

Abstract
Pre-steady-state kinetic studies of Escherichia coli glutaminyl-tRNA synthetase conclusively demonstrate the existence of long-distance pathways of communication through the protein-RNA complex. Measurements of aminoacyl-tRNA synthesis reveal a rapid burst of product formation followed by a slower linear increase corresponding to kcat. Thus, a step after chemistry but before regeneration of active enzyme is rate-limiting for synthesis of Gln-tRNAGln. Single-turnover kinetics validates these observations, confirming that the rate of the chemical step for tRNA aminoacylation (kchem) exceeds the steady-state rate by nearly 10-fold. The concentration dependence of the single-turnover reaction further reveals that the glutamine Kd is significantly higher than the steady-state Km value. The separation of binding from catalytic events by transient kinetics now allows precise interpretation of how alterations in tRNA structure affect the aminoacylation reaction. Mutation of U35 in the tRNA anticodon loop decreases kchem by 30-fold and weakens glutamine binding affinity by 20-fold, demonstrating that the active-site configuration depends on enzyme-tRNA contacts some 40 Å distant. By contrast, mutation of the adjacent G36 has very small effects on kchem and Kd for glutamine. Together with x-ray crystallographic data, these findings allow a comparative evaluation of alternative long-range signaling pathways and lay the groundwork for systematic exploration of how induced-fit conformational transitions may control substrate selection in this model enzyme-RNA complex.
Recognition between proteins and RNA generally occurs by an induced-fit mechanism, in which the protein, the RNA, or both undergo conformational changes en route to the final bound complex (1, 2). Although induced fit necessarily incurs an entropic penalty compared with a preformed and rigid interaction, the built-in flexibility nonetheless may help to increase the kinetic association rate, thus lowering the free-energy barrier for complex formation. In enzymatic reactions, it is also established that induced fit can provide a means to increase substrate specificity: Noncognate substrates may induce partial or incorrect rearrangements, leading to misalignment of reactive moieties along the pathway to the transition state (3). Thus, enzymes that modify RNA may employ induced fit for substrate discrimination at both binding and catalytic steps of the reaction.
Aminoacyl-tRNA synthetases are superb model systems for investigating the operation of induced fit in enzyme-RNA complexes. In all organisms, these enzymes maintain fidelity of the genetic code by catalyzing the synthesis of cognate aminoacyl-tRNAs for use in protein synthesis (4). This synthesis occurs by means of a two-step reaction in which the amino acid is first activated to form an aminoacyl adenylate intermediate with release of pyrophosphate (PPi). In the second step, the nucleophilic oxygen from the 2′-OH or 3′-OH group on the 3′-terminal tRNA ribose sugar attacks the mixed anhydride intermediate, yielding aminoacyl-tRNA with concomitant production of AMP. Crystallographic studies of many tRNA synthetases in different ligand-binding states have revealed protein architectures in which interspersed rigid and flexible polypeptide segments facilitate rearrangements of both enzyme and tRNA in the course of complex formation (4, 5).
We are investigating Escherichia coli glutaminyl-tRNA synthetase (GlnRS) as a well studied monomeric class I tRNA synthetase that makes use of an induced-fit catalytic mechanism. GlnRS is one of four class I enzymes that require the presence of tRNA for catalysis of adenylate formation, suggesting that tRNA is required for proper active-site assembly (6). Supporting this notion, fluorescence studies have revealed clear conformational changes in GlnRS upon ATP and tRNA binding (7, 8). Further, comparisons of the unliganded and tRNA-bound GlnRS structures show that tRNA binding drives an extensive set of conformational changes through all four domains of the enzyme (9, 10). The conformational rearrangements converge in the active-site domain, where portions of both the amino acid- and ATP-binding sites are misoriented with respect to each other in the absence of tRNA (9, 11, 12). Because the tRNA also is rearranged compared with its presumed solution structure, formation of the GlnRS-tRNA complex is best described as a mutual induced fit involving conformational changes of both macromolecular partners.
Several studies have implicated nucleotides in the anticodon loop and acceptor end as GlnRS specificity determinants (13-15). A peptide loop binding the extreme inner corner of the tRNA L-shape and a β-ribbon emanating from a distal β-barrel domain are identified as potential mediators of pathways by which the distal tRNA contacts may be communicated to the active site (16, 17). These two structural elements are indeed plausibly positioned in the tertiary architecture to play such a role (10). However, the hypotheses of long-distance signal propagation rely exclusively on steady-state kinetic measurements. For example, tRNA anticodon mutants alter both the catalytic rate constant (kcat) for aminoacylation and the Michaelis constant (Km) for glutamine (14), but these data cannot be interpreted in terms of individual rate or equilibrium constants, and thus they do not provide evidence for the communication of signals to the distant active site.
To directly address the influence of distal tRNA contacts on the mechanism of specific aminoacylation, we have developed chemical-quench methodologies for the isolation of specific rate and equilibrium constants along the GlnRS reaction pathway. The highly sensitive assay employs 32P-labeled tRNA and involves separation of substrate and product by TLC after P1 nuclease digestion (18). Analysis of data collected from native and anticodon-modified complexes conclusively demonstrates the existence of a long-range signaling pathway and provides a necessary foundation for further intensive examination of how induced fit functions to provide substrate specificity.
Methods
Enzyme and tRNA Preparation. T7 RNA polymerase was purified from E. coli BL21 cells containing the plasmid pAR1219, as described in refs. 12 and 19. A (His)6 C-terminal-tagged GlnRS was constructed by inserting the glnS gene after the T7 promoter of the expression vector pSJW1 (20). The His-tagged GlnRS was expressed in strain BL21-DE3 (pLysS) by induction with 1 mM isopropyl β-D-thiogalactoside at OD600 between 0.4 and 0.6. Cells were resuspended in a buffer containing 0.5 M NaCl, 50 mM Hepes (pH 7.2), and 10 mM imidazole and were disrupted by French press. Then, 100 mM PMSF, 100 mM benzamidine, 50 μl of RQ1 DNase (Promega), and 15 mM MgCl2 were added to the lysate, and the mixture was stirred at room temperature for 20 min. The enzyme was purified on a 5-ml nickel column (Amersham Pharmacia) and equilibrated in 10 mM imidazole/20 mM Hepes (pH 7.2)/1 mM 2-mercaptoethanol/0.5 M NaCl; the elution buffer was identical to the equilibration buffer except for the inclusion of 120 mM imidazole. His-tagged GlnRS was recovered at >99% purity as judged by SDS/PAGE and was stored at high concentration at -20°C, as described for the native enzyme (6). The enzyme was quantitated based on E280 of 1.06 for a 1 mg/ml solution, determined by computation from the protein sequence. Both steady-state and microscopic kinetic parameters of the His-tagged GlnRS were indistinguishable from those of native GlnRS within experimental error.
E. coli 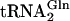 and tRNA variants (each containing a catalytically neutral U1G mutation to promote efficient transcription initiation) were transcribed in high yield from a synthetic DNA template, as described in refs. 12, 21, and 22.
and tRNA variants (each containing a catalytically neutral U1G mutation to promote efficient transcription initiation) were transcribed in high yield from a synthetic DNA template, as described in refs. 12, 21, and 22.
Aminoacylation Assay Using 32P-Labeled tRNA. tRNA was 32P-labeled at the 3′-terminal internucleotide linkage by using the exchange reaction of tRNA nucleotidyltransferase, as described in refs. 12 and 18. To ensure maximal substrate activity, labeled and unlabeled tRNA was mixed to the appropriate final concentration and heated at 65°C for 3 min, followed by the addition of MgCl2 to 7.5 mM and cooling to ambient temperature. tRNA concentrations were determined spectrophotometrically at 260 nm based on A260 = 1.0 corresponding to 40 μg/ml. All reactions were quenched in a buffer containing 0.1% SDS and 0.15 M sodium acetate (pH 5.2). P1 nuclease digestions were performed by adding 1-5 μl of the reaction mixture to a microtiter well containing 3-5 μl of 0.1 mg/ml P1 nuclease (Fluka) and 0.15 M sodium acetate (pH 5.2) and incubating for 10 min at ambient temperature. Aminoacylated tRNA (as 3′-aminoacylated A76) and nonreacted substrate (as unmodified AMP) were separated by TLC and quantitated by using phosphorimaging analysis, as described in ref. 12.
Plateau aminoacylation levels were determined as described in ref. 12. The plateau values for native tRNAGln and for both anticodon mutants were in the range of 70 ± 10%. Steady-state kinetic reactions to determine the glutamine Km were performed at 37°C under the following conditions: 1-3 nM GlnRS, 3-5 μM tRNAGln, 10 mM ATP, 5 mM DTT, 50 mM Tris (pH 7.3), 15 mM MgCl2, and 0.01-10 mM glutamine. Km values for tRNAGln were determined under identical conditions with 10 mM glutamine and tRNAGln concentrations ranging from 0.03 to 3 μM. Product formation was linearly fit to derive Vo and replotted by using Eadie-Hofstee analysis to derive kcat and Km.
Rapid Chemical-Quench Kinetics. Single-turnover measurements to determine kchem were performed by using a rapid chemical-quench flow apparatus (RQF-3, Kin Tek Instruments, University Park, PA) at saturating substrate concentrations (3 μM tRNAGln, 10 mM glutamine, and 10 mM ATP) under the same conditions as the steady-state reactions. The temperature was maintained at 37°C for all experiments. Enzyme and substrates in two 20-μl sample loops were mixed rapidly into a single reaction loop of specified dimensions to control the time of the reaction. The molar ratio of GlnRS to tRNAGln was maintained at 5:1 for all experiments. Reactions were quenched in 50 μl of 400 mM sodium acetate (pH 5.2) and 0.1% SDS. Eight to 10 timepoints were collected for each kchem determination.
The concentration dependencies of the single-turnover reactions were determined by titrating the concentration of one substrate while maintaining saturating levels of the other two. To determine the glutamine dissociation constant (Kd), reaction mixtures contained 15 μM GlnRS, 3 μM tRNAGln, 5 mM ATP, 13.75 μM MgCl2, 50 mM Tris (pH 7.5), 5 mM DTT, and 0.1-10 mM glutamine. The tRNAGln concentration dependence was determined under identical conditions except that 10 mM glutamine and 10 nM to 3 μM tRNAGln were used. For all experiments, the concentration of 3′ end-labeled tRNAGln was negligible compared with the unlabeled concentration. kchem values determined at each substrate concentration were fit to a hyperbolic binding curve (Y = S0·kchem/S0 + Kd) to derive the Kd under aminoacylation conditions. All single-turnover data under both saturating and subsaturating conditions were acquired in triplicate, fit to a first-order exponential equation (Y = A1·e-kchem·t), and plotted by using kaleidagraph or scientist software.
Pre-steady-state burst experiments were performed in the rapid quench instrument by using the above protocol at saturating concentrations of glutamine and ATP and with enzyme and tRNA concentrations maintained between 0.3 and 1 μM GlnRS and 10 and 21 μM tRNAGln. Data were fit to the equation Y = A1·e-kchem·t + kcat·t.
Results
Assay for Steady-State and Single-Turnover Aminoacylation Kinetics. A highly sensitive and generalizable assay for tRNA aminoacylation was adapted for rigorous measurement of steady-state and microscopic rate constants by E. coli GlnRS. The assay is based on 32P-labeling at the 3′-internucleotide linkage of the tRNA (18). After aminoacylation, the labeled tRNA is degraded by P1 nuclease, resulting in a mixture of labeled aminoacyl-AMP (product) and labeled AMP (unreacted substrate). These two compounds are then separated by TLC and quantified by image analysis. Previously, we have used this assay to determine kcat/Km for cognate glutaminylation and for formation of misacylated Glu-tRNAGln by GlnRS (10). As an initial step, we extended this analysis to the determination of individual kcat and Km parameters of the steady-state reaction with respect to glutamine and tRNAGln (Table 1). These parameters are nearly identical to those determined with the conventional assay, further verifying that this experimental approach is suitable for quantitative kinetic studies (14, 23, 24).
Table 1. Steady-state and microscopic kinetic parameters.
| tRNAGln | kcat,* sec−1 | Km (Gln),* mM | Km (tRNA), μM | kcat/Km (Gln), sec−1·M−1 | kcat/Km tRNA sec−1·M−1 | kchem, sec−1 | Kd (Gln), mM | Kd (tRNA), μM |
|---|---|---|---|---|---|---|---|---|
| Wild type | 3.20 ± 0.48 | 0.26 ± 0.04 | 0.31 ± 0.09 | 1.2 × 104 | 1.0 × 107 | 28 ± 2 | 1.10 ± 0.04 | ND |
| U35A | 0.16 ± 0.03 | 17.8 ± 3.8 | 5.25 ± 1.25 | 9.0 | 3.0 × 104 | 1.04 ± 0.28 | 19.8 ± 5.1 | 8.5 ± 4.0 |
| G36U | 1.45 ± 0.13 | 1.43 ± 0.37 | 3.0 ± 0.1 | 1.0 × 103 | 4.8 × 105 | 19.8 ± 0.7 | 4.25 ± 0.53 | ND |
ND, not determined.
For U35A, the previously published values are kcat = 0.15 s−1; Km (glutamine) = 5.62 mM. For G36U, the previously published values are kcat = 6.2 s−1, Km (glutamine) = 1.1 mM (14).
The assay then was used to measure the rate of the chemical step (kchem) for tRNAGln aminoacylation by single-turnover kinetics by using a rapid chemical-quench instrument. In GlnRS, formation of the activated aminoacyl adenylate intermediate requires the presence of tRNA (4), making isolation of the second tRNA transfer step difficult. Therefore, for all reactions, we measured a composite kchem that represents the two-step reaction by using ATP, glutamine, and tRNAGln substrates and generating Gln-tRNAGln, AMP, and PPi. GlnRS was maintained at 5-fold molar excess over tRNA to ensure single-turnover conditions, and the concentrations of ATP, glutamine, and tRNAGln were varied systematically to establish full substrate saturation. A set of mixing controls were performed to assess whether observed reaction rates might depend on premixing of reactants in the two syringes. Under saturating substrate conditions, each substrate was in turn sequestered in one syringe while the other reactants, including enzyme, were isolated in the other syringe. No dependence of the reaction rates on premixing was observed. Reactions were fit to a first-order exponential to determine a kchem of 28 sec-1 (Fig. 1). This rate encompasses all reaction steps up to and including the formation of Gln-tRNAGln on the enzyme and is nearly 10-fold greater than the steady-state kcat (Table 1). Thus, the rate-limiting step determining kcat represents a physical step after aminoacylation and does not correspond to bond-breaking and -forming reactions in the active site.
Fig. 1.

Time course of the two-step glutaminylation reaction monitoring formation of Gln-tRNAGln by rapid chemical quench. ATP, glutamine, and tRNAGln are each present at saturating concentration with enzyme in 5-fold molar excess relative to tRNA (see Methods). (Inset) TLC plate indicating separation of AMP (unreacted substrate) from Gln-AMP (product) after P1 nuclease digestion of the quenched reaction. Data to 30 sec were included in the fit to derive kchem.
Measurement of Burst Kinetics. To further examine the relationship between steady-state and transient kinetics, we measured aminoacylation by GlnRS under pre-steady-state conditions. We mixed 100-300 nM GlnRS with 20 μM tRNAGln in the presence of saturating concentrations of glutamine and ATP and monitored the reaction time course in the rapid-quench instrument. The enzyme and tRNA substrate concentrations were chosen to allow monitoring of a single turnover on the enzyme followed by steady-state product accumulation. The time course of product formation indeed revealed a rapid burst of Gln-tRNAGln synthesis followed by a steady-state rate, confirming that the rate-limiting step follows both substrate binding and aminoacyltRNA synthesis (Fig. 2). This step likely corresponds to the release of glutaminyl-tRNA. Fitting the burst data yields kchem = 45 sec-1 and kcat = 2.2 sec-1, similar to the values determined by the separate Eadie-Hofstee steady-state analysis and the single-turnover exponential plots (Table 1). Extrapolation of the burst plots to the ordinate yielded an estimate for the fraction of active sites at 65-80%. This value corresponds closely to plateau values determined for aminoacylation of tRNAGln transcripts prepared by the methods used here (12).
Fig. 2.

Time course of glutaminylation under pre-steady-state conditions (see text), showing a rapid burst of Gln-tRNAGln product in the first enzyme turnover followed by a slower steady-state rate.
Examination of Glutamine and tRNAGln Binding. The application of transient kinetics to the GlnRS reaction opens new possibilities for examining binding equilibria and kinetics. As an initial exploration of substrate binding, we examined the dependencies of the single-turnover reaction rates on the concentrations of glutamine and tRNAGln. Both of these substrates showed curvature in the concentration dependence of the reaction rate in the presence of saturating levels of the other two, indicating that the kinetics follows a two-step mechanism that is limited by first-order isomerization of the enzyme-substrate complex (25). The titrations of glutamine showed single-exponential kinetics at all concentrations tested, indicating that this substrate is in rapid equilibrium with the enzyme-tRNAGln-ATP complex and that Kd determined from the replot of kchem vs. substrate concentration ([S]) to a hyperbolic equation is equivalent to the equilibrium binding constant (Fig. 3). The kinetic Kd value determined for glutamine is 1 mM, ≈4-fold higher than the steady-state Km. The discrepancy between Km and Kd for glutamine is consistent with the finding that the rate-limiting step in the steady-state reaction occurs after product formation on the enzyme. Given glutamine affinity at 1 mM and kchem of 28 sec-1, rapid equilibrium binding (koff >> kchem) would hold for kon > 106 M-1·sec-1, well below diffusion control limits (kon and koff are the rate constants for binding and dissociation of glutamine, respectively, to the GlnRS-tRNAGln-ATP ground-state ternary complex).
Fig. 3.

Determination of glutamine affinity. (A) Time course of glutaminylation performed at various concentrations of glutamine and at saturating concentrations of ATP and tRNAGln. The glutamine concentrations used are 5 (○), 1 (□), 0.5 (⋄), 0.1 (×), 0.05 (+), and 0.01 (▵) mM. (B) Replot of the data by hyperbolic fit to derive the Kd for glutamine (see Table 1). Data to 30 sec were taken and included in the fits for each concentration of glutamine.
tRNA titrations revealed single-exponential kinetics at high concentrations, but at low concentrations (50 nM and below), a distinct lag was observed in the accumulation of product (data not shown). In general, the time dependence of a single-turnover enzymatic reaction shows a lag at low [S] if the rates of binding and dissociation at those concentrations (kon[S] and koff) are comparable to the rate of the chemical step (kchem) on the enzyme (the rate for the reverse of the chemical step is assumed negligible for aminoacylation of tRNA) (25, 26). The presence of a lag under these conditions demonstrates that tRNAGln is not in rapid equilibrium with GlnRS. Thus, the Kd for tRNA cannot be determined by fitting the kchem vs. [S] plot to a hyperbolic curve, as was possible for the glutamine substrate. Further transient kinetics experiments together with simulations and global fitting will be required to derive Kd in these circumstances (25, 27). Determination of koff for tRNA binding to the unliganded enzyme by fluorescence showed that this parameter is comparable in magnitude to the kchem determined here, consistent with the notion that the tRNA is not in rapid equilibrium (24).
A Signaling Pathway from Anticodon to Active Site. The crystal structure of the GlnRS-tRNAGln complex reveals hydrogen-bonding interactions with base-specific moieties at all three positions of the CUG anticodon (28). Among these, both U35 and G36 are important recognition determinants as judged by significant decreases in the steady-state rates of glutaminylation and increases in the glutamine Km upon replacement with other bases (13, 14). These data suggested the possibility that recognition of the anticodon is signaled some 40 Å across the structure to influence events in the active site. To examine whether intracomplex communication indeed occurs, we determined kchem and Kd (glutamine) for tRNA mutants U35A and G36U and compared these with the steady-state glutaminylation parameters for these species (Table 1 and Fig. 4). Both mutants showed single-exponential kinetics at all concentrations of glutamine, indicating rapid equilibrium. The rate of the chemical steps in aminoacylation is decreased 30-fold in the U35A mutant, whereas glutamine affinity is weakened by nearly 20-fold. Because both kchem and Kd (glutamine) are microscopic constants that identify molecular events directly in the active site, these data unambiguously demonstrate the existence of a signaling pathway originating with enzyme interactions at the central base of the anticodon (Fig. 5). Interestingly, whereas tRNA is not in rapid equilibrium for G36U, no lag was found at low tRNA concentrations for U35A (data not shown), allowing determination of Kd at 8.5 μM for this mutant. Rapid-equilibrium conditions hold for the U35A mutant presumably because kchem is decreased, whereas koff (the rate at which tRNA dissociates from the ternary GlnRS-ATP-glutamine complex) is likely increased to account for the weakened tRNA affinity.
Fig. 4.

Glutamine affinities of mutant complexes. (A) Plot of the kchem for glutaminylation vs. glutamine concentration for the G36U mutant of tRNAGln. The glutamine concentrations used are 40, 20, 10, 5, 2.5, and 1 mM. (B) Plot of the kchem for glutaminylation vs. glutamine concentration for the U35A mutant of tRNAGln. The glutamine concentrations used are 79, 41, 20.5, 10, 4, and 2 mM.
Fig. 5.

Structural basis for long-distance signaling. (A) Structure of the GlnRS-tRNAGln complex showing possible signaling pathways. The active site is marked by a glutaminyl adenylate analog (light blue). Red, Rossmann fold active-site domain and distal β-barrels; yellow, acceptor-stem binding domain; orange, nucleotides U35 and G36. Two alternative signaling pathways are shown in purple (helix-loop-helix motif) and in dark blue (extended β-ribbon). (B) Close-up view of the anticodon-binding site and alternative signaling pathways. Hydrogen bonds made at U35 and G36 are shown as dashed lines.
Whereas kcat/Km with respect to tRNA for G36U is decreased by >20-fold compared with the wild-type reaction, Kd for glutamine is weakened by only 4-fold, and kchem is nearly unaffected. Thus, the communication pathway from anticodon to active site is strongly triggered by enzyme interactions with U35 and is activated only weakly by those made with G36.
Discussion
tRNA Aminoacylation by Rapid Chemical Quench. We have applied a previously described chemical quench-based assay for tRNA aminoacylation to the measurement of rate constants for glutaminyl-tRNA synthesis and equilibrium constants for substrate binding (18). In contrast to the conventional acid-precipitation filter-binding assay (29), the high sensitivity of this approach allows very weak reactions to be reliably monitored (12). The chemical-quench methodology should be generalizable to all tRNA synthetases and likely will require only minor modifications to optimize TLC separations of different aminoacyl adenylates from AMP (18).
Most pre-steady-state kinetic studies of tRNA synthetases have addressed the adenylate synthesis step, which proceeds independently of tRNA for most of the enzymes (30-34). However, early studies of MetRS, PheRS, TyrRS, ArgRS, and TrpRS used chemical-quench flow to examine aminoacylation with 14C-labeled amino acid (35-39). More recently, intrinsic protein tryptophan fluorescence was used as a probe to examine aminoacyl-tRNA synthesis by TyrRS and TrpRS (26, 40-42). Among these studies, rate constants for the isolated second step by TrpRS, ArgRS, and TyrRS are in the range of 25-45 sec-1, similar to the kchem value of 28 sec-1 measured here for the two-step reaction by GlnRS (37, 39, 40, 42), whereas the experiments with MetRS and PheRS returned lower values of 2-6 sec-1 for the transfer step (35-36). The rate of transfer in GlnRS is thus similar to that found in other tRNA synthetases. Burst kinetics also has been observed previously for both ArgRS and TrpRS enzymes (37, 39).
Communication from the Anticodon to the Active Site. It is well known that kcat and Km for aminoacylation are composite terms reflecting interactions in enzyme-substrate, enzyme-intermediate, and enzyme-product complexes (43). Thus, alteration in these constants for modified tRNAs or enzyme mutants cannot be assigned to particular steps in the reaction pathway. The correlation of altered steady-state parameters with crystal structures is consequently ambiguous, because kinetic effects cannot be assigned to particular structural regions. Clearly, an understanding of the component parts and operation of signaling pathways within the GlnRS-tRNA complex demands measurement of microscopic kinetic parameters for the separate binding and chemical steps.
The existence of a long-range communication pathway originating from the anticodon is definitively established by analysis of the U35A tRNA mutant (Table 1). Both the glutamine-binding affinity and the rate of the aminoacylation reaction are significantly affected by this mutation, despite the nearly 40-Å separation between the enzyme-anticodon interface and the active site. In the crystal structure of the GlnRS-tRNAGln complex, all three anticodon bases are unstacked to bind in complementary pockets formed by the C-terminal β-barrel domains of the enzyme (28) (Fig. 5). Discriminating side-chain interactions with the Watson-Crick moieties of U35 are made by Arg-341 and Gln-517 of the enzyme, whereas similar hydrogen bonds with G36 are made by Arg-402.
Although no definitive data regarding the existence of the signaling pathway were previously reported, several proposals to address the functioning of such a (formerly hypothetical) pathway nonetheless have been made based on steady-state kinetics and on several provocative features of the crystal structure. One proposal invokes communication by means of a long antiparallel β-ribbon that emanates from the proximal β-barrel domain to pack on sequences adjacent to the MSK active-site motif (10, 17) (Fig. 5). Alternatively, it was suggested that the anticodon-binding signal might be propagated through a helix-loop-helix motif that binds the extreme inner corner of the tRNA L-shape (16). The crystal structure of unliganded GlnRS showed that both of these protein elements indeed undergo structural rearrangements upon tRNA binding (9). A clear pathway from the inner corner of the tRNA to the active site could be traced; this pathway includes the peptide loop segment that links the two long antiparallel strands of the proximal β-barrel. However, comparison of the tRNA-bound and unliganded structures did not reveal specific, clear rearrangements in response to binding of the anticodon bases. Instead, it appears that the binding sites for all three bases are largely preformed in the enzyme. The β-barrels rotate as a rigid unit by ≈4° upon tRNA binding, corresponding to an outward displacement of ≈1 Å, but no pathway propagating structural changes between the anticodon nucleotides and either the inner corner of the tRNA or the antiparallel β-ribbon could be discerned.
Although results from these crystallographic experiments have not been immediately illuminating, some insight nonetheless may be gleaned from the finding that signal propagation is much stronger from U35 than from G36 (Table 1), even though these two bases are directly adjacent to each other in the anticodon loop (Fig. 5). The base of U35 is bound directly by the guanidinium group of Arg-341, which emanates from the C-terminal portion of a long α-helix that runs along the entire vertical stem of the tRNA from the inner elbow to the anticodon. By contrast, all of the contacts with G36 are made by amino acids in the distal β-barrel domain located farthest from the active site. Thus, signal propagation is more effective when the recognition contact is from an amino acid that has a more direct connection, through a single α-helix, to the surface loop that rearranges in response to binding at the tRNA inner elbow (9). Although further experimentation clearly is required, these findings do suggest that signal propagation along the enzyme-RNA interface, through the helix-loop-helix motif at the tRNA inner elbow, may be the preferred pathway. Such a signaling pathway may require a covalent connection of the tRNA backbone from anticodon to 3′ end, as suggested based on tRNAGln microhelix experiments (44) and on the observation that kinetic defects in tRNA mutants are generally larger than in enzyme mutants that disrupt the same interaction (45).
Long-range signaling clearly is implicated in many tRNA synthetases based on crystallographic and biochemical experiments (4). However, the coupling of pre-steady-state analysis with x-ray crystallography to directly elucidate specific pathways of intramolecular communication has not been reported previously. Application of these techniques to additional modified complexes with alterations on either side of the protein-RNA interface ultimately should allow precise dissection of the molecular switches controlling tRNA selectivity by GlnRS. This information may in turn prove crucial to protein engineering experiments designed to alter either the amino acid or the tRNA selectivities of the enzyme. The general applicability of the chemical-quench methodology should make this approach useful in many if not all other tRNA synthetases.
Acknowledgments
We thank Olke Uhlenbeck and Alexey Wolfson for encouragement and helpful discussions, and Tim Bullock for construction of the His-tagged GlnRS expression vector. This work was supported by National Institutes of Health Grant GM63713 (to J.J.P.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: GlnRS, glutaminyl-tRNA synthetase.
References
- 1.Williamson, J. R. (2000) Nat. Struct. Biol. 7, 834-837. [DOI] [PubMed] [Google Scholar]
- 2.Leulliot, N. & Varani, G. (2001) Biochemistry 40, 7947-7956. [DOI] [PubMed] [Google Scholar]
- 3.Post, C. B. & Ray, W. J., Jr. (1995) Biochemistry 34, 15881-15885. [DOI] [PubMed] [Google Scholar]
- 4.Ibba, M. & Söll, D. (2000) Annu. Rev. Biochem. 69, 617-650. [DOI] [PubMed] [Google Scholar]
- 5.Francklyn, C., Perona, J. J., Puetz, J. & Hou, Y.-M. (2002) RNA 8, 1363-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibba, M, Becker, H. D., Stathopoulos, C., Tumbula, D. L. & Söll, D. (2000) Trends Biochem. Sci. 25, 311-316. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharyya, T. & Roy, S. (1993) Biochemistry 32, 9268-9273. [DOI] [PubMed] [Google Scholar]
- 8.Mandal, A. K., Bhattacharyya, A., Bhattacharyya, S., Bhattacharyya, T. & Roy, S. (1998) Protein Sci. 7, 1046-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherlin, L. D. & Perona, J. J. (2003) Structure (London) 11, 591-603. [DOI] [PubMed] [Google Scholar]
- 10.Rould, M. A., Perona, J. J., Söll, D. & Steitz, T. A. (1989) Science 246, 1135-1141. [DOI] [PubMed] [Google Scholar]
- 11.Rath, V. L., Sylvian, L. F., Beijer, B., Sproat, B. S. & Steitz, T. A. (1998) Structure (London) 6, 439-448. [DOI] [PubMed] [Google Scholar]
- 12.Bullock, T. L., Uter, N. T., Nissan, T. A. & Perona, J. J. (2003) J. Mol. Biol. 328, 395-408. [DOI] [PubMed] [Google Scholar]
- 13.Jahn, M., Rogers, J. M. & Söll, D. (1991) Nature 352, 258-260. [DOI] [PubMed] [Google Scholar]
- 14.Ibba, M., Hong, K., Sherman, J. M., Sever, S. & Söll, D. (1996) Proc. Natl. Acad. Sci. USA 93, 6953-6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McClain, W. H., Schneider, J., Bhattacharya, S. & Gabriel, K. (1998) Proc. Natl. Acad. Sci. USA 95, 460-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers, M. J., Adachi, T., Inokuchi, H. & Söll, D. (1994) Proc. Natl. Acad. Sci. USA 91, 291-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weygand-Durasevic, I., Rogers, M. J. & Söll, D. (1994) J. Mol. Biol. 240, 111-118. [DOI] [PubMed] [Google Scholar]
- 18.Wolfson, A. D., Pleiss, J. A. & Uhlenbeck, O. C. (2002) Proc. Natl. Acad. Sci. USA 99, 5965-5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grodberg, J. & Dunn, J. J. (1988) J. Bacteriol. 170, 1245-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winder, S. J. & Kendrick-Jones, J. (1995) Anal. Biochem. 231, 271-273. [DOI] [PubMed] [Google Scholar]
- 21.Bullock, T. L., Sherlin, L. D. & Perona, J. J. (2000) Nat. Struct. Biol. 7, 497-504. [DOI] [PubMed] [Google Scholar]
- 22.Sherlin, L. D., Bullock, T. L., Nissan, T. A., Perona, J. J., LaRiviere, F. J., Uhlenbeck, O. C. & Scaringe, S. A. (2001) RNA 7, 1671-1678. [PMC free article] [PubMed] [Google Scholar]
- 23.Nissan, T. A. & Perona, J. J. (2000) RNA 6, 1585-1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong, K.-W., Ibba, M., Weygand-Durasevic, I., Rogers, M. J., Thomann, H.-U. & Söll, D. (1996) EMBO J. 15, 1983-1991. [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson, K. A. (1991) in The Enzymes, ed. Sigman, D. S. (Academic, San Diego), 3rd Ed., Vol. 20, pp. 1-61. [Google Scholar]
- 26.Avis, J. M., Day, G. A., Garcia, G. A. & Fersht, A. R. (1993) Biochemistry 32, 5312-5320. [DOI] [PubMed] [Google Scholar]
- 27.Beebe, J. A. & Fierke, C. A. (1994) Biochemistry 33, 10294-10304. [DOI] [PubMed] [Google Scholar]
- 28.Rould, M. A., Perona, J. J. & Steitz, T. A. (1991) Nature 352, 213-218. [DOI] [PubMed] [Google Scholar]
- 29.Loftfield, R. B. (1972) Prog. Nucleic Acids Res. Mol. Biol. 12, 87-128. [DOI] [PubMed] [Google Scholar]
- 30.Wells, T. C. & Fersht, A. R. (1986) Biochemistry 25, 1881-1886. [DOI] [PubMed] [Google Scholar]
- 31.Fersht, A. R., Knill-Jones, J. W., Bedouelle, H. & Winter, G. (1988) Biochemistry 27, 1581-1587. [DOI] [PubMed] [Google Scholar]
- 32.Pope, A. J., Lapointe, J., Mensah, L., Benson, N., Brown, M. J. B. & Moore, K. J. (1998) J. Biol. Chem. 273, 31680-31690. [DOI] [PubMed] [Google Scholar]
- 33.Merle, M., Trezeguet, V., Graves, P. V., Andrews, D., Muench, K. H. & Labouesse, B. (1986) Biochemistry 25, 1115-1123. [DOI] [PubMed] [Google Scholar]
- 34.Bovee, M. L., Pierce, M. A. & Francklyn, C. S. (2003) Biochemistry 42, 15102-15113. [DOI] [PubMed] [Google Scholar]
- 35.Mulvey, R. S. & Fersht, A. R. (1978) Biochemistry 17, 5591-5597. [DOI] [PubMed] [Google Scholar]
- 36.Lin, S. X., Baltzinger, M. & Remy, P. (1984) Biochemistry 23, 4109-4116. [DOI] [PubMed] [Google Scholar]
- 37.Trezeguet, V., Merle, M., Gandar, J.-C. & Labouesse, B. (1986) Biochemistry 25, 7125-7136. [DOI] [PubMed] [Google Scholar]
- 38.Fersht, A. R. & Jakes, R. (1975) Biochemistry 14, 3350-3356. [DOI] [PubMed] [Google Scholar]
- 39.Fersht, A. R., Gangloff, J. & Dirheimer, G. (1978) Biochemistry 17, 3740-3746. [DOI] [PubMed] [Google Scholar]
- 40.Avis, J. M. & Fersht, A. R. (1993) Biochemistry 32, 5321-5326. [DOI] [PubMed] [Google Scholar]
- 41.Xin, Y., Li, W., Dwyer, D. S. & First, A. R. (2000) J. Mol. Biol. 303, 287-298. [DOI] [PubMed] [Google Scholar]
- 42.Ibba, M., Sever, S., Praetorius-Ibba, M. & Söll, D. (1999) Nucleic Acids Res. 27, 3631-3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fersht, A. R. (1999) Structure and Mechanism in Protein Science (Freeman, New York).
- 44.Wright, D. J., Martinis, S. A., Jahn, M., Söll, D. & Schimmel, P. (1993) Biochimie 75, 1041-1049. [DOI] [PubMed] [Google Scholar]
- 45.Sherman, J. M., Thomann, H.-U. & Söll, D. (1996) J. Mol. Biol. 256, 818-828. [DOI] [PubMed] [Google Scholar]