

Abstract
Many secretory proteins are synthesized as proforms that become biologically active through a proteolytic cleavage in the trans-Golgi complex or at a later stage in the secretory pathway. Haptoglobin (Hp) is unusual in that it is cleaved in the endoplasmic reticulum before it enters the Golgi. Here, we present evidence that the recently discovered complement C1r-like protein (C1r-LP) mediates this cleavage. C1r-LP has not previously been shown to possess proteolytic activity, despite its homology to trypsin-like Ser proteinases. We demonstrate that coexpression of the proform of Hp (proHp) and C1r-LP in COS-1 cells effected cleavage of proHp in the endoplasmic reticulum. This cleavage depended on proteolytic activity of C1r-LP because mutation of the putative active-site Ser residue abolished the reaction. Furthermore, incubation of affinity-purified C1r-LP and proHp led to the cleavage of the latter protein. ProHp appeared to be cleaved at the expected site because substitution of Gly for Arg-161 blocked the reaction. C1r-LP showed specificity for proHp, in that it did not cleave the proform of complement C1s, a protein similar to Hp particularly around the cleavage site. C1r-LP accounts for at least part of the endogenous proHp-cleavage activity because suppression of the C1r-LP expression by RNA interference reduced the cleavage of proHp by up to 45% in the cells of a human hepatoma cell line (HepG2).
Haptoglobin (Hp) is an abundant plasma glycoprotein whose concentration increases severalfold during inflammation (1). The best understood function of Hp is its ability to bind hemoglobin released upon hemolysis, protecting the tissues from oxidative damage (2). Hp has also been shown to play a role in immune response of T cells (3), regulation of cell proliferation (4), angiogenesis, and arterial restructuring (5). Hp has long been known to be synthesized in the liver, but other tissues, e.g., skin, lung, and kidney, have recently been shown to express low amounts (6, 7).
The primary translation product of the Hp mRNA is a polypeptide that cotranslationally dimerizes and is proteolytically cleaved while still in the endoplasmic reticulum (ER) (8). As a result, a tetrameric molecule is formed consisting of two short and two long polypeptides; the α- and β-chains, respectively. The β-chain, with a molecular mass of ≈35 kDa, is related to trypsin-like Ser proteinases but does not possess the essential catalytic amino acid residues. In humans, the α-chain exists in two major allelic forms of 9 and 18 kDa, respectively, the latter arising from partial gene duplication (9). The proteolytic cleavage of the proform of Hp (proHp) seems to increase severalfold the affinity of the molecule for hemoglobin (10). The enzyme responsible for the cleavage of proHp has not yet been identified, although a cleaving activity has been detected in the luminal content of rat liver microsomes (11), in the medium of cultured primary rat hepatocytes, and in rat plasma (12).
Many functionally and structurally diverse proteins, such as cell-surface receptors, enzymes, and hormones, are synthesized as proforms that are cleaved late in the secretory pathway: in the trans-Golgi apparatus and/or in the secretory vesicles or granules (13). A group of Ser proteinases called proprotein convertases (PCs) mediate this process. There are currently nine known eukaryotic PCs: furin, PC1, PC2, PC4, PACE4, PC6, PC7, SKI-1, and NARC-1. The first seven of these cleave proproteins after a basic residue in the motif (K,R)-(X)n-(K,R)↓, where n = 0, 2, 4, or 6 (14). SKI-1, on the other hand, cleaves after nonbasic residues in the consensus sequence (K,R)-(X)2-(L,T)↓ (14). The natural substrates for NARC-1 have yet to be identified; however, the enzyme can undergo two autocatalytic cleavages after the sequences (Y,I)-V-V-(V,L)-(L,M)↓ (15) or L-V-FA-Q↓ (16). ProHp is cleaved after an Arg residue in the sequence P-V-Q-R↓ (17) and, therefore, does not seem to be a substrate for any known PC. We wanted to identify the proHp-cleaving enzyme to see whether it is a new type of enzyme involved in the proteolytic maturation of proproteins or an already known PC having an atypical substrate specificity.
In the present study, we isolated a proHp-cleaving enzyme from human serum and identified it as the recently discovered complement C1r-like protein (C1r-LP) (18). Through coexpression and RNA interference experiments, we obtained evidence that C1r-LP is a PC that cleaves proHp in the ER of cells of both hepatic and nonhepatic origin. C1r-LP, the synthesis of which has been detected only in liver so far, is structurally similar to the activators of the classical complement pathway: C1r and C1s. In the blood, these proteins occur in a complex consisting of two C1r, two C1s, and six C1q molecules called the first complement factor (C1). Upon binding of C1 to antigen-clustered antibodies, C1r undergoes autoproteolytic activation and cleaves C1s, leading to activation of the complement system (19). In contrast, no function, to our knowledge, has previously been assigned to C1r-LP.
Materials and Methods
Materials. All chromatography media were from Amersham Pharmacia Biosciences (Uppsala). Normal human serum was obtained from the Blood Centre of Uppsala University Hospital, Uppsala. Samples from late steps in the commercial production of an albumin solution for clinical use were obtained from Octapharma (Stockholm). Rabbit antibodies to Hp and goat antibodies to C1r and C1s were from DAKO. Antibodies to the myc epitope were raised in rabbit by using the peptide CEQKLISEEDLN conjugated to keyhole limpet hemocyanin.
Cell Culture. Green monkey kidney endothelial cells (COS-1), BHK-21, Chinese hamster ovary cells, human embryonic kidney (HEK) 293 cells, and human hepatocarcinoma cells (HepG2) were grown in DMEM supplemented with 10% FBS, L-glutamine, and antibiotics.
Construction of Expression Vectors. cDNA for human proHp 2 (a gift from A. Bollen, University of Brussels, Brussels) was cloned by PCR into pcDNA3 (Invitrogen); for the sake of simplicity, we refer to proHp2 and Hp2 as proHp and Hp, respectively. An Arg-161Gly mutation was introduced in proHp by the overlap extension method using PCR (20). ProHp lacking the signal peptide (amino acids 1-17) was cloned in-frame with the N-terminal IgG light-chain κ-signal peptide and the C-terminal myc-His6 tag of pSecTag (Invitrogen). cDNAs for human proC1r and proC1s were kindly provided by N. Thielens (Institut de Biologie Structurale Jean-Pierre Ebel, Grenoble, France) and were cloned into pcDNA3.1 (Invitrogen).
Preparation of Radiolabeled proHp. Hp expressed in COS-1 cells is not cleaved upon secretion and was used as substrate in the cleavage assay. For this purpose, the cells were transfected by electroporation with human proHp cDNA in pcDNA3. The cells were then metabolically labeled with [35S]Met as described (21). The medium was collected and centrifuged, and the supernatants were transferred to -20°C.
Standard Assay for proHp Cleaving Activity. The pH of medium of cells transfected with human proHp cDNA and labeled with [35S]Met ([35S]Met medium) was adjusted by addition of 1.5 M Tris·HCl (pH 8.8) to a final concentration of 150 mM. Then, 8-25 μl of the medium was mixed with 1-5 μl of the sample to be analyzed and incubated at 37°C. Depending on the level of cleaving activity, the length of incubation was 30 min to 12 h. Subsequently, the reaction mixture was subjected to immunoprecipitation, followed by SDS/PAGE and PhosphorImaging as described (21).
Purification of proHp Convertase (HpC) from Human Serum. HpC was purified from an albumin solution, which, after heat treatment, is used clinically for infusion. A sample containing 65-72 g of protein was diluted with 20 mM Tris·HCl (pH 8.0) containing 0.5 M NaCl to a final protein concentration of 20 mg/ml and was then loaded onto a concavalin A Sepharose column equilibrated with the same buffer. Bound proteins were eluted with 30 mM α-D-mannopyranoside and dialyzed against 10 mM Tris·HCl (pH 8.0). They were then applied onto a Q Sepharose column equilibrated with 10 mM Tris·HCl (pH 8.0), and bound proteins were eluted with a linear gradient of 0-1 M NaCl. Fractions with HpC activity were pooled and mixed with a saturated ammonium sulfate solution to a final concentration of 1 M. This solution was loaded onto a phenyl Sepharose column, and bound proteins were eluted with a linear gradient of 1-0 M (NH4)2SO4 in 10 mM Tris·HCl (pH 8.0). Fractions with HpC activity were pooled, dialyzed against 10 mM Tris·HCl (pH 8.0), and loaded onto a Mono Q column. Bound proteins were eluted with a gradient of 0-1 M NaCl. The fraction with the highest HpC activity was then loaded onto a Superose 12 column, which was eluted with 20 mM Tris·HCl (pH 8.0) containing 1 M NaCl. The obtained fractions were stored at +4°C. Because of the small dynamic range of the enzyme assay, the specific activity was not monitored during the purification procedure.
MS Analysis. Two-dimensional gel electrophoresis and matrix-assisted laser desorption ionization-MS analysis were performed at the Expression Proteomics Laboratory in Uppsala. Briefly, proteins were precipitated with cold acetone and subjected to isoelectric focusing in a gel strip with an immobilized pH gradient of 3-6 (Amersham Pharmacia Biosciences) followed by SDS/12.5% PAGE. The separated proteins were stained with colloidal Coomassie brilliant blue, and spots were excised. After trypsin treatment, peptide masses were determined with a matrix-assisted laser desorption ionization/time-of-flight mass spectrometer and analyzed with the Matrix Science (London) mascot search engine, which can be accessed at www.matrixscience.co.uk.
Cloning of C1r-LP. The coding part of the cDNA of the human C1r-LP was amplified from a human placenta cDNA library (Marathon-Ready, Clontech) by PCR. The primers were 5′-GATGCCTGGACCCAGAGTGT-3′ (forward) and 5′-CTCACTCCAGGCCCTCTGTT-3′ (reverse), and a protocol of 40 cycles was used: 95°C for 1 min, 61°C for 1 min, and 72°C for 2 min. The DNA fragment corresponding to nucleotides 17-1481 in the GenBank mRNA sequence of C1r-LP (GenBank accession no. gi|7706082|) was cloned in-frame with the C-terminal myc-His6 tag of pcDNA6b-myc-his (Invitrogen) by PCR. A Ser-436Ala mutation in the putative active site of C1r-LP was introduced by the overlap extension method by using PCR.
In Vivo Cleavage of Precursor Proteins. Cells grown in six-well plates were transfected with 2 μg of DNA by using Lipofectamine (Invitrogen) according to the manufacturer's protocol. For the detection of protein by immunoblotting, the cells were transferred to a serum-free medium (Optimem I, Invitrogen) 1 day after transfection and were grown for an additional 24 h. Subsequently, the media and cells were subjected to SDS/PAGE, followed by immunoblotting with antibodies to the respective protein. Detection was performed by using an enhanced chemiluminescence kit (Amersham Pharmacia Biosciences) and a cooled charge-coupled device camera (Intelligent Dark Box Device, Fuji, Japan).
Pulse-Chase Experiments. Two days after transfection, the cells were rinsed twice with PBS and the labeling medium was added. After 30 min, new labeling medium containing 200 μCi (1 Ci = 37 GBq) of [35S]Met was added, and the cells were labeled for 5 or 7.5 min (pulse period). The medium was then withdrawn, and the cells were incubated for various periods of time in labeling medium supplemented with 200 μM methionine (chase period). In some experiments, brefeldin A (Sigma) was present in the medium at a concentration of 10 μg/ml. After the chase, the media were collected, and the cells were solubilized. After centrifugation, the supernatants were subjected to immunoprecipitation with antibodies to Hp as described above for the proHp-cleaving assay. For endo-β-N-acetylglucosaminidase H (EndoH) treatment, the pellets obtained from the immunoprecipitation were suspended in 0.5% SDS and 1% 2-mercaptoethanol and boiled for 10 min. The samples were then centrifuged, the supernatants were mixed with 1,000 units of EndoH (New England Biolabs), and the buffer was supplied with the enzyme. After 16 h of incubation at 37°C, sample buffer was added, and the proteins were analyzed by SDS/PAGE, followed by PhosphorImaging.
In Vitro Cleavage of proHp by Recombinant C1r-LP. HEK293 and COS-1 cells were plated onto 150-mm cell-culture dishes and transiently transfected with C1r-LP cDNA cloned into pcDNA6-myc-his or with proHp cDNA cloned into pSecTag. After 24 h, the cells were rinsed twice with PBS and grown for 48 h in a serum-free medium (Optimem I). The cell media were then collected and centrifuged for 30 min at 4,000 × g. The supernatants (40 ml) were loaded onto a Cu2+ chelating Sepharose gel (2 ml), and bound protein was eluted with 250 mM imidazole. Recombinant proHp and C1r-LP obtained in this way were stored at +4°C. The cleavage reactions were incubated at 37°C overnight, and proHp cleavage was assessed by Western blotting with anti-Hp antibodies.
RNA Interference Experiments. The target sequence, starting at position 901 in the C1r-LP mRNA: 5′-GAACCAGAGTGTGAATGTG-3′, was selected by using the irnai′ program, which can be accessed at www.mekentosj.com/irnai. The target sequences in C1r mRNA (5′-GGCUGGCUUCUAUGAUUAUG-3′) and Ext2 mRNA [used as irrelevant short interfering RNA (siRNA), 5′-GGUGUAUAUCUAUGCUCUG-3′] were designed by Ambion using an algorithm developed by Cenix Bioscience (Dresden, Germany).
HepG2 cells were seeded in 24-well plates and transfected 24 h later with 50 nM annealed siRNA duplexes (Ambion Europe, Huntingdon, U.K.) by using 3 μl/ml Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After another 36 h, the transfection procedure was repeated, and the cells were grown for 12 h in serum-free medium (Optimem I). Finally, proHp cleavage was assessed by Western blotting.
The DNA oligonucleotides for the cloning of the hairpin siRNA (DNA Technology, Aarhus, Denmark) were annealed, and the resulting dsDNA fragment was cloned into the pSilencer 3.0-H1 vector (Ambion). HEK293 cells were seeded on six-well plates and transfected 24 h later with proHp cDNA in pcDNA3, and with pSilencer containing either C1r-LP-specific siRNA or irrelevant siRNA (provided with the vector). proHp cleavage was analyzed 24-48 h after transfection by Western blotting.
Results
Isolation of HpC Activity from Human Serum. Rat liver microsomes (11), medium from primary rat hepatocytes, and rat plasma (8) have earlier been reported to contain HpC activity. Previous attempts to purify an enzyme with the corresponding activity from these sources failed due to the low level of activity and instability of the enzyme(s) (12). We therefore looked for another source of HpC and found that it could also be detected in human serum in which it was stable for several weeks at 4°C. To obtain the substrate for the activity assay, we transfected COS-1 cells with a plasmid expressing human proHp, labeled the cells with [35S]Met, and collected the medium. Aliquots of this medium were then incubated with different samples, and Hp was detected through immunoprecipitation, followed by SDS/PAGE and PhosphorImaging. The major band migrated as a 55-kDa protein in the absence of HpC activity: proHp (Fig. 1, lane 1). In the presence of cleaving activity, however, part of the proHp was converted into α- and β-chains (of 18 and 35 kDa, respectively; Fig. 1, lane 2). Initial experiments with various chromatographic procedures proved the levels of enzyme in serum to be too low for purification. We therefore tested various fractions from a commercial production of different plasma proteins for the presence of HpC activity. This analysis showed that the product of the second-to-last step in the preparation of an albumin solution for clinical use had a specific activity similar to that of serum (lane 3); this activity was abolished by the commercially used heat treatment (lane 4). We found that the nonheated albumin solution was more suitable as starting material than serum because it contained fewer protein species and a larger proportion of albumin (>98%), which could readily be removed by lectin affinity chromatography. Thus, this material was first applied to a concavalin A Sepharose column and was then further fractionated as described in Materials and Methods. In each purification step, fractions containing HpC activity were identified through the standard cleaving assay (data not shown). The specific activity of the final fraction was 2 × 103 to 10 × 103 times higher than that of the starting material (data not shown).
Fig. 1.

Detection of proHp cleaving activity in human serum and in the material from the last two steps of a commercial production of an albumin solution. Medium from cells expressing proHp and labeled with [35S]Met (12 μl) was mixed with the sample to be analyzed (1 μl) and incubated at 37°C for 30 min. After immunoprecipitation and SDS/PAGE, radiolabeled Hp was detected by PhosphorImaging. The samples analyzed were buffer (lane 1), human serum (lane 2), material from the second-to-last step in the preparation of a commercial albumin solution (lane 3), and from the final, heat-treated albumin solution (lane 4); the protein concentrations were 75-150 mg/ml. PHp, α,and β, mark the positions of the proform, and the α- and β-chains of Hp, respectively. The molecular masses (in kilodaltons) of reference proteins are shown on the left.
Identification of C1r-LP as HpC. Analysis by SDS/PAGE showed that the fraction with the highest HpC activity in the last purification step contained several proteins (data not shown). To see whether any of these might be a proteinase, we turned to 2D gel electrophoresis and MS (Fig. 2). None of the identified proteins were identical to a known proteinase, but one of them, C1r-LP, showed sequence homology to a proteinase.
Fig. 2.

Analysis by 2D gel electrophoresis of the fraction with the highest proHp cleaving activity obtained in the final step of purification starting from the albumin solution described in Fig. 1 (lane 3). Proteins were separated by isoelectric focusing in a pH gradient of 3-6, followed by SDS/12.5% PAGE. Shown is the part of the gel that contained proteins visualized by staining with Coomassie brilliant blue. Protein spots were excised, treated with trypsin, and analyzed by MS. Spots marked with numbers produced high Mowse scores for the corresponding proteins: 1, 2, complement C1r-like protein (GenBank accession no. gi|7706083|); 3, acid sphingomyelinase-like phosphodiesterase 3a (GenBank accession no. gi|18202617|); 4, 5, 6, soluble IL-1 receptor accessory protein-beta (GenBank accession no. gi|33286873|); 7, 8, 9, 10, leucine-rich α2-glycoprotein (GenBank accession no. gi|72059|); 11, 12, 13, Zn-α2-glycoprotein chain B (GenBank accession no. gi|4699583|); and 14, a protein similar to the 40 S ribosomal protein S17 (GenBank accession no. gi|20554971|). Those spots marked with an asterisk did not produce any hit in the database search. The molecular masses (in kilodaltons) of reference proteins are shown on the left.
To determine whether C1r-LP had the capacity to cleave proHp, we wanted to coexpress these two proteins in COS-1 cells. The cDNA of human C1r-LP was first cloned, and the protein was expressed with a myc-His6 tag. Analysis of the cells and medium by SDS/PAGE followed by immunoblotting with antibodies to the myc epitope revealed a band of ≈75 kDa (Fig. 3A). The large difference between the calculated molecular mass (52 kDa) and the apparent molecular mass upon SDS/PAGE indicates that the protein was heavily glycosylated. Indeed, when expressed in Escherichia coli, C1r-LP had an apparent molecular mass of 52 kDa (unpublished observations). As described above, proHp expressed alone in COS-1 cells was secreted almost exclusively in the precursor form (Fig. 1, lane 1). Upon coexpression with C1r-LP, however, the medium contained mostly the cleaved form of Hp (50-80%, Fig. 3B, compare lanes 1 and 2). Cleavage of proHp by C1r-LP was also observed in BKH-21 and Chinese hamster ovary cells (data not shown).
Fig. 3.

Cleavage of proHp by recombinant C1r-LP. (A) Cells were transfected with vector expressing myc-His tagged C1r-LP or mock vector, and cells and media were analyzed by Western blotting with antibodies to the myc epitope. The molecular masses (in kilodaltons) of reference proteins are shown on the right. (B) Cells were transfected with vector expressing proHp, and either mock vector, or vector expressing either C1r-LP, or a C1r-LP S436A mutant (C1r-LP*). Subsequently, cell media were analyzed by Western blotting with antibodies to Hp (α-Hp) or to myc-tag (α-myc). (C) myc-His6-tagged proHp and C1r-LP were isolated as described in Materials and Methods. ProHp was mixed with C1r-LP or buffer only and incubated for 16 h at 37°C. Subsequently, proHp cleavage was analyzed by Western blotting.
It has been suggested, based on comparison of amino acid sequences, that C1r-LP might not possess proteolytic activity (18). To assess whether proteolytic activity of C1r-LP was responsible for the observed cleavage of proHp, we mutated the conserved Ser residue from the hypothetical active site (Ser-436) to Ala. The resulting protein was then coexpressed with proHp, and the cleavage of the latter protein was examined by Western blotting. As shown in Fig. 3B, the mutated C1r-LP (lane 3, top) was unable to cleave proHp although it was secreted as efficiently as the wild-type protein (lanes 2 and 3, bottom).
To test whether the observed cleavage of proHp was mediated directly by C1r-LP, and not by another enzyme activated by C1r-LP, we expressed proHp and C1r-LP with His6 tags in mammalian cells and then isolated them by affinity chromatography. Subsequent incubation of the two proteins resulted in cleavage of proHp (Fig. 3C, lane 1), and the degree of processing depended on the amount of C1r-LP (data not shown).
C1r-LP Cleaves proHp Early in the Secretory Pathway. As described above, coexpression of proHp and C1r-LP in COS-1 cells led to the appearance of cleaved Hp in the medium. To determine at what point this cleavage occurred during secretion, we conducted pulse-chase experiments with COS-1 cells coexpressing the proteins. Approximately half of the radiolabeled Hp was cleaved during a 5- or 7.5-min pulse (41-47%; Fig. 4A, lanes 1 and 2). After a subsequent chase of 20 min, most of the protein (72%) was converted to the mature form (lane 3). At longer times, the intracellular amount decreased due to secretion into the medium. Treatment of the 0- and 20-min cell samples with Endo H, an enzyme removing N-linked oligosaccharides present on proteins in the ER and cis-Golgi, modified proHp and the β-chain (Fig. 4B, pHp to pHpΔ and βc to 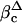 ). As expected, the β-chain in the medium (βm) was Endo H-resistant. Together, these observations demonstrate that C1r-LP cleaves proHp in the ER, and possibly to some extent in the cis-Golgi. This notion was further supported by the fact that brefeldin A, a fungal metabolite inhibiting protein transport from the ER to the trans-Golgi, had no effect on the proHp cleavage (data not shown).
). As expected, the β-chain in the medium (βm) was Endo H-resistant. Together, these observations demonstrate that C1r-LP cleaves proHp in the ER, and possibly to some extent in the cis-Golgi. This notion was further supported by the fact that brefeldin A, a fungal metabolite inhibiting protein transport from the ER to the trans-Golgi, had no effect on the proHp cleavage (data not shown).
Fig. 4.

Time course of intracellular cleavage of proHp by C1r-LP. (A) COS-1 cells expressing proHp and C1r-LP were pulse-labeled for 5 or 7.5 min with [35S]Met and chased for the indicated times. Cells were then solubilized and subjected to immunoprecipitation with antibodies to Hp. After SDS/PAGE, radiolabeled Hp was visualized by PhosphorImaging. Only the top part of the gel is shown. PHp and β mark the positions of the proform and the β-chain of Hp, respectively. (B) Some of the samples described in A, as well as the medium obtained after 240 min of chase, were treated with EndoH before electrophoresis. PHp, βc, and βm mark the positions of the proform, cellular β-chain, and (Endo H-resistant) extracellular β-chain of Hp, respectively. PHpΔ and 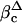 indicate the respective deglycosylated forms appearing on Endo H treatment.
indicate the respective deglycosylated forms appearing on Endo H treatment.
Substrate Specificity of C1r-LP. Hp is closely related to C1r and C1s (22), Ser proteinases that are activated by a proteolytic cleavage. The amino acid sequence similarity around the cleavage/activation sites of all three proteins (Fig. 5A) prompted us to ascertain whether the inactive forms of C1r and C1s, here referred to as proC1r and proC1s, could also be cleaved by C1r-LP. proC1s, when expressed in COS-1 cells, was secreted in its precursor form (of 85 kDa; Fig. 5B, lane 1), and there was no cleavage upon coexpression with C1r-LP (lane 2). In contrast, C1r was secreted mostly in the cleaved/active form when expressed in COS-1 cells (data not shown), as reported earlier for another expression system (23). Therefore, this protein could not be tested as substrate for C1r-LP. Furthermore, we found that both proC1s and proHp were efficiently cleaved when coexpressed with proC1r (Fig. 5 B, lane 3, and C, lane 2, respectively).
Fig. 5.

Specificity of proHp cleavage by C1r-LP. (A) Alignment of amino acid sequences around cleavage site in proHp, proC1s, and proC1r. For comparison, the corresponding sequence of C1r-LP is also shown. Identical amino acid residues are bold and the position of the cleavage site is marked with an arrowhead. (B) Media of COS-1 cells expressing proC1s alone, or proC1s with either C1r-LP or proC1r were analyzed by Western blotting with antibodies to C1s (α-C1s). pC1s, A, and B mark the positions of proC1s, and the A and B chains of C1s, respectively. Expression of C1r-LP was assessed by Western blotting with antibodies to the myc epitope (α-myc). (C) Media of cells transfected with vector expressing proHp, and either mock vector, or vector expressing C1r were analyzed by Western blotting with antibodies to Hp. pHp, α, and β mark the positions of proHp, and the α- and β-chains of Hp, respectively. (D) Cells were transfected with vector expressing a proHp R-161G mutant, and either mock vector, or vector expressing C1r-LP. Subsequently, cell media were analyzed by Western blotting with antibodies to Hp (α-Hp). pHp*, α, and β mark the positions of the mutated proform, and the α- and β-chains of Hp, respectively. Expression of C1r-LP was assessed by Western blotting with antibodies to the myc epitope (α-myc).
In hepatocytes, proHp has been reported to be cleaved after an Arg residue (Arg-161 in human Hp2), which is subsequently removed by a plasma carboxypeptidase (8). To test whether C1r-LP cleaved proHp at the same site, we mutated Arg-161 to Gly, and the resulting protein was coexpressed with C1r-LP in COS-1 cells. As shown in Fig. 5D, the mutation abolished cleavage (>98% Hp uncleaved).
siRNA Inhibition of C1r-LP Activity in Human Cells. To investigate whether C1r-LP is a genuine proHp convertase, we used siRNA to suppress the synthesis of this protein in cells of a human hepatocarcinoma cell line (HepG2), which secrete Hp. As shown in Fig. 6A, lane 1, most of the Hp produced by these cells was cleaved (60-80%). Upon treatment with the C1r-LP siRNA, the degree of cleavage was markedly reduced (Fig. 6A, lane 2, by 25-45%). siRNA specific for C1r inhibited proHp cleavage only slightly (Fig. 6A, lane 3, 2-12%), and no inhibition was observed with an irrelevant siRNA (Fig. 6A, lane 4).
Fig. 6.

Inhibition of proHp cleavage by siRNA specific for C1r-LP. (A) HepG2 cells were transfected with 50 nM siRNA specific for either C1r-LP, C1r, or irrelevant siRNA, as described in Materials and Methods. Subsequently, cell media were analyzed by Western blotting with antibodies to Hp. (B) HEK293 cells were transiently transfected with vector expressing proHp, and vector expressing either irrelevant siRNA, C1r-LP siRNA, or C1r-LP. Cell extracts were then analyzed by Western blotting with antibodies to Hp.
Hp has recently been shown to be expressed at low levels by various nonhepatic tissues, e.g., kidney (6). We used HEK293 cells, which are derived from embryonic kidney, to test whether C1r-LP siRNA would inhibit cleavage of proHp in such cells. We could not detect any Hp production in these cells (data not shown), but when they were transfected with a vector expressing proHp cDNA, the synthesized Hp was partially cleaved (Fig. 6B, lane 1). When these cells were additionally transfected with a plasmid coding for C1r-LP siRNA, the cleavage of proHp was effectively abolished (Fig. 6B, lane 2, by 60-66%). No inhibition was observed when a plasmid expressing irrelevant siRNA was used (Fig. 6B, lane 3). Upon coexpression with C1r-LP, all proHp was converted into the mature form (>90%, Fig. 6B, lane 4), showing that the amount of endogenous enzyme was the limiting factor for proHp cleavage in these cells.
Inhibitors and pH Optimum of Cleavage of proHp by C1r-LP. Because the enzymatic activity of C1r-LP has not previously been studied, we characterized the effect of various inhibitors on the cleavage of proHp by this protein. Three general inhibitors for Ser proteinases, diisopropyl fluorophosphate (0.2 mM), phenylmethanesulfonyl fluoride (2 mM), and 4-(2-aminoethyl)benzenesulfonyl fluoride (4 mM), were found to inhibit the enzyme by 59-85%. In contrast, N-(trans-epoxysuccinyl)-L-lecine 4-guanidinobutylamide (0.2 mM), a general inhibitor of cysteine proteinases, was without effect, as was leupeptin (0.1 mM), which inhibits both cysteine and some Ser proteinases. EDTA (20 mM), which chelates divalent cations, was found to stimulate the enzyme by 10%. The pH optimum of the reaction was 8.0, with negligible activity below pH 6.5 (data not shown).
Discussion
In this study, we identified a protein in human plasma, C1r-LP, which has the capacity to specifically cleave proHp. Normally, this cleavage occurs shortly after the synthesis of proHp in the ER, and we obtained compelling evidence that C1r-LP accounts for at least part of this activity. First, we found that coexpression of the two proteins in COS-1 cells led to secretion of cleaved proHp. Second, approximately half the proHp labeled during a 5-min incubation with [35S]Met was cleaved, and the cleavage product was Endo H-sensitive. Third, transfection of a hepatoma cell line (HepG2) with siRNA specific for C1r-LP reduced cleavage of endogenous proHp by up to 45%.
It is possible that all cleavage of proHp in HepG2 cells is mediated by C1r-LP and that the partial reduction by C1r-LP siRNA that we observed was due to incomplete elimination of this enzyme. Alternatively, the result can be accounted for by the presence of another proHp-cleaving enzyme. C1r, which is also synthesized by HepG2 cells, cleaves proC1s at a sequence site very similar to that of proHp (Fig. 5A). Indeed, we found that upon coexpression of proHp and proC1r in COS-1 cells, proHp was cleaved (Fig. 5C). However, in hepatocytes, as well as in HepG2 cells, C1r is secreted as an inactive proprotein (24), which is activated extracellularly through the classical complement pathway (25). Consistently, suppression of C1r by siRNA in HepG2 cells reduced proHp cleavage by only 3-12% (Fig. 6A). Furthermore, we found that incubation of the luminal contents of microsomes prepared from these cells led to an increased cleavage of proHp but not of proC1s (unpublished observations). Hp is synthesized at low levels in tissues other than liver, e.g., kidney. We found that the cells of the kidney-derived cell line HEK293, while not producing detectable amounts of Hp, were able to partially cleave recombinant proHp. This cleavage was effectively abolished by C1r-LP siRNA (Fig. 6B). Further work is required to determine the role C1r-LP plays in the cleavage of proHp in different tissue types.
Comparison of the genes for C1r and C1r-LP suggests that C1r-LP arose through a duplication of C1r, or of a common ancestral gene, followed by an internal deletion (18). Thus, C1r-LP contains the domain corresponding to the Ser proteinase domain of C1r but lacks some of its regulatory parts. It had been noted that C1r-LP lacks an Arg residue at the position where the related Ser proteinases have a site for cleavage/activation (18). This observation suggests that C1r-LP is active directly after its synthesis and explains why it could cleave proHp in the ER (Fig. 4). The apparent lack of activation makes C1r-LP unique among PCs; the other members require at least one tightly regulated proteolytic cleavage to become active (14).
Trypsin-like Ser proteinases, such as C1r, have an Asp residue at the bottom of the primary specificity pocket, which interacts electrostatically with an Arg residue in the P1 site of the substrate (26). We show that proHp cleavage by C1r-LP required an Arg residue at position P1 (Fig. 5D). Remarkably, C1r-LP appears to have a Ser rather than an Asp residue in its specificity pocket, indicating that it binds its substrate differently than its homologues. Furthermore, we found that C1r-LP did not cleave proC1s (Fig. 5B), whose sequence around the cleavage site is very similar to that of proHp (Fig. 5A). Also, C1r-LP cleaved a proHp mutant that had the four amino acid residues preceding the cleavage site exchanged to the corresponding residues in C1s (K.B.W. and E. F., unpublished observations), showing that the specificity of the enzyme is determined also by structures outside the cleavage site. The physiological function of C1r-LP in the circulation is unknown. One possible function would be to convert proHp that has escaped intracellular cleavage. Indeed, a fraction of the Hp secreted by rat hepatocytes has been shown to be uncleaved (8, 27).
Acknowledgments
We thank Å. Engström and J. Forsberg for performing 2D gel electrophoresis and MS; S. Winge (Octapharma) for providing samples from the production of plasma proteins; K. Nilsson Ekdahl (Uppsala University) for antibodies to C1r and C1s; and B. Tomkinson, R. Robinson, and I. Björk for critical comments on the manuscript. This work was supported by the Swedish Research Council and the E. O. and Edla Johansson Scientific Foundation, and by the Wallenberg Consortium North.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: C1r-LP, complement C1r-like protein; Hp, haptoglobin; proHp, proform of Hp; ER, endoplasmic reticulum; PC, proprotein convertase; HpC, proHp convertase; HEK, human embryonic kidney; EndoH, endo-β-N-acetylglucosaminidase H; siRNA, short interfering RNA.
References
- 1.Dobryszycka, W. (1997) Eur. J. Clin. Chem. Clin. Biochem. 35, 647-654. [PubMed] [Google Scholar]
- 2.Lim, Y. K., Jenner, A., Ali, A. B., Wang, Y., Hsu, S. I., Chong, S. M., Baumman, H., Halliwell, B. & Lim, S. K. (2000) Kidney Int. 58, 1033-1044. [DOI] [PubMed] [Google Scholar]
- 3.Arredouani, M., Matthijs, P., Van Hoeyveld, E., Kasran, A., Baumann, H., Ceuppens, J. L. & Stevens, E. (2003) Immunology 108, 144-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Ghmati, S. M., Arredouani, M., Van Hoeyveld, E. M., Ceuppens, J. L. & Stevens, E. A. (2002) Scand. J. Immunol. 55, 352-358. [DOI] [PubMed] [Google Scholar]
- 5.Smeets, M. B., Pasterkamp, G., Lim, S. K., Velema, E., van Middelaar, B. & de Kleijn, D. P. (2002) FEBS Lett. 529, 221-224. [DOI] [PubMed] [Google Scholar]
- 6.D'Armiento, J., Dalal, S. S. & Chada, K. (1997) Gene 195, 19-27. [DOI] [PubMed] [Google Scholar]
- 7.Lee, M. Y., Kim, S. Y., Choi, J. S., Lee, I. H., Choi, Y. S., Jin, J. Y., Park, S. J., Sung, K. W., Chun, M. H. & Kim, I. S. (2002) J. Cereb. Blood Flow Metab. 22, 1176-1180. [DOI] [PubMed] [Google Scholar]
- 8.Hanley, J. M., Haugen, T. H. & Heath, E. C. (1983) J. Biol. Chem. 258, 7858-7869. [PubMed] [Google Scholar]
- 9.Bowman, B. H. & Kurosky, A. (1982) Adv. Hum. Genet. 12, 189-261. [DOI] [PubMed] [Google Scholar]
- 10.Heinderyckx, M., Jacobs, P. & Bollen, A. (1988) Mol. Biol. Rep. 13, 225-232. [DOI] [PubMed] [Google Scholar]
- 11.Wassler, M. & Fries, E. (1993) J. Cell Biol. 123, 285-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanley, J. M. & Heath, E. C. (1985) Arch. Biochem. Biophys. 239, 404-419. [DOI] [PubMed] [Google Scholar]
- 13.Thomas, G. (2002) Nat. Rev. Mol. Cell Biol. 3, 753-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seidah, N. G. & Chretien, M. (1999) Brain Res. 848, 45-62. [DOI] [PubMed] [Google Scholar]
- 15.Seidah, N. G., Benjannet, S., Wickham, L., Marcinkiewicz, J., Jasmin, S. B., Stifani, S., Basak, A., Prat, A. & Chretien, M. (2003) Proc. Natl. Acad. Sci. USA 100, 928-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naureckiene, S., Ma, L., Sreekumar, K., Purandare, U., Lo, C. F., Huang, Y., Chiang, L. W., Grenier, J. M., Ozenberger, B. A., Jacobsen, J. S., et al. (2003) Arch. Biochem. Biophys. 420, 55-67. [DOI] [PubMed] [Google Scholar]
- 17.Yang, F., Brune, J. L., Baldwin, W. D., Barnett, D. R. & Bowman, B. H. (1983) Proc. Natl. Acad. Sci. USA 80, 5875-5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Circolo, A., Garnier, G. & Volanakis, J. E. (2003) Mol. Immunol. 39, 899-906. [DOI] [PubMed] [Google Scholar]
- 19.Janeway, C., Travers, P., Walport, M. & Shlomchik, M. (2001) Immunobiology: The Immune System in Health & Disease (Garland Publishing, New York).
- 20.Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K. & Pease, L. R. (1989) Gene 77, 51-59. [DOI] [PubMed] [Google Scholar]
- 21.Kaczmarczyk, A., Thuveson, M. & Fries, E. (2002) J. Biol. Chem. 277, 13578-13582. [DOI] [PubMed] [Google Scholar]
- 22.Tosi, M., Duponchel, C., Meo, T. & Couture-Tosi, E. (1989) J. Mol. Biol. 208, 709-714. [DOI] [PubMed] [Google Scholar]
- 23.Dobo, J., Gal, P., Szilagyi, K., Cseh, S., Lorincz, Z., Schumaker, V. N. & Zavodszky, P. (1999) J. Immunol. 162, 1108-1112. [PubMed] [Google Scholar]
- 24.Reboul, A., Bensa, J. C. & Colomb, M. G. (1986) Biochem. J. 233, 559-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arlaud, G. J., Gaboriaud, C., Thielens, N. M. & Rossi, V. (2002) Biochem. Soc. Trans. 30, 1001-1006. [DOI] [PubMed] [Google Scholar]
- 26.Budayova-Spano, M., Grabarse, W., Thielens, N. M., Hillen, H., Lacroix, M., Schmidt, M., Fontecilla-Camps, J. C., Arlaud, G. J. & Gaboriaud, C. (2002) Structure (London) 10, 1509-1519. [DOI] [PubMed] [Google Scholar]
- 27.Misumi, Y., Tanaka, Y. & Ikehara, Y. (1983) Biochem. Biophys. Res. Commun. 114, 729-736. [DOI] [PubMed] [Google Scholar]