown function. For various reasons, some of which are genetic and some of which are unknown, these peptides are greatly over-produced in AD. The two overproduced peptides, Aβ1-40 and Aβ1-42, when liberated from their parent APP, refold (“misfold”) and aggregate spontaneously to a neurotoxic, β-sheet-containing fibrillar form. This structure attacks particular neurons in the CNS, causing them to become dysfunctional and, eventually, die in large numbers. This neurotoxicity (for Aβ1-40) was first described by Yankner et al. (3). Shortly afterward, Lorenzo and Yankner (4) described an organic compound, Congo red, that inhibits Aβ fibril formation and neurotoxicity. The involved peptides, Aβ1-40 and Aβ1-42, will hereafter be referred to as Aβ40 and Aβ42, respectively.
The plaques are extracellular. One result of the consequent dysfunction is thought to be the hyperphosphorylation of the microtubule-associated tau protein (5), which then aggregates to form the tangles that fill the cell body and neurites, making them dysfunctional.
A currently popular view is that the misfolded (6-8) Aβ peptides produce a novel conformation with new and toxic properties. The notion that the cytotoxicity of Aβ peptides involves the disturbance of cytosolic Ca2+ ion homeostasis has been stated (9, 10). It is thought that the misfolded Aβ peptide, especially Aβ42, is an oligomer of that peptide (11-13). Our proposed mechanism (6) (Fig. 1) involves a deleterious influx of external Ca2+ ions. The consequences are severe: dysfunction and cell death. Our molecular mechanism for immediate Ca2+ influx involves the activation of Ca-permeant α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in the neurons that have these particular receptors (i.e., it is not due to release of Ca2+ ions from internal storage). This influx is inhibited completely by 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline (NBQX), a specific AMPA-receptor antagonist (see Results). Earlier experiments with 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), another AMPA antagonist, confirm this observation. These observations prove the direct and crucial involvement of Ca2+-permeant AMPA receptors in this mechanism. This mechanism requires the β-sheet-containing fibrils and protofibrils formed by Aβ42; it is the molecular basis of our proposed AD therapy.
Fig. 1.

Schematic representation of neurotoxicity. Misfolding of peptide Aβ42 produces a neurotoxic molecule.
In our experience, Aβ42 must be preincubated for at least 24 h before the preparation will promote Ca2+ ion influx, as described in Results. By this time, the early-stage oligomers, also called “donuts,” have turned into “protofibrils” and fibrils. This transition into protofibrils and fibrils is expected to occur in vivo as in vitro. Donuts, the early oligomeric forms, do not contain β-sheet structures, as do protofibrils and fibrils (13). Such a scheme (Fig. 1) could explain the neurotoxicity (deleterious Ca2+ ion influx) as well as the singular distribution of cell loss. Only certain neurons have the particular receptors that are sensitive to Aβ42 fibrils and that conduct Ca2+ ions. It is known that the distribution of, for example, Ca-permeant AMPA-receptors is quite restricted. Our proposed therapy is based on this model.
A high-throughput screen of a library of >3,000 small molecules with known “biological activity” was set up to find compounds that efficiently decrease the β-sheet content of aggregating Aβ42. Lead compounds were characterized by using thioflavin T (ThT) as a β-sheet assay. The most effective of six compounds that were found was 4,5-dianilinophthalimide (DAPH). Here, we describe the properties of DAPH that eliminate the toxic properties of Aβ42 and make it a very promising candidate for the treatment of AD.
Materials and Methods
Peptide and Compound Preparation. Stock solutions of Aβ42 (special TFA preparation, catalog no. 03-112, BioSource International, Camarillo, CA) were prepared in autoclaved water with the addition of 1 M NaOH to pH 10-11.
Aβ42Total is unfractionated synthetic Aβ42 that was dissolved in water at pH 8-9 by using NH3 or at pH 10-11 by using dilute NaOH and stored at -40°C.
Aβ4230K is seedless Aβ42 made according to the protocols of Fezoui et al. (15), except that we needed to filter the dissolved peptide through a 30,000-kDa spin filter instead of the 10,000-kDa filter that they used. However, they had described only the procedure for the shorter peptide Aβ40. The method, as used by us, involves dissolving the total Aβ42 peptide in water at pH 10.5 by using NaOH. There was considerable loss of peptide; the concentration of peptide in the filtrate was determined by the intrinsic fluorescence of the single tyrosine residue [Ex = 280 nm; Em = 310 nm, where Ex, and Em are the excitation and emission wavelengths, respectively, by using tyrosinamide as the standard.
Experimental samples were prepared by diluting the stock solution of Aβ42 to 10 μM (unless noted otherwise) in Tyrode's solution 2 mM Ca (150 mM NaCl/3mMKCl/10 mM Hepes, pH 7.4/2 mM CaCl2/10 mM d-glucose, pH 7.4/0.02% Na azide). DAPH (D210; Sigma) was prepared in DMSO.
Fura-2 Fluorescence. To measure Ca, the ratiometric Ca dye fura-2 was loaded into a mouse neuronal cell line, CATH.a (CRL-11179; American Type Culture Collection) and plated on acid-washed poly-d-lysine-coated glass coverslips. Cytosolic Ca concentrations were measured as described (8) in Tyrode's solution/2 mM Ca. Rapidly alternating measurements of fura-2 (excited at 340 and 380 nm) were performed by measuring emission at 516 nm using the special Fura2-Fluo3 filter set 7400 (Chroma Technology, Brattleboro, VT), an Axiovert 100 inverted microscope (Zeiss), and a photometry system (DeltaRam model, Photon Technology International, Lawrenceville, NJ).
In our experience, the variance of peak heights in these cell cultures is always large, which we believe is because of the variation from cell to cell of the density of receptors on the surface of the cell.
ThT Experiments. To measure β-sheet formation, ThT was added to samples and fluorescent measurements were read as described. Each sample was set up in a 1.5-ml Eppendorf tube and incubated in a 37°C water bath. If stirring of the sample during incubation was required, it was set up in a 2.0-ml screw-cap vial (Corning) with a magnetic stir bar and placed on a stirrer in the 37°C room. In “coincubated” samples, the Aβ42 was added first, then DAPH was added, and the sample was incubated for the designated amount of time (usually 48 h). If it was an “added after aggregation” sample, DAPH was added after the Aβ42 was incubated. The ThT dye was always added last and after incubation. For measurement, each sample was split into four wells (100 μl per well) of a 96-well black-bottom plate (catalog no. 35-3943, VWR International, Westchester, PA). ThT fluorescence was measured at room temperature in a Fluoroskan II at Em = 444 nm and Ex = 510 nm or a FLUOstar Optima plate-reader (BMG Lab Technologies, Durham, NC) at Ex = 440 nm and Em = 480 nm. The ThT fluorescence spectrum was measured in an f4500 spectrofluorimeter (Hitachi, Tokyo) at Ex = 435 nm and Em = 450-550 nm.
DiBAC4 (3) Fluorescence. To measure membrane potential of cultured neuronal cells in the original high-throughput screen, the voltage-sensitive dye DiBAC4 (3) was added to solutions containing Aβ42Total in 96-well plates and measured in a Fluoroskan II at Em = 485 nm and Ex = 510 nm or FLUOstar Optima plate-reader (BMG Lab Technologies) at Em = 480 nm and Ex = 505 nm. It turned out later that compounds that lowered the DiBAC4 (3) fluorescence did so by reducing the fluorescent signal for β-sheet content of aggregated Aβ42Total; the cells were not involved.
Electron Microscopy (EM). Negative-staining EM was used to visualize the kinetics and morphology of Aβ42 fibrillization over time, with and without the presence of inhibitors. Samples of 10 μM seedless Aβ42 (Aβ4230k) were incubated for the indicated amount of time at 37°C and vigorously vortex mixed immediately before and after incubation. Very small volumes, typically 5 μl, of each sample were absorbed for 2-4 min onto glow-discharged, carbon-coated, Formvar-filmed 400 mesh copper grids and gently wicked away with filter paper. A total of 5 μl of freshly filtered 2% uranyl acetate staining solution was then absorbed for 2 min onto the grid and gently wicked off. Grids were allowed to dry in a light-protected environment overnight before being viewed in a 1200 EXII EM (JEOL) operated at 80 kV. Images were captured on EM film, and positives were printed.
EM Image Analysis. Images were scanned for fiber measurement in NIH IMAGE 1.62. Fiber measurements were calibrated by comparison with the pixel lengths of T4 phage tails, which are known to be 100 nm long (16). Measurements were analyzed, and width-distribution histograms were produced by using EXCEL 98 (Microsoft).
Results
Aggregation Kinetics of the Aβ42 Peptide. The longer of the two AD peptides, Aβ42, is thought to be involved more directly in the neurotoxicity underlying the disease. Certainly, the Aβ42 peptide aggregates more readily to form fibrils than Aβ40 (17). However, it is not clear whether that property correlates with the ability of the peptide to form toxic oligomers. Our experiments used two different preparations of the synthetic AD Aβ42 peptide, which we called Aβ42Total and Aβ4230k (see Materials and Methods). The synthetic Aβ42 was dissolved in water at a pH of ≈8 and then stored at -20°C until diluted into Tyrode's solution/2 mM Ca (pH 7.4). We refer to this preparation as Aβ42Total. For our experiments, it was preincubated for 24 or 48 h at 37°C without stirring. The seedless Aβ4230K peptide, prepared as described in Materials and Methods, was also pre-incubated before experiments at pH 7.4.
Fig. 2 A and B shows the time course of aggregation of Aβ4230k (10 μM in Tyrode's solution/2 mM Ca, pH 7.4) by using negative-staining EM. Under these conditions, several stages can be distinguished. At 0 h, many very small objects are seen, but characteristically, we also see many small circular structures of ≈5 nm in diameter (so-called donuts). They are made up of five globular subunits, as reported in ref. 8. At 6 h, we see only flexible fibrous structures, consisting of globular structures similar to those seen at 0 h (“beads-on-a-string”). These beads-on-a-string are referred to as protofibrils. Next, at 16 h, these globular units form the beginning of a long smooth fibril, still somewhat flexible. They appear to be growing in one direction, as judged by the globular subunits seen at one end of nascent fibrils. Finally, at 24 and 48 h, the fibrils are smooth, very long, and straight, but intertwined to form “nests.” These images emphasize the role of small globular subunits forming donuts at 0 h and forming protofibrils at slightly later times. During this time, there is very little β-sheet structure detectable by ThT; it is the lag phase if one follows β-sheet content (18). However, the description does not allow us to deduce the possible structure of the neurotoxic oligomers.
Fig. 2.

Time course of aggregation of Aβ4230k at 10 μM in Tyrode's solution/2 mM Ca (pH 7.4) incubated at 37°C for the indicated times; negative-staining EM is shown. (A) t = 0 h. Arrow indicates one of the many donuts shown, which is enlarged in E. A larger and rarer aggregate is also shown. (B) t = 6 h. The “beads” are roughly the same size as the beads that make up the donuts in A and E. (C) t = 16 h. (D) t = 48 h. (E) t = 0 h. A portion of A (arrow) is enlarged to show a donut. The image is black/white-inverted for clarity.
A High-Throughput Screen Yielded Compounds, for Example, DAPH, That Destabilize β-Amyloid Fibrils (see Fig. 10). A library of 3,780 compounds with known biological activity was kindly provided by Brent Stockwell (Columbia University, New York), as well as robotic equipment. We were interested in the possible effect of aggregated Aβ42 on the membrane potential of neuronal cells because we knew that aggregated Aβ42 causes large Ca2+ influx into certain neuronal cells. We used PC12 cells and exposed them to aggregated Aβ42Total (10 μM, 48 h at 37°C, pH 7.4) in the presence of the slow polarization-sensitive dye DiBAC4 (3), which fluoresces† strongly when membranes are depolarized. It is known that this (anionic) dye can enter depolarized cell membranes and forms reversible fluorescent complexes with intracellular proteins. The PC12 cells, when exposed to preincubated Aβ42Total, fluoresced strongly at 530 nm (Ex = 485 nm). We looked for, and occasionally found, a strong decrease in fluorescence in the presence of certain compounds. The compound DAPH gave the strongest decrease, but we found five other compounds with robust decreases (data not shown). Further experiments showed that the observed fluorescence had nothing to do with the cells but was caused by the formation of a fluorescent complex between the aggregated peptide and the dye (21). We found that the fluorescent behavior of DiBAC4 (3) and its decrease by certain compounds was seen also when ThT was used. ThT is a well known reagent used to measure β-sheet conformation in peptides and proteins; apparently, DiBAC4 (3) behaves similarly. We concluded that we had found several small molecules, including DAPH, that effectively interfered with the prominent β-sheet conformation of Aβ42 protofibrils and fibrils.
Fig. 10.

DAPH eliminates Ca influx of Aβ42Total in CATH.a cells. (A) As described in the legend to Fig. 8, solutions were sequentially exchanged with 10 μM aggregated Aβ42Total, then Aβ42Total plus DAPH (coincubated at 10 μM plus 10 μM), and then Aβ42Total. Because DAPH is soluble only in DMSO, DMSO was present in all solutions. This experiment was repeated three times. (B) Effect of DAPH (30-0.25 μM) on Ca influx. The protocol used was as in A, except with DAPH concentrations ranging from 30-0.25 μM. (Variance is high; see Materials and Methods.) The corresponding concentration of DMSO was used for the controls.
DAPH Reverses the Fibrils Formed by Aβ4230k or Aβ42Total. A very striking effect of DAPH is the ability to reverse completely the fibril formation of Aβ4230k (Fig. 3). The peptide was allowed to aggregate under the usual conditions at pH 7.4 for 24 h and form fibrils. The incubation was either continued for another 24 h in the presence of the vehicle (1% DMSO) or in the presence of equimolar DAPH that had been dissolved in DMSO. The usual fibrils are formed in the control and are seen with negative staining in the EM, albeit not as crisply as without DMSO. Clearly, DAPH completely reverses the fibrils, and only amorphous masses are observed that may include the globular protein units seen at 0 h (Fig. 2).
Fig. 3.

DAPH reverses Aβ4230k fibrils. (A) Aβ4230k at 10 μM in Tyrode's solution/2 mM Ca (pH 7.4), incubated at 37°C for 24 h. (B) Incubation continued for another 24 h in the presence of vehicle (1% DMSO). (C) Incubation continued for another 24 h in the presence of 10 μM DAPH in DMSO.
Fibrils of aggregated Aβ42Total give a strong ThT fluorescence signal, indicating high β-sheet content. Reversal of the fibrils, as detected by the ThT assay, takes only very few minutes and depends on the concentration of DAPH (Fig. 4).
Fig. 4.

Time course of reversal of fibril, formed by incubating 10 μM Aβ42Total in Tyrode's solution/2 mM Ca (pH 7.4) at 37° for 24 h. The β-sheet content was measured with ThT (see Materials and Methods). To obtain net values, the fluorescence of ThT in Tyrode's buffer was subtracted. ▪, Aggregated Aβ42Total plus 10 μM DAPH; ○, aggregated Aβ42Total plus 20 μM DAPH. The 0-h time points shown were Aβ42Total with only DMSO (the vehicle).
DAPH Blocks Fibril Formation by Aβ4230k. Coincubation of Aβ4230k with equimolar DAPH (10:10 μM) resulted in almost complete elimination of the usual fibrils, as seen by EM (Fig. 5). The vehicle DMSO (1%), the solvent for the water-insoluble DAPH, was present in the test and control experiments. EM images (Fig. 5) reveal a nearly complete elimination of higher-order structure upon 24 h incubation with DAPH; very rarely, small “protofibrillar” and amorphous structures appear against a backdrop of tiny, 2- to 3-nm objects, which are not observed in control samples without DAPH.
Fig. 5.
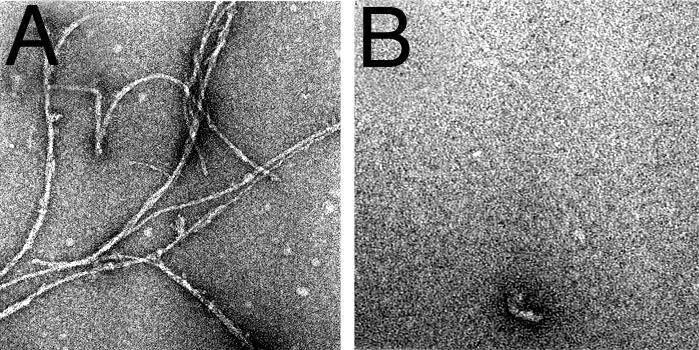
DAPH prevents Aβ4230k fibril formation. Aβ4230k was incubated without (A) and with (B) DAPH at a 10:10 μM dilution in Tyrode's solution/2 mM Ca (pH 7.4) at 37°C for 24 h. Samples were prepared by using negative-staining EM.
DAPH Profoundly Alters the β-Sheet Conformation of Aggregated Aβ42Total. When Aβ42Total (10 μM) is preincubated with equimolar DAPH for 48 h at 37°C pH 7.4, the usual sharp maximum-emission peak of the ThT complex is greatly reduced, indicating a profound structural change in β-sheet conformation (Fig. 6). In addition, a second and new emission peak appears at ≈510-530 nm; we do not know what the structure of the new ThT complex is. Aβ4230k behaves similarly.
Fig. 6.

ThT spectrum of Aβ42Total plus DAPH suggests a new β-sheet conformation. To measure β-sheet content, ThT was added to Aβ42Total plus DAPH and coincubated at a dilution of 10:10 μM for 48 h at 37°C. An emission spectrum at Ex = 435 nm was measured with an f4500 spectrofluorimeter.
The IC50 value for the inhibition of β-sheet content of aggregated Aβ42Total by DAPH is ≈15 μM (Fig. 7) when ThT is the fluorescent reagent. However, we have reported (19) that the dye DiBAC4 (3) also forms a highly fluorescent complex with aggregated (preincubated) Aβ42Total. In fact, this was the dye used by us to conduct the high-throughput screen that led to the characterization of DAPH as an effective inhibitor of β-sheet conformation. We tested the inhibition of β-sheet formation by using DiBAC4† (3, 23), and we found a much lower IC50 value (4.5 μM) than we found with ThT (Fig. 7). Apparently, less DAPH is required to displace Aβ from DiBAC4 (3) than from ThT because Aβ binds less avidly to DiBAC4 (3).
Fig. 7.

The IC50 value for the inhibition by DAPH of Aβ42Total, measured with ThT fluorescence, is 15.2 μM DAPH. Each data point is the mean of four experiments of 10 μM aggregated Aβ42Total plus DMSO, with varying DAPH concentrations. A logistics curve was fitted and then used to obtain the IC50 value. Parallel experiments using DiBAC4 (3) fluorescence are also shown; this IC50 value is ≈4.5 μM DAPH.
Treatment of Neuronal Cells with Preincubated Aβ42 Causes Ca Influx via Ca-Permeant AMPA Receptors. Preincubated Aβ42Total was applied to CATH.a neuronal cells that had been loaded with fura-2 (see Materials and Methods). We measured cytosolic Ca2+ concentration as a function of time and saw an immediate and dramatic influx of Ca (Fig. 8A). This Ca influx decayed spontaneously and slowly, but not to zero. In the presence of CNQX, an AMPA receptor antagonist with some specificity toward NMDA receptors, a peak of Ca influx was prevented. When NBQX was present (Fig. 8B), there was also no new Ca spike, but the level of cytosolic Ca dropped abruptly to a value close to and somewhat below the value found in the Tyrode's solution control. This inhibition by NBQX was at least partially reversible. NBQX is a highly specific antagonist for AMPA receptors. Its effective shut down of Ca influx argues very strongly for the involvement of a Ca-permeant AMPA receptor, activated by preincubated Aβ42, as one important mechanism for the disruption of neuronal Ca homeostasis in AD.
Fig. 8.

AMPA antagonists NBQX and CNQX eliminate Ca influx. CATH.a cells on a coverslip were loaded with the Ca-sensitive fluorescent dye fura-2 and measured at Ex = 340 and 380 nm and Em = 510 nm by using a microscope-photometry system obtained from Photon Technology International. Solutions were exchanged sequentially with 10 μM aggregated Aβ42Total, then Aβ42Total plus 50 μM CNQX (A) or 50 μM NBQX (B), and then Aβ42Total again. The NBQX effect is partially reversible.
Ca Influx Depends on Preincubation and on the Concentration of Aβ42. The Aβ42 peptide must be preincubated at pH 7.4 and 37°C for 24-48 h to modulate the immediate Ca influx into these neuronal cells maximally. This Ca influx is true for both Aβ42Total (Fig. 9A) and Aβ4230k (data not shown). Apparently, unincubated Aβ42Total already contains some Aβ42 oligomer/protofibril that is able to activate the AMPA receptors to a limited extent; further incubation increases the amount of this component or, perhaps, allows another, more active form to appear.
Fig. 9.

Ca influx by aggregated Aβ42Total into CATH.a cells is concentration-dependent. (A) We incubated 10 μM aggregated Aβ42 Total at 37°C for 0, 24, and 48 h. (B) Aβ42Total at 5,10, and 20 μM (n = 1, 15, and 3, respectively) and Aβ4230k at 10 and 20 μM (n = 5 and 4, respectively) were incubated for 48 h at 37°C, then applied to neuronal CATH.a cells, and cytosolic Ca was measured by fura-2 fluorescence (n = 1, 15, and 3, respectively).
The rate of immediate Ca2+ influx (i.e., the peak height in Fig. 8) depends on the Aβ42 concentration, particularly when Aβ42Total is used (Fig. 9B). It must also depend on the density of these AMPA receptors on the neuronal cell surface, perhaps accounting for the large variance observed between cells and, therefore, between dishes. The Aβ42 peptide used in this experiment has been preincubated for 48 h under the usual conditions. A similar dependence on concentration is seen also when Aβ4230k is used, but the actual Ca2+ influx is much less. The reason for this difference is not clear.
Inhibition of Ca Influx by DAPH. The immediate Ca2+ ion influx caused by preincubated Aβ42Total (10 μM) is shown clearly in Fig. 10A. Dramatically, the Ca2+ ion influx is abolished by the presence of DAPH, also at 10 μM, during the preincubation. This effect is at least partly reversible when the cells are next exposed to preincubated Aβ42Total alone. Similar inhibition is seen at 10, 5, 2.5, and 1 μM DAPH, but it is greatly reduced at 0.5 and 0.25 μM (Fig. 10B). The suggested IC50 value for this part of the curve is ≈0.7 μM, which is much less than the IC50 value for the effect of DAPH on the β-sheet content of aggregated Aβ42Total (≈15 μM ThT). This value suggests that the neurotoxic “oligomer” of Aβ42Total is much more sensitive to DAPH than the β-sheet-containing mature fibrils seen in the EM.
At higher DAPH concentrations (20 or 30 μM), the cytosolic Ca2+ concentrations in treated CATH.a cells dips well below the control values seen in buffer alone (see Discussion). This observation is not due to the increased concentrations of DMSO in these experiments, because the net Ca2+ concentrations are recorded in Fig. 10. We have no explanation for this effect.
Discussion
We have reported (7) the development of “decoy peptides” that complex with the aggregated misfolded Aβ42 peptide and, thereby, prevent their toxic interaction with neuronal cells. The decoy peptides remain promising candidates for AD therapy‡ and are ready for testing in animals.
As we pursue an approach to AD therapy that is designed to inhibit the mechanism of neuronal toxicity described above, we are aware that others are pursuing different paths to therapy (see ref. 14 for review). These approaches are very welcome but are described as being only partially effective in correcting cognitive deficits in mice or humans. Clearly, there is a need for an additional and different therapeutic approach, such as the decoy peptides mentioned above and the approach described here. Ultimately, the AD patient will benefit from a combination of different drugs.
Our high-throughput screen discovered a small molecule that possessed the potential to eliminate the neurotoxicity characteristic of the disease. Whereas other research groups have concentrated their efforts on developing an AD therapy that prevents or at least reduces the production of the neurotoxic Aβ peptides Aβ40 and Aβ42, we have chosen to develop small molecules that block the toxic activities of these misfolded peptides.
Our hypothesis states (Fig. 1) that, in the first instance, the misfolded peptide aggregates spontaneously, eventually forming fibrils. One or several stages in this process possess new properties that allow them to interact with particular neurons, which then become dysfunctional and eventually die. Our experiments indicate that the first event in this toxicity is the activation by aggregated Aβ peptides of Ca-permeant AMPA receptors on particular sets of neurons (Fig. 1). We hypothesize that those neurons that possess these particular receptors will be affected, perhaps explaining the very particular distribution of cell-type specificity in early AD. The dysfunction and death of such neurons causes the disease.
Our aim is to study in vitro the molecular mechanism of this interaction, especially in the first few seconds, and then to find means of preventing this interaction. We believe that we have found such a molecule, which has become a candidate for AD therapy.
We find that during incubation, an increase in β-sheet content of the Aβ42 peptide parallels the increase in neurotoxicity, as expressed in Ca2+ ion influx (Fig. 9A). Here, we report on a class of small molecules discovered in a robotic high-throughput screen that searched for compounds that are effective in altering the β-sheet conformation that is typical of aggregated misfolded Aβ42. We had observed that preincubation of the Aβ42 peptide was necessary before neurotoxicity (Ca2+ influx) was seen (Fig. 9A) and that this effective change was paralleled by an increase in β-sheet content (18). The particular representative of this class that we describe here is DAPH, which shows considerable promise.
The set of libraries that we searched contained ≈3,780 molecules with known biological functions. It was put together for another purpose by our colleague Brent Stockwell. For historical reasons, the original screen used the dye DiBAC4 (3) as an indicator for the degree of β-sheet conformation in the aggregated misfolded Aβ42 peptide. Half a dozen compounds from the library showed a great reduction in fluorescence when aggregated Aβ42 at 10 μM was exposed to unknown compounds at 5 μg/ml. The use of DiBAC4 (3) to measure β-sheet conformation was validated with the usual ThT fluorescence test for β-sheet content. DAPH was the most effective at lowering the fluorescence associated with binding to β-sheets.
Three in vitro assays have been used to investigate and quantitate the effect of DAPH on the properties of aggregated Aβ42. These are (i) the ability of DAPH to block the toxic influx of Ca2+ ions into neuronal cells, (ii) the great reduction in β-sheet content of aggregated Aβ42 by using ThT, and (iii) the dramatic inhibition and reversal of fibril formation when Aβ42 aggregates.
DAPH was included in the library of biologically active compounds because it is a tyrosine kinase inhibitor with specificity for the epidermal growth factor receptor kinase. The additional activities described for DAPH are likely to reside in different structural aspects of this molecule, but where these are remains to be discovered.
It is unlikely that the kinase-inhibitor property of DAPH plays a role in its effect on Aβ fibrils and neurotoxicity because cells are not present in our in vitro assays. Moreover, seven other tyrosine kinase inhibitors that we tried were inactive, with one exception (data not shown).
Coincubation of DAPH with the peptide Aβ42 eliminates the Ca-influx potential, presumably because it changes the β-sheet conformation of the fibrils or protofibrils drastically. This observation by itself does not solve the question of whether the neurotoxicity of Aβ42 aggregates resides in protofibrils that are intermediates or final fibrils or some other oligomeric aggregate that is formed as a side product in vitro (and in vivo). In our experience, the decrease in neurotoxicity correlates with loss of β-sheet conformation. The stoichiometry of the process is interesting; it seems that complete reversal of fibril formation, as seen in the EM, occurs at equimolar peptide/DAPH ratio (for example, 10:10 μM) (Figs. 3 and 5) and that the IC50 value for β-sheet elimination is ≈15 μM (Fig. 7). We can assume that we are looking at the behavior of the bulk peptide. However, much less DAPH is needed to eliminate the neurotoxicity; the Ca2+-influx effect, when Aβ42 is 10 μM, reduces the effect with an IC50 of ≈0.7 μM DAPH (Fig. 10B). This observation supports the notion that the active Aβ oligomer species has a high-affinity binding site that controls the Ca2+-influx effect and a low-affinity site that is involved in the reversal of β-sheet-containing fibrils (Fig. 7). In our experience, preincubation of Aβ42 for >24 h is needed for the full Ca2+-influx effect (Fig. 9A), suggesting that the active oligomer might not be an early intermediate, as suggested by Wang et al. (20) but is perhaps a novel product that is formed during aggregation. This observation raises the interesting possibility, discussed by Lansbury and coworkers (17), that there might be an equilibrium between mature fibrils (as in plaques) and active oligomers. If such oligomers existed around mature plaques, they might account for the heavily stained neuritis surrounding the plaques. The plaques themselves might indeed be “harmless.”
DAPH is an interesting compound for studying the stability and interactions of β-sheet-containing fibrils. Structural analogs of DAPH would enable us to define the function of each part of the molecule. We already know from Traxler et al. (21) the effect that substitutions in different parts of DAPH have on its ability to inhibit tyrosine kinase and other protein kinases. We will eliminate from the DAPH molecule its kinase-inhibitor capacity to avoid possible toxicity complications.
DAPH is a remarkably efficient molecule for eliminating the neuronal cell toxicity that lies at the root of AD pathology. It is also extraordinarily efficient at destroying the underlying β-sheet structure of the characteristic AD fibrils (plaques). It has not escaped our notice that DAPH has the potential to reverse the β-sheet-containing fibrils that are characteristic of other protein-misfolding diseases.
Acknowledgments
We thank Dr. Brent Stockwell for invaluable help in performing the high-throughput screen that yielded DAPH. We also thank Prof. Jonathan King and David Colby (Massachusetts Institute of Technology) and Prof. Bruce Yankner (Children's Hospital, Harvard Medical School, Boston) for helpful discussions. This work was supported by the Kurt and Johanna Immerwahr Fund for Alzheimer Research at the Massachusetts Institute of Technology and the Massachusetts Institute of Technology Undergraduate Research Opportunities Program.
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 30, 2002.
Abbreviations: AD, Alzheimer's disease; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; DAPH, 4,5-dianilinophthalimide; NBQX, 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; ThT, thioflavin T; Ex, excitation wavelength; Em, emission wavelength; EM, electron microscopy/microscope.
See accompanying Biography on page 14323.
Footnotes
Emission spectra of fluorescent complexes of aggregated Aβ42 are as follows: DiBAC4 (3), Ex = 485 nm and Em = 530 nm; and ThT, Ex = 435 nm and Em = 485 nm.
The fact that they are made entirely of D-amino acids makes them physiologically stable.
References
- 1.Glenner, G. G. & Wong, C. W. (1984) Biochem. Biophys. Res. Commun. 122, 1131-1135. [DOI] [PubMed] [Google Scholar]
- 2.Sisodia, S. S., Koo, E. H., Beyreuther, K., Unterbeck, A. & Price, D. L. (1990) Science 248, 492-495. [DOI] [PubMed] [Google Scholar]
- 3.Yankner, B. A., Duffy, L. K. & Kirschner, D. A. (1990) Science 250, 279-282. [DOI] [PubMed] [Google Scholar]
- 4.Lorenzo, A. & Yankner, B. A. (1994) Proc. Natl. Acad. Sci. USA 91, 12243-12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferreira, A., Lu, Q., Orecchio, L. & Kosik, K. S. (1997) Mol. Cell. Neurosci. 9, 220-234. [DOI] [PubMed] [Google Scholar]
- 6.Ingram, V. M. (2004) in Subcellular Biochemistry: Alzheimer's Disease: Cellular and Molecular Aspects of Amyloid, eds. Harris, R. & Fahrenholz, F. (Plenum, London), in press.
- 7.Blanchard, B. J., Konopka, G., Russell, M. & Ingram, V. M. (1997) Brain Res. 776, 40-50. [DOI] [PubMed] [Google Scholar]
- 8.Blanchard, B. J., Hiniker, A. E., Lu, C. C., Margolin, Y., Yu, A. S. & Ingram, V. M. (2000) J. Alzheimers Dis. 2, 137-149. [DOI] [PubMed] [Google Scholar]
- 9.Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I. & Rydel, R. (1992) J. Neurosci. 12, 376-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattson, M. P. & Furukawa, K. (2003) in Alzheimer's Disease and Related Disorders: Research Advances, eds. Iqbal, K. & Winblad, B. (Ana. Asian Intl. Acad. Aging, Bucharest, Romania).
- 11.Klein, W. L., Stine, W. B., Jr., & Teplow, D. B. (2004) Neurobiol. Aging 25, 569-580. [DOI] [PubMed] [Google Scholar]
- 12.Bitan, G., Kirkitadze, M. D., Lomakin, A., Vollers, S. S., Benedek, G. B. & Teplow, D. B. (2003) Proc. Natl. Acad. Sci. USA 100, 330-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh, D. M., Klyubin, I., Fadeeva, J. V., Rowan, M. J. & Selkoe, D. J. (2002) Biochem. Soc. Trans. 30, 552-557. [DOI] [PubMed] [Google Scholar]
- 14.Selkoe, D. J. & Schenk, D. (2003) Annu. Rev. Pharmacol. Toxicol. 43, 545-584. [DOI] [PubMed] [Google Scholar]
- 15.Fezoui, Y., Hartley, D. M., Harper, J. D., Khurana, R., Walsh, D. M., Condron, M. M., Selkoe, D. J., Lansbury, P. T., Jr., Fink, A. L. & Teplow, D. B. (2000) Amyloid 7, 166-178. [DOI] [PubMed] [Google Scholar]
- 16.Williams, R. C. & Fraser, D. (1953) J. Bacteriol. 66, 458-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harper, J. D., Wong, S. S., Lieber, C. M. & Lansbury, P. T., Jr. (1999) Biochemistry 38, 8972-8980. [DOI] [PubMed] [Google Scholar]
- 18.Blanchard, B. J., Thomas, V. L. & Ingram, V. M. (2002) Biochem. Biophys. Res. Commun. 293, 1197-1203. [DOI] [PubMed] [Google Scholar]
- 19.Blanchard, B. J., Stockwell, B. R. & Ingram, V. M. (2002) Biochem. Biophys. Res. Commun. 293, 1204-1208. [DOI] [PubMed] [Google Scholar]
- 20.Wang, Q., Walsh, D. M., Rowan, M. J., Selkoe, D. J. & Anwyl, R. (2004) J. Neurosci. 24, 3370-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Traxler, P., Trinks, U., Buchdunger, E., Mett, H., Meyer, T., Muller, M., Regenass, U., Rosel, J. & Lydon, N. (1995) J. Med. Chem. 38, 2441-2448. [DOI] [PubMed] [Google Scholar]