

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of severe immune activation with macrophage and T-cell infiltration resulting in, multi organ damage. HLH may be primary or secondary in etiology. A high index of suspicion is essential for early diagnosis and treatment. Diagnostic criteria need to be refined and newer treatment options to be explored in order to improve survival especially in adult HLH and malignancy-associated HLH (M-HLH).
We report a case of malignancy associated HLH (M-HLH) in adult treated on one of the only FDA-approved protocols for adult HLH to highlight the diagnostic and therapeutic challenges of this disease entity.
Keywords: Hemophagocytosis, Lymphohistiocytosis, Ferritin, Adults
1. Background
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of severe immune activation and deregulation characterized by hyperactive macrophages and lymphocytes, pro-inflammatory cytokine hypersecretion, hemophagocytosis, and organ damage. HLH can be primary (genetic) presenting in children [1] or secondary to malignancy, infections and rheumatologic disorders (macrophage activation syndrome) [2], [3], [4]. In addition to a high index of suspicion leading to early diagnosis and intervention there remains a need to refine current diagnostic criteria and treatment options [5]. We report a case of malignancy-associated hemophagocytic lymphohistiocytosis (M-HLH) treated on an FDA-approved protocol to highlight the diagnostic and therapeutic challenges of this disease entity in adults.
2. Clinical case
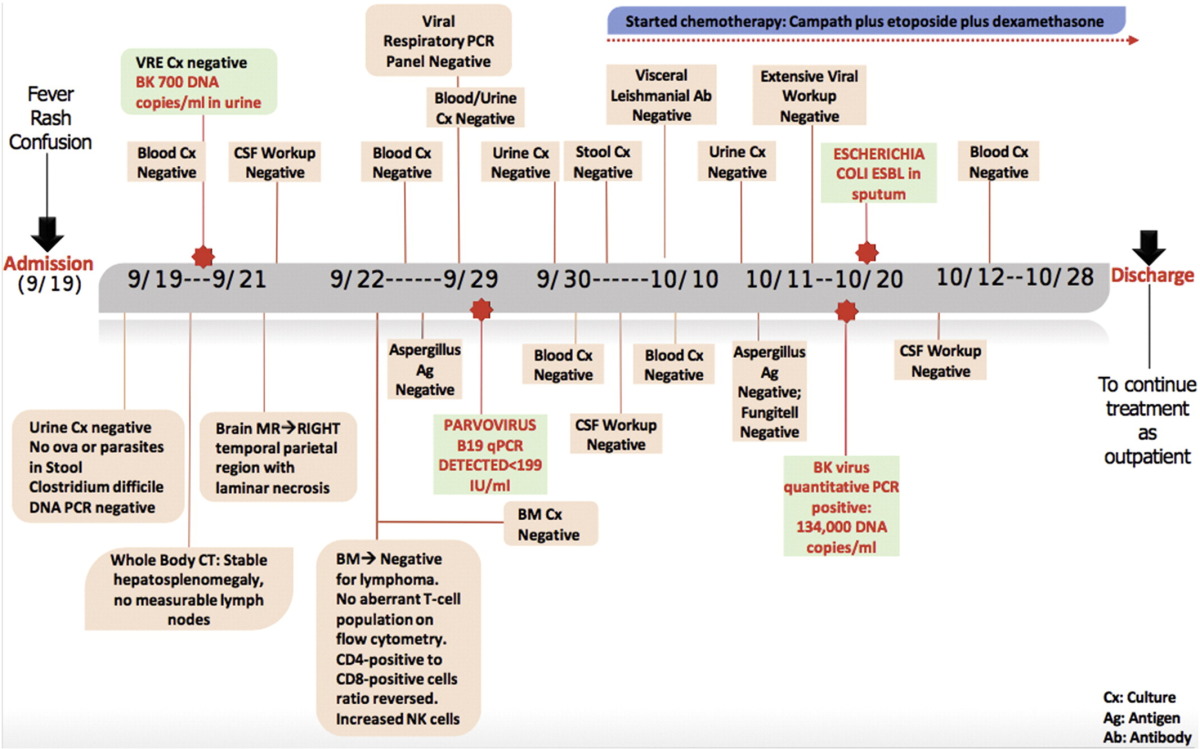
Our patient is a 25-year-old male with history of recurrent hospital admissions since 2010 for a constellation of constitutional symptoms, generalized skin rash, respiratory or gastrointestinal infections indicating potential underlying infectious or an immunological disorder. Despite of extensive laboratory tests and imaging studies done periodically, a definitive diagnosis could not be reached. In February 2015 he presented at UT MD Anderson Cancer Center (UTMDACC) with fever, skin lesions including multiple erythematous and crusted papules, eschar and large necrotic plaques, weight loss and disseminated lymphadenopathy. Axillary lymph node and skin punch biopsies demonstrated a cutaneous T-cell lymphoma with cytotoxic features-gamma-delta positive. PET-CT demonstrated extensive disease in the skin and sub-cutaneous soft-tissue. Bone marrow was not involved by lymphoma. He received 4 cycles of cyclophosphamide, hydroxydaunorubicin, vincristine (Oncovin), etoposide and prednisone (CHOEP) and had partial response with significant decrease in bulk and activity of skin lesions per follow up PET-CT. He progressed approximately one month later with new skin lesions, FDG-avid on PET and was treated with 2 cycles of dexamethasone, high-dose cytarabine, cisplatin (DHAP) chemotherapy with a partial response again per PET. He was supposed to proceed for allogenic stem cell transplant from matched related donor (sister) but developed severe weakness and in September 2015 was admitted with severe fatigue, fever, diarrhea and new onset confusion. He was hypotensive and had decreased oxygen saturation on presentation in ER. Clinical examination revealed a skin rash with tiny, clear, non-pruritic pustules on bilateral upper extremities, hepato-splenomegaly (16 cm spleen on ultrasound). CBC revealed Hb 8.1 g/dL, WBC 0.3 × 109/L, ANC 0.27 × 109/L, platelets 13 × 109/L. Further workup revealed hepatitis (bilirubin 1.5 mg/dL, ALT 156 IU/L, and AST 473 IU/L), pancreatitis (amylase 539 IU/L and lipase 1999 IU/L, disseminated intravascular coagulation fibrinogen 117 mg/dL, prothrombin time 12.3 s (reference range 12.7–15.0), partial thromboplastin time 38.0 s (24.7–35.9), and d-dimer 0.92 μg/mL (0.00–0.40), LDH 4108 IU/L, ferritin 66,027 ng/mL (30–400 ng/mL), and triglycerides 623 mg/dL.
Fig. 1.

(A) Fluid attenuated inversion recovery image demonstrates diffuse white matter signal involving the right temporal, occipital and parietal lobes. There is mass effect and midline shift to the left, with narrowing of the paramesencephalic cistern and compression of the uncus into the right cerebral peduncle. (B) Susceptibility image demonstrates a left anterior capsular/caudate focus of hemorrhage (arrow) with surrounding edema. (C) The equivalent FLAIR image shows surrounding edema.
Fig. 2.

(A) H&E section of clot shows an infiltrate of histiocytes (arrow). (B) Immunohistochemical studies for CD68 demonstrate markedly increased histiocytes in bone marrow. (C) & (D) Aspirate smears demonstrate scattered histiocytic cells containing phagocytized red blood cells.
Fig. 3.

Baseline and on treatment trends of ferritin (A), lactate dehydrogenase (B), fibrinogen (C) and triglycerides (D). Protocol treatment start date was 10/2/15.
Table 1.
Flow diagram.
3. Discussion
Adult HLH is a rare and almost universally fatal disease entity without treatment with published median survival of 1.8–2.2 months. Its incidence is uncertain, as the diagnosis is difficult to make due to poor recognition, low index of suspicion, and lack of definitive diagnostic criteria. Diagnostic and treatment approaches are extrapolations from retrospective databases and clinical trials in childhood HLH. These knowledge gaps have led to under-diagnosis and delayed diagnosis with poor outcomes [3], [4], [6]. Traditional diagnostic criteria (HLH-2004) require one or both of the following: (i) molecular diagnosis consistent with HLH (familial type), (ii) 5 out of 8 diagnostic criteria, including fever, cytopenias (affecting two or more lineages), splenomegaly, hypertriglyceridemia, hypofibrinogenemia, elevated ferritin, elevated IL-2R (soluble CD-25), hemophagocytosis in bone marrow, spleen or lymph nodes, low or absent NK cell activity [7]. Further studies and expert opinions suggest that additional clinical and laboratory features may be useful diagnostic tools. We have developed and validated 18-variable extended diagnostic criteria to identify M-HLH in adults [5]. Adding laboratory variables that are more easily available than in HLH-2004 appeared to be a good surrogate and may allow us to consider a diagnosis of M-HLH at an earlier stage with greater sensitivity.
Adult HLH is frequently a secondary process incited by an underlying inflammatory disorder, infections, immunological disorder or malignancy that may need to be treated concurrently for a favorable outcome. A study of 62 adult HLH patients reported malignancies (52%), infections (34%), autoimmune disorders (8%), idiopathic (6%) as underlying disorders and among malignancies, 59% were T-cell lymphomas and 19% were diffuse large B-cell lymphomas [8]. Another study of 162 adult HLH patients reported malignancy (60%), infections (25%) and autoimmune disorders (3%) as associated conditions with B-cell lymphoma (22%) as the most common malignancy [6]. In our series the most common malignancies associated with M-HLH were acute myeloid leukemia/myelodysplastic syndrome (21%) and T-lymphomas (16%) [5].
Our patient had underlying T-cell cutaneous lymphoma with at least partial response to salvage therapy and a possible immunological disorder in view of his past history, with eventual progression to HLH. The familial type was not considered likely as the patient had no such symptoms till about 20 years of age although it is indeed possible that he had hypomorphic mutations predisposing to HLH. Adult (secondary) HLH has a poor prognosis, left untreated survival ranges from a few days to weeks owing to progressive organ failure with overall mortality ranging from 41%–75% [3]. Because of high morbidity and mortality of this disease rapid diagnosis and treatment is necessary. Given the success of the earlier HLH-94 protocol, the HLH-2004 protocol moved cyclosporine dosing to the beginning of induction and hydrocortisone was added to methotrexate as part of the intrathecal therapy [9], [10]. In the adult HLH protocol at UTMDACC we have added alemtuzumab, a monoclonal antibody directed against CD52 (an antigen expressed on the surface of mature T-, NK-cells and macrophages) to etoposide and dexamethasone. This was based on reports indicating that alemtuzumab as a single agent can be successful as a bridge to allogeneic stem cell transplant in pediatric HLH patients who had failed frontline etoposide, cyclosporine or etoposide with anti-thymocyte globulin (ATG) based standard regimes [11]. We also added intrathecal methotrexate and hydrocortisone upfront in our patient since he had central nervous system involvement. Our patient is one of the first adults to be identified and treated on an FDA approved adult specific HLH protocol in the United States and has responded well. The protocol is open and we have enrolled few more patients at this time. We believe that a non-myelosuppressive and less immunosuppressive therapy would be a better approach to treat M-HLH adult patients who are already immunocompromised and cytopenic. In this group are novel therapies including anti-IL6 based therapies such as tocilizumab that has widely been used for post chimeric antigen receptor (CAR)-T cell cytokine release syndrome and a human anti-interferon gamma (IFN-gamma) monoclonal antibody that has shown outstanding results in primary pediatric HLH. These novel therapies may be better tolerated and improve outcomes in adult patients with M-HLH either in combination with etoposide or ATG and steroid based regimens or in patients who fail such standard regimens and should be evaluated systematically in these scenarios [12].
Conflict of interest
No relevant COI to disclose.
Funding source
This study was conducted following the guidelines of The University of Texas MD Anderson Cancer Center after local IRB approval. It was supported in part by the MD Anderson Cancer Center Support Grant (CCSG) CA016672.
Author contributions
AS, TM, RA, DS, ZV, GT, HK, ND treated the patient, reviewed and acquired the pathologic and radiologic images, participated in the discussion, have reviewed and approved the current version of the manuscript.
Naval Daver is responsible for the overall content as guarantor.
Transparency document
Transparency document.
Acknowledgements
None.
Footnotes
The Transparency document, associated with this article can be found, in online version.
References
- 1.Janka G.E., Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol. Am. Soc. Hematol. Educ. Program. 2013:605–611. doi: 10.1182/asheducation-2013.1.605. [DOI] [PubMed] [Google Scholar]
- 2.Chandrakasan S., Filipovich A.H. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J. Pediatr. 2013;163(5):1253–1259. doi: 10.1016/j.jpeds.2013.06.053. [DOI] [PubMed] [Google Scholar]
- 3.Ramos-Casals M., Brito-Zerón P., López-Guillermo A. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–1516. doi: 10.1016/S0140-6736(13)61048-X. [DOI] [PubMed] [Google Scholar]
- 4.Machaczka M., Vaktnäs J., Klimkowska M., Hägglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk. Lymphoma. 2011;52(4):613–619. doi: 10.3109/10428194.2010.551153. [DOI] [PubMed] [Google Scholar]
- 5.Tamamyan G.H.K., Ning J., Jain P. Malignancy-associated HLH in adults: relation to hemophagocytosis, characteristics, and outcomes. Cancer. 2016;122(18):2857–2866. doi: 10.1002/cncr.30084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivière S., Galicier L., Coppo P., Marzac C., Aumont C., Lambotte O., Fardet L. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am. J. Med. 2014 Nov;127(11):1118–1125. doi: 10.1016/j.amjmed.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 7.Henter J.I., Horne A., Aricó M. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 8.Parikh S.A., Kapoor P., Letendre L. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin. Proc. 2014;89(4):484–492. doi: 10.1016/j.mayocp.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 9.George M.R. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J. Blood Med. 2014;5:69–86. doi: 10.2147/JBM.S46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campo M., Berliner N. Hemophagocytic lymphohistiocytosis in adults. Hematol. Oncol. Clin. North Am. 2015;29(5):915–925. doi: 10.1016/j.hoc.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Jordan M.B., Allen C.E., Weitzman S., Filipovich A.H., McClain K.L. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–4052. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan M.B., Locatelli F., Allen C. A novel targeted approach to the treatment of hemophagocytic lymphohistiocytosis (HLH) with an anti-interferon gamma (IFNγ) monoclonal antibody (mAb), NI-0501: first results from a pilot phase 2 study in children with primary HLH. Blood. 2015;126(23) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.