

Abstract
Background
DNA and RNA fractions from whole blood, serum and plasma are increasingly popular analytes that are currently under investigation for their utility in the diagnosis and staging of disease. Small non-coding ribonucleic acids (sRNAs), specifically microRNAs (miRNAs) and their variant isoforms (isomiRs), and transfer RNA (tRNA)-derived small RNAs (tDRs) comprise a repertoire of molecules particularly promising in this regard.
Results
In this designed study, we compared the performance of various methods and kits for isolating circulating extracellular sRNAs (ex-sRNAs). ex-sRNAs from one healthy individual were isolated using five different isolation kits: Qiagen Circulating Nucleic Acid Kit, ThermoFisher Scientific Ambion TRIzol LS Reagent, Qiagen miRNEasy, QiaSymphony RNA extraction kit and the Exiqon MiRCURY RNA Isolation Kit. Each isolation method was repeated four times. A total of 20 small RNA sequencing (sRNAseq) libraries were constructed, sequenced and compared using a rigorous bioinformatics approach. The Circulating Nucleic Acid Kit had the greatest miRNA isolation variability, but had the lowest isolation variability for other RNA classes (isomiRs, tDRs, and other miscellaneous sRNAs (osRNA). However, the Circulating Nucleic Acid Kit consistently generated the fewest number of reads mapped to the genome, as compared to the best-performing method, Ambion TRIzol, which mapped 10% of the miRNAs, 7.2% of the tDRs and 23.1% of the osRNAs. The other methods performed intermediary, with QiaSymphony mapping 14% of the osRNAs, and miRNEasy mapping 4.6% of the tDRs and 2.9% of the miRNAs, achieving the second best kit performance rating overall.
Conclusions
In summary, each isolation kit displayed different performance characteristics that could be construed as biased or advantageous, depending upon the downstream application and number of samples that require processing.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3470-z) contains supplementary material, which is available to authorized users.
Background
Biomarkers come in many forms—proteins, nucleic acids, metabolites, small molecules—and can be evaluated as indicators of specific metabolic, physiologic, or pathologic states or conditions. Biomarkers have been used in numerous clinical assays to detect the presence or risk of developing disease. Biomarker assays should be conducted from an easily accessible source with minimally invasive medical procedures. One class of molecules, e.g., small RNAs (sRNAs), with great potential for biomarker utility is micro RNAs (miRNAs). miRNAs are 20-25nts in length and are one class of sRNAs that play vital roles in multiple cellular and developmental processes, primarily via post-transcriptional regulation of gene expression [1]. Currently, over 1880 annotated miRNAs (miRBase v21) have been reported in the human transcriptome, targeting >60% of coding genes in the genome [2]. These miRNAs are primarily transcribed by RNA polymerase II either as standalone transcription units or as part of the non-coding intronic sequence within a host gene. They typically function through interactions with Argonaute family proteins, which lead to the formation of a RNA-induced silencing complex (RISC) and suppressed gene expression [3]. At certain stages of the cell cycle, miRNAs have also been reported to assume an activating role in gene expression [4]. Mature miRNAs are also released from cells into circulation, and are therefore detectable in serum [5, 6], plasma [7] and all biological fluids tested. As such, extracellular RNA, particularly ex-sRNAs, have great potential as disease biomarkers in non-invasive assays.
miRNAs are becoming increasingly recognized for their potential to diagnose and stage disease, with cancer being a great example [8–13]. The utility of these miRNAs, in part, is due to their relatively high copy number, stable biochemical properties under clinical conditions, and discriminating transcription that can characterize unique physiological abnormalities. Despite the wide-range of studies that have been conducted to find sRNAs and disease associations, technical challenges continue to deter the utilization of sRNAs in clinical applications. One of the biggest issues for sRNA-based studies is the relatively low concentration of sRNAs present in serum and plasma samples. Currently, there are several miRNA extraction kits that are able to work with low input amounts and extract sRNAs from blood products. Previous sRNA studies [14–16] used a variety of extraction approaches, each with their own advantages and disadvantages. Yet, no consensus exists on the best approach.
Methods for RNA characterization can be classified into two major categories: hybridization-based microarray or synthesis/base-extension-based. Earlier sRNA studies mostly consisted of real-time quantitative polymerase chain reaction (RT-PCR) or hybridization-based assays. However, with the advancement of high-throughput sequencing technology, high-throughput sRNA screening has shifted from hybridization-based microarray technology to sRNAseq technology. One of the most considerable advantages that sRNAseq offers over microarrays is that it does not limit the detection of sRNA to a set of previously known targets. sRNAseq begins by constructing a cDNA sequence library reversely transcribed from short sRNA selected via different methods, e.g., size-selected gel electrophoresis. The prepared, indexed and pooled cDNA library can then be sequenced on different massive parallel sequencing platforms. Subsequent bioinformatic analysis of sRNAseq data provides the identification, quantification and differential expression of sRNAs. Since size-selection is agnostic to sRNA class (excluding potential chemical modifications), it has the potential to capture many species of sRNAs short in length, including miRNAs, miRNA isoforms (isomiRs) [17, 18], transfer RNA (tRNA)-derived small RNAs (tDRs) [19, 20], and other miscellaneous sRNAs (osRNAs) [21, 22]. IsomiRs are the isoforms of miRNA. The isomiRs usually have alternative seed sequences as compared to reference miRNA sequences [23]. The altered seed sequence can cause substantial differences in the repertoire of predicted mRNA targets. tDRs are the product of either active cleavage or an artifact of small RNA library construction. The parent tRNAs are adaptor molecules with a length typically ranging from 73 to 94 nucleotides. It is speculated that the cleavage of tRNAs by an RNAse III enzyme, angiogenin, may occur in a number of reactive conditions to produce tRNA-derived halves (tRHs) [24, 25]. The osRNAs we tried to detect include rRNA, snoRNAs, snRNA, lincRNA, and other miscellaneous sRNAs.
Although sRNA isolation and sRNAseq have primarily been used to quantify miRNAs, it is not completely understood as to what extent RNA isolation and sRNA library kits capture other types of sRNAs. Furthermore, there are many commercially available sRNA extraction kits, and the field would greatly benefit from a carefully designed study to evaluate and compare kit efficiency and reliability. Motivated by these reasons, we benchmarked five popular extraction kits for performance in aiding ex-sRNA analysis.
Methods
Reagents
Five miRNA extraction kits were obtained: Qiagen Circulating Nucleic Acid Kit (NAK), ThermoFisher Scientific Ambion TRIzol LS Reagent, Qiagen miRNEasy, QiaSymphony RNA extraction kit and the Exiqon MiRCURY RNA Isolation Kit. Highly pure diethyl pyrocarbonate (DEPC)-free and nuclease-free water were purchased from Qiagen.
Sample handling
After obtaining informed consent, blood from a single subject was collected in pre-chilled tubes containing ethylenediamine-tetra-acetate and placed on ice. Samples were centrifuged at 3,000 x g for 10 min, aliquoted and stored at -80 °C until further analyses were performed. sRNAs from the serum of a single subject were extracted four times per kit. For each replicate, 200 μl of serum was used. RNA was extracted with the miRNEasy Serum/Plasma Kit, QIAamp Circulating NAK, miRCURY RNA Isolation—BioFluids Kit, and with Ambion TRIzol LS reagent according to manufacturer’s instructions. RNA was extracted on the QIAsymphony SP (Qiagen Corporation, Germany) using the QIAsymphony RNA Kit (Qiagen, 931636) and protocol RNA_CT_400_V7, which incorporates DNase treatment. The resulting RNA was eluted with RNase free water and stored at −80 °C until use. Samples were initially quantified using a Qubit fluorometric RNA assay (Life Technologies, Grand Island, NY). Additional analyses of purity and total RNA quantification were performed using a NanoDrop spectrophotometer (Thermo Scientific) and Agilent RNA 6000 Pico chip (Agilent) according to the manufacturer’s protocol using the reagents, chips, and ladder provided in the kit. The RNA concentration were measured using Qubit. However, since the targets are extracellular miRNA, the concentration were often below the detection threshold. Additionally, we verified the RNA concentration using Agilent Bioanalyzer. The concentrations are reported in Additional file 1: Table S1.
Next-generation small RNA sequencing
RNAseq was performed by the Vanderbilt Technologies for Advanced Genomics core (VANTAGE). Libraries were prepared using the TruSeq Small RNA sample preparation kit (Illumina, San Diego, CA). The sRNA protocol specifically ligates RNA adapters to mature miRNAs harboring a 5’-phosphate and 3’-hydroxyl group as a result of enzymatic cleavage by RNase III processing enzymes, e.g., Dicer. In the first step, RNA adapters were ligated onto each end of the sRNA, and reverse transcription was used to create single-stranded cDNA. This cDNA was then PCR amplified for 18 cycles with a universal primer and a second primer containing one of 20 uniquely indexed tags to allow multiplexing. Size-selection of the cDNA constructs was performed using a 3% gel cassette on the Pippin Prep (Sage Sciences, Beverly, MA) to include only mature miRNAs and other sRNAs in the 5–40 bp size range and to remove adapter-adapter products. The resulting cDNA libraries then underwent a quality check on the Agilent Bioanalyzer HS DNA assay (Agilent, Santa Clara, CA) to confirm the final library size and on the Agilent Mx3005P quantitative PCR machine using the KAPA library quantification kit (Illumina, San Diego, CA) to determine concentration. A 2 nM stock was created, and samples were pooled by molarity for equimolar multiplexing. From the pool, 10 pM of the pool was loaded into each well of the flow cell on the Illumina cBot for cluster generation. The flow cell was then loaded and sequenced on the Illumina NextSeq500 to obtain at least 15 million single end (1x50 bp) reads per sample. The raw sequencing reads in BCL format were processed through CASAVA-1.8.2 for FASTQ conversion and de-multiplexed. The RTA chastity filter was applied, and only PF (pass filter) reads were retained for further analysis.
Bioinformatics and data analyses
We implemented a custom in-house data analysis pipeline [19] for sRNAseq data processing. We categorized ex-sRNAs into four major categories: miRNAs, isomiRs, tDRs, and osRNAs. Cutadapt [26] was used to trim 3’ adapters for raw reads. Multi-perspective quality control [27] on raw data was performed using QC3 [28]. All reads with lengths less than 16nts in length were discarded. The adaptor-trimmed reads were formatted into a non-redundant FASTQ file, where the read sequence and copy number was recorded for each unique tag. The usable unique reads were mapped to the whole genome by Bowtie1 [29] allowing only one mismatch. In addition, our pipeline takes into consideration non-templated nucleotide additions [30–33] at the 3’ end of miRNAs during alignment, resulting in a more accurate miRNA expression quantification. The miRNA coordinates were extracted from miRBase. The tRNA (tDR) coordinates were prepared by combining the latest UCSC tRNA database GtRNAdb [34] with the tRNA loci of mitochondria from the Ensembl database [35]. The osRNA coordinates were extracted from the Ensembl database. The tDR reads were used not only for tDR quantification, but also for tRNA mapping position coverage analysis. ex-sRNAs were divided into three major categories: miRNAs, tDRs, and osRNAs (including sRNAs-derived from parent long non-coding RNAs (lncRNAs), snoRNAs, snRNAs, and miscellaneous RNAs in the Ensembl database). IsomiRs were detected by matching alignment of the reads at +1 or +2 positions from the start of the 5’ annotation of miRNAs.
The map and cluster analysis were performed using Heatmap3 [36] to evaluate the relationship between repeated samples based on the sRNA reads aligned. Furthermore, we computed intra-class correlation (ICC), a statistical measure of the homogeneity between more than two groups [37]. ICC (R package “irr”) was used to assess each kit’s agreement for sRNA expression measured from the replicates. ICC is a numerical value ranging from 0 to 1, where a higher value indicates more agreement among repeats. Additionally, we used Levene’s test [38] (R lawstat package) to assess the quality of variances for the sRNAs detected among repeats. Each kit was also evaluated based on the number of sRNA detected.
Results
The yields of small RNA were measured by Agilent Bioanalyzer and varied greatly (by at least one order of magnitude) (Table S1). We computed the coefficients of variance (CV) of yields within each kit as assessments of the repeatability of yields. MiRCURY kit achieved the lowest CV of 0.04, followed by Ambio TRIzol (CV = 0.29), QiaSymphony (CV = 0.31), Circulating NAK (CV = 1.05) and miRNAEasy had the highest CV of 1.31.
The performance of each kit was first evaluated for sequencing quality and aligned reads. The factors we considered included: total number of reads sequenced, number of reads aligned to each of the sRNA classes, variation in number of reads, multiple alignment issues (reads perfectly mapped to multiple) genomic locations, and unmapped reads (Fig. 1, Additional file 1: Table S2). In regards to the total number of reads sequenced, Circulating NAK produced the most reads with an average of 32.9 million over the four replicates. However, the majority of the reads (83%) in samples isolated by the Circulating NAK were not mapped to the human genome. In contrast, QiaSymphony produced the fewest number of reads with 6.6 million; yet, 37% were mapped to the human genome. Ambion TRIzol yielded 11 million reads with 54% mapping, which was the highest of any method tested. All kits had at least 50% of reads not mapped to human genome which suggested that these reads were not RNA reads. For reads mapped to sRNAs, Ambion TRIzol consistently produced the most reads for miRNAs (23.1%), tDRs (7.2%), and osRNAs (23.1%). Circulating NAK performed poorly for all sRNAs species. Because equal amounts of the sequencing library from each replicate were pooled onto the same lane of Illumina NextSeq500, ideally, the number of reads sequenced for each sample should be roughly equal. However, variability in sequencing depth can be caused by many factors, including sample quality and statistical variation [39]. Since we used 20 technical replicates of the same sample, the observed variation likely reflects the efficiency and native difference of the kits rather than the sample quality. The completed read count table for the four types of sRNA are provided as supplementary data (Additional file 1: Table S3-S6).
Fig. 1.
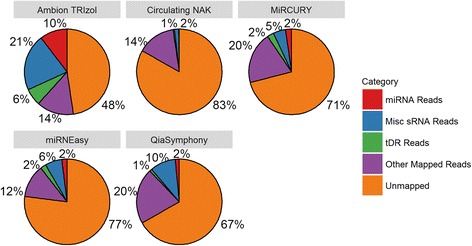
Pie chart that depicts the percentage of number of reads in different alignment categories: miRNA (include isomiR), tDR, osRNA, other mapped reads, unmapped. For any sRNA sequencing project on tissue, the unmapped rate will be at least 50%. The unmapped rate will be even higher for extracellular sRNA sequencing because low sRNA content in serum
We conducted unbiased cluster analysis using Heatmap3 [40] for each of the four major sRNA categories (Fig. 2). The cluster analysis essentially measures the repeatability of each kit, and inherent differences among the kits will cause some variability in the sRNA data. In contrast, replicate analyses of the same kit are expected to perform similarly and tightly cluster. For miRNAs, replicates using Ambion TRIzol, QiaSymphony and Circulating NAK clustered together; for isomiRs, replicates using Circulating NAK clustered together; for tDRs, replicates using Ambion TRIzol and Circulating NAK clustered together; for osRNA, Ambion TRIzol, miRCURY, and QiaSymphony clustered together. Overall, Ambion TRIzol and Circulating NAK produced the best cluster results for replicates.
Fig. 2.
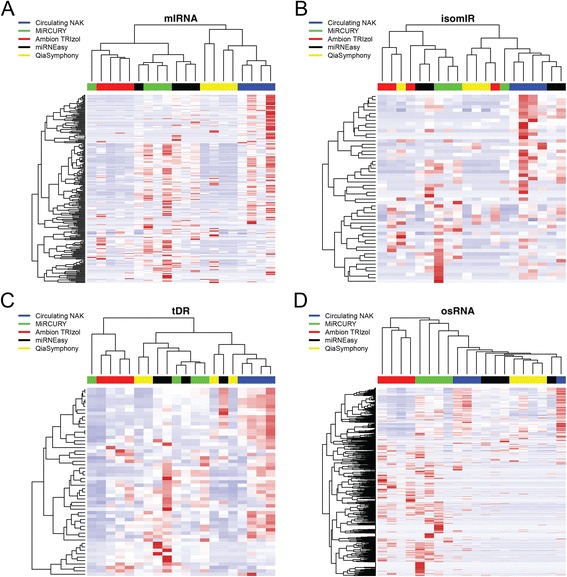
a Cluster and heatmap results for miRNA. b Cluster and heatmap results for isomiRs. c Cluster and heatmap results for detected tRNAs. d Cluster and heatmap results for miscellaneous sRNAs
We used ICC within each kit (based on four repeats per kit) to measure the repeatability of the extraction kits. For miRNAs, QiaSymphony achieved the highest ICC of 0.74; for isomiRs, tDRs, and osRNA, Circulating NAK achieved the highest ICCs of 0.79, 0.97 and 0.96, respectively. We also computed the ICC across all 20 samples to capture the overall homogeneity. It is worth noting that miRNAs had the worst overall ICC of 0.28, followed by isomiRs and osRNA. The mean ICC for tDRs across all isolated methods was 0.9 (Fig. 3, Additional file 1: Table S7). Using Levene’s test we found no significant difference in the equality of variances for the sRNAs detected among repeats (p = 1).
Fig. 3.
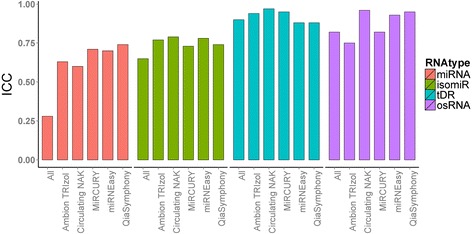
Intra-class correlation coefficient ranges from 0 to 1. Higher number indicates more agreement among replicates
Next, we examined the number of detected sRNAs by each kit (Fig. 4). To determine if a sRNA was detected, we selected several detection thresholds which have been commonly used for assessing sRNA detection (read counts >1, >5, >10, >15, >20) [41]. Circulating NAK consistently detected the greatest number of miRNAs and isomiRs at all detection thresholds. For tDRs, the miRNEasy kit detected the most sRNAs at all thresholds, but Circulating NAK also performed equally well for the lowest and highest detection thresholds. For osRNAs, the miRCURY kit performed the best at all detection thresholds. We also performed the tRNA alignment pattern analysis (5’ end on the left, 3’ end on the right), color coded by anticodon type, which showed some difference in alignment patterns of tDRs among the kits (Fig. 5). For Ambion TRIzol and miRNEasy, a higher percentage of the reads were aligned to parent tRNAs, and Circulating NAK and QiaSymphony had the lowest percentage of reads aligned to parent tRNAs. The read distributions by anticodon type of tRNA were also different among the kits.
Fig. 4.
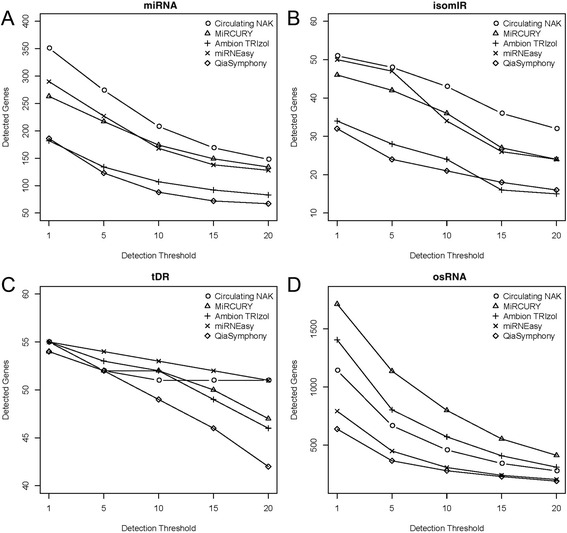
a Number of detected miRNA. b Number of detected isomiR. c Number of detected tRNA. d Number of detected miscellaneous RNA. X-axis denotes the read count threshold used for detection. As the detection threshold increased, less RNAs were identified
Fig. 5.
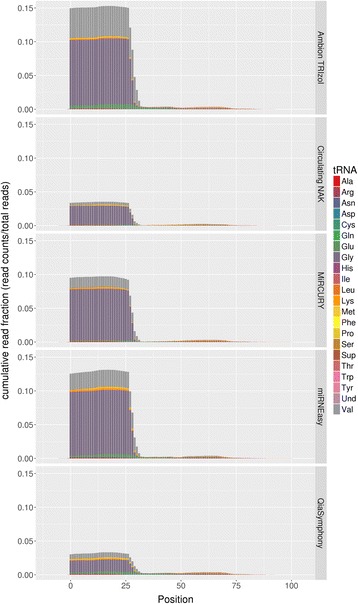
tRNA positional alignment distribution. Color indicates the tRNA by anticodon type. The x-axis denotes the position of tRNA from 0 to the end of tRNA. The y-axis denotes the cumulative read fractions. Visible quantity difference can be seen in the tRNA type detected by different kits. This suggests that there are selection bias of tRNA in by the kits
However, the kit that detects the most sRNAs might also detect the most singleton miRNAs. We define a singleton as a species that is detected by one of the five isolation kits. Circulating NAK detected the most singleton miRNAs, which explains why Circulating NAK also detected the most miRNAs. Very few singleton isomiRs and no singleton tDRs were detected. miRCURY detected the most singleton osRNAs (Fig. 6). A list of the top singleton miRNAs, computed and ranked by the differences between one kit and the other four kits, is available in the Additional file 1: Table S8. Using Levene’s test, we found that the commonly detected sRNAs have no significant amount of difference in level of expression among kits. The presence of singleton sRNA may represent each kit’s uniqueness and can be interpreted as either advantageous or biased. A list of the top other sRNAs, computed and ranked by the differences between one kit and the other four kits, is available in the Additional file 1: Table S9.
Fig. 6.
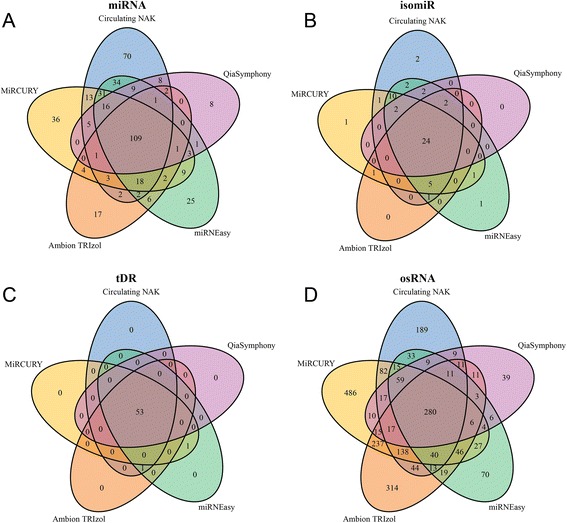
Venn diagrams to represent the intersection and uniquely detected sRNAs, detection threshold used was read count > 10. a The intersection of detected miRNA by all kits. b The intersection of detected isomiRs by all kits. c The intersection of detected tRNA by all kits. d The intersection of detected miscellaneous sRNA by all kits
Discussion
ex-sRNAs have the potential to make a strong impact in the field of biomarker research. The ability to detect ex-sRNA from easily obtainable medium, such as serum or liquid biopsies, can greatly enhance the capability of molecular diagnose [42–44]. A reproducible biomarker also provides valuable insights into specific biological traits at the molecular and cellular level. While having great potential, the analysis of ex-sRNAs from high throughput sequencing pose serious bioinformatics challenges [45]. Currently, one of the difficulties preventing the full fruition of ex-sRNA as a reliable biomarker analysis is the consistency of detection. Another potential issue clouding the future of ex-sRNAs is the obscurity of the origin of these nucleic acids in human serum/plasma. Some arguments have been made that ex-miRNAs can be released into the blood stream directly from blood cells [46] and/or other tissue cells [47].
Our study was not designed to study the origin of sRNAs detected in serum. Instead, our study was motivated by the lack of consensus over the best approach for ex-sRNA isolation. To tackle this problem, we designed a thorough experiment to evaluate the performance of five sRNA isolation kits that have been previously used for isolating ex-miRNAs. Through careful examination of the sequencing data, we can safely conclude that not only were ex-miRNAs detectable in serum, but other species of sRNA, including, ex- isomiRs, ex-tDRs, and ex-osRNAs, were also detectable. We primarily examined the performance of the isolation kits from three aspects: 1) the number of reads sequenced and aligned to each species of sRNA; 2) the repeatability of the replicates within each kit; 3) the number of sRNAs detected. While each kit was used per manufacturer’s instructions, and the amount of nucleic acid input into each replicate for each kit was the same, there were notable kit-specific differences in RNA yield. Each kit designated a different volume of water for elution which is reflected in the concentration and total amount of RNA yielded, and may impart a certain level of bias in our results.
All five kits had poor mapping rate to human genome (<50%), suggesting all kits captured a large amount of miscellaneous material. The phenomenon of high unmapped rates is quite common for high throughput sequencing experiments, however the scale of the unmapped rate depends on the sample quality, type of sequencing, and the abundance of the source RNA. In exome sequencing, it had been shown that the unmapped rate is anywhere from 5 to 19% [48]. For small RNA sequencing on tissue samples, we have previously reported the unmapped rate between 30 and 40% [20]. Our current study focused on ex-sRNAs from serum which is of much less abundance compared to tissue samples. The high unmapped rate is a reflection of the nature of the experiment.
Ambion TRIzol had the most reads annotated as sRNAs; however, the observed increase in genomic alignments did not translate into a higher number of detected sRNAs, suggesting either there was selection bias for Ambion TRIzol or the reads were dominated by miRNAs with high expression. Reproducibility was evaluated using cluster analysis and ICC, and Circulating NAK had the highest repeatability overall. In terms of the number of ex-sRNAs detected, Circulating NAK detected the most ex-osRNAs, ex-isomiRs and also performed well for ex-tDRs. miRCURY detected the highest number of ex-osRNAs. However, currently, ex-osRNAs are the least studied sRNAs and their biological functions remain largely unknown.
In sRNA sequencing, especially extracellular sequencing, the detection threshold of miRNA can significantly affect the number of sRNAs detected. In our study, for demonstration purposes, we have used detection threshold of read counts >1, >5, >10, >15, >20. In reality, sRNA detected with less than 10 read counts are difficult to replicate by RT-PCR. For reliable sRNA detection, it is recommended to set a detection threshold with read count > 10 [49].
Conclusions
In conclusion, our data suggest that each isolation kit displays inherent performance characteristics that may be construed as biased, or advantageous, depending upon the downstream application and number of samples that require processing. The Circulating NAK consistently generated the fewest number of reads mapped to the genome, in comparison to the best performing method, Ambion TRIzol, where 10% of the detected miRNAs, 7.2% of the tDRs and 23.1% of osRNAs were mapped. The performance of the other methods was intermediary, with QiaSymphony mapping 14% of osRNAs and miRNEasy mapping 4.6% of tDRs and 2.9% of miRNAs, making it the second best performing kit in terms of sRNA extraction efficiency. However, the Circulating NAK kit detected the highest number of miRNAs. These data suggest that the choice of sRNA isolation kits for ex-sRNA analysis is not trivial and may introduce significant bias that must be addressed when interpreting outcomes.
Acknowledgements
Special thanks to Tia Hughes and Cara Sutcliffe in the VANTAGE core at Vanderbilt University for expert assistance with RNA isolation and sample quality assessments. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
YG, SZ, QS, ZP were supported by P30 CA68485. YX and CRF were supported by R01 DK105847 (CRF), P30 DK020593 (CRF), and P30 DK058404 (CRF). KCV were supported by NIH awards R01 HL128996, R01 HL127173, and P01 HL116263; as well as American Heart Association award CSA20660001.
Availability of the data and materials
The data used in this study will be uploaded to Gene Expression Omnibus upon acceptance.
Authors’ contributions
YG and CF designed the study. YG, WZ, KV, CF contributed to the writing of the manuscript. YX prepared the sample and extracted RNA. SZ, QS, PZ and YG conducted data analysis. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
NA.
Ethics approval and consent to participate
The subject gave informed written consent before participating in this study, which was approved by the Internal Review Board of Vanderbilt University (090657) and registered at ClinicalTrials.gov (NCT00983463). All studies were conducted in accordance with NIH and institutional guidelines for human subject research. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki, as reflected in a priori approval by Vanderbilt University Medical School.
Abbreviations
- ex
extracellular
- isomiR
Micro RNA isoform
- lncRNA
long non-coding RNAs
- miRNA
micro RNA
- NAK
Nucleic acid kit
- RT-PCR
Quantitative polymerase chain reaction
- sRNA
small RNA
- sRNAseq
small RNA sequencing
- tDR
transfer RNA derived small RNA
- tDR
transfer RNA derived small RNAs
- tRNA
transfer RNA
Additional file
Supplementary Tables S1-S9. (XLSX 576 kb)
Contributor Information
Yan Guo, Email: yan.guo@vanderbilt.edu.
Kasey Vickers, Email: kasey.vickers@vanderbilt.edu.
Yanhua Xiong, Email: yanhua.xiong@vanderbilt.edu.
Shilin Zhao, Email: shilin.zhao@vanderbilt.edu.
Quanhu Sheng, Email: Quanhu.sheng@vanderbilt.edu.
Pan Zhang, Email: pan.zhang@vanderbilt.edu.
Wanding Zhou, Email: zhouwanding@vanderbilt.edu.
Charles R. Flynn, Phone: (615) 343-8329, Email: robb.flynn@vanderbilt.edu
References
- 1.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 2.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–24. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 3.Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and function. Thromb Haemost. 2012;107(4):605–10. doi: 10.1160/TH11-12-0836. [DOI] [PubMed] [Google Scholar]
- 4.Vasudevan S, Tong YC, Steitz JA. Switching from repression to activation: MicroRNAs can up-regulate translation. Science. 2007;318(5858):1931–4. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 5.Fang C, Zhu DX, Dong HJ, Zhou ZJ, Wang YH, Liu L, Fan L, Miao KR, Liu P, Xu W, et al. Serum microRNAs are promising novel biomarkers for diffuse large B cell lymphoma. Ann Hematol. 2012;91(4):553–9. doi: 10.1007/s00277-011-1350-9. [DOI] [PubMed] [Google Scholar]
- 6.Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, Benjamin H, Kushnir M, Cholakh H, Melamed N, et al. Serum microRNAs are promising novel biomarkers. PLoS One. 2008;3(9) doi: 10.1371/journal.pone.0003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, et al. Circulating microRNAs as stable blood-based markers for cancer detection. P Natl Acad Sci USA. 2008;105(30):10513–8. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sai S, Ichikawa D, Tomita H, Ikoma D, Tani N, Ikoma H, Kikuchi S, Fujiwara H, Ueda Y, Otsuji E. Quantification of plasma cell-free DNA in patients with gastric cancer. Anticancer Res. 2007;27(4C):2747–51. [PubMed] [Google Scholar]
- 10.Vinayanuwattikun C, Winayanuwattikun P, Chantranuwat P, Mutirangura A, Sriuranpong V. The impact of non-tumor-derived circulating nucleic acids implicates the prognosis of non-small cell lung cancer. J Cancer Res Clin Oncol. 2013;139(1):67–76. doi: 10.1007/s00432-012-1300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin VY, Siu JM, Cheuk I, Ng EKO, Kwong A. Circulating cell-free miRNAs as biomarker for triple-negative breast cancer. Brit J Cancer. 2015;112(11):1751–9. doi: 10.1038/bjc.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathe A, Scott RJ, Avery-Kiejda KA. miRNAs and other epigenetic changes as biomarkers in triple negative breast cancer. Int J Mol Sci. 2015;16(12):28347–76. doi: 10.3390/ijms161226090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu ZB, Chen X, Zhao Y, Tian T, Jin GF, Shu YQ, Chen YJ, Xu L, Zen K, Zhang CY, et al. Serum MicroRNA signatures identified in a genome-wide serum MicroRNA expression profiling predict survival of Non-small-cell lung cancer. J Clin Oncol. 2010;28(10):1721–6. doi: 10.1200/JCO.2009.24.9342. [DOI] [PubMed] [Google Scholar]
- 14.Page K, Guttery DS, Zahra N, Primrose L, Elshaw SR, Pringle JH, Blighe K, Marchese SD, Hills A, Woodley L, et al. Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0077963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsen AC, Mikkelsen LH, Borup R, Kiss K, Toft PB, von Buchwald C, Coupland SE, Prause JU, Heegaard S. MicroRNA expression profile in conjunctival melanoma. Invest Ophthalmol Vis Sci. 2016;57(10):4205–12. doi: 10.1167/iovs.16-19862. [DOI] [PubMed] [Google Scholar]
- 16.Mraz M, Malinova K, Mayer J, Pospisilova S. MicroRNA isolation and stability in stored RNA samples. Biochem Biophys Res Commun. 2009;390(1):1–4. doi: 10.1016/j.bbrc.2009.09.061. [DOI] [PubMed] [Google Scholar]
- 17.Cloonan N, Wani S, Xu QY, Gu J, Lea K, Heater S, Barbacioru C, Steptoe AL, Martin HC, Nourbakhsh E, et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011;12(12):R126. doi: 10.1186/gb-2011-12-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan GC, Chan E, Molnar A, Sarkar R, Alexieva D, Isa IM, Robinson S, Zhang SC, Ellis P, Langford CF, et al. 5 ’ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014;42(14):9424–35. doi: 10.1093/nar/gku656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Y, Bosompem A, Mohan S, Erdogan B, Ye F, Vickers KC, Sheng QH, Zhao SL, Li CI, Su PF, et al. Transfer RNA detection by small RNA deep sequencing and disease association with myelodysplastic syndromes. BMC Genomics. 2015;16. [DOI] [PMC free article] [PubMed]
- 20.Guo Y, Xiong Y, Sheng Q, Zhao S, Wattacheril J, Flynn CR. A micro-RNA expression signature for human NAFLD progression. J Gastroenterol. 2016;51(10):1022–30. doi: 10.1007/s00535-016-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vickers KC, Roteta LA, Hucheson-Dilks H, Han L, Guo Y. Mining diverse small RNA species in the deep transcriptome. Trends Biochem Sci. 2015;40(1):4–7. doi: 10.1016/j.tibs.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han L, Vickers KC, Samuels DC, Guo Y. Alternative applications for distinct RNA sequencing strategies. Brief Bioinform. 2014. [DOI] [PMC free article] [PubMed]
- 23.Morin RD, O’Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao YJ, McDonald H, Zeng T, Hirst M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells (vol 18, pg 610, 2008) Genome Res. 2009;19(5):958. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Silva MR, Cabrera-Cabrera F, Guida MC, Cayota A. Hints of tRNA-derived small RNAs role in RNA silencing mechanisms. Genes (Basel) 2012;3(4):603–14. doi: 10.3390/genes3040603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu H, Feng J, Liu Q, Sun F, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009;583(2):437–42. doi: 10.1016/j.febslet.2008.12.043. [DOI] [PubMed] [Google Scholar]
- 26.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–2. [Google Scholar]
- 27.Sheng Q, Vickers K, Zhao S, Wang J, Samuels DC, Koues O, Shyr Y, Guo Y. Multi-perspective quality control of Illumina RNA sequencing data analysis. Brief Funct Genomics. 2016. [DOI] [PMC free article] [PubMed]
- 28.Guo Y, Zhao S, Sheng Q, Ye F, Li J, Lehmann B, Pietenpol J, Samuels DC, Shyr Y. Multi-perspective quality control of Illumina exome sequencing data using QC3. Genomics. 2014;103(5-6):323–8. doi: 10.1016/j.ygeno.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res. 2011;21(2):203–15. doi: 10.1101/gr.116657.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Gene Dev. 2006;20(24):3407–25. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westholm JO, Ladewig E, Okamura K, Robine N, Lai EC. Common and distinct patterns of terminal modifications to mirtrons and canonical microRNAs. RNA. 2012;18(2):177–92. doi: 10.1261/rna.030627.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23(11):1635–48. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 34.Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37(Database issue):D93–7. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. Ensembl 2014. Nucleic Acids Res. 2014;42(Database issue):D749–55. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao S, Guo Y, Sheng Q, Shyr Y. Advanced heat map and clustering analysis using heatmap3. Biomed Res Int. 2014;2014:986048. doi: 10.1155/2014/986048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nam JM. Homogeneity score test for the intraclass version of the kappa statistics and sample-size determination in multiple or stratified studies. Biometrics. 2003;59(4):1027–35. doi: 10.1111/j.0006-341X.2003.00118.x. [DOI] [PubMed] [Google Scholar]
- 38.Levene H. Contributions to probability and statistics: essays in honor of Harold hotelling. Palo Alto: Stanford University Press; 1960. [Google Scholar]
- 39.Guo Y, Cai Q, Li C, Li J, Courtney R, Zheng W, Long J. An evaluation of allele frequency estimation accuracy using pooled sequencing data. Int J Comput Biol Drug Des. 2013;6(4):279–93. doi: 10.1504/IJCBDD.2013.056709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao SL, Guo Y, Sheng QH, Shyr Y. Advanced heat Map and clustering analysis using Heatmap3. Biomed Res Int. 2014. [DOI] [PMC free article] [PubMed]
- 41.Guo Y, Zhao SL, Sheng QH, Guo MS, Lehmann B, Pietenpol J, Samuels DC, Shyr Y. RNAseq by total RNA library identifies additional RNAs compared to poly(A) RNA library. Biomed Res Int. 2015. [DOI] [PMC free article] [PubMed]
- 42.Karachaliou N, Mayo-de-Las-Casas C, Molina-Vila MA, Rosell R. Real-time liquid biopsies become a reality in cancer treatment. Ann Transl Med. 2015;3(3):36. doi: 10.3978/j.issn.2305-5839.2015.01.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10(8):472–84. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 44.Alix-Panabieres C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem. 2013;59(1):110–8. doi: 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- 45.Buschmann D, Haberberger A, Kirchner B, Spornraft M, Riedmaier I, Schelling G, Pfaffl MW. Toward reliable biomarker signatures in the age of liquid biopsies - how to standardize the small RNA-Seq workflow. Nucleic Acids Res. 2016;44(13):5995–6018. doi: 10.1093/nar/gkw545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chin LJ, Slack FJ. A truth serum for cancer - microRNAs have major potential as cancer biomarkers. Cell Res. 2008;18(10):983–4. doi: 10.1038/cr.2008.290. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, Guo J, Zhang Y, Chen J, Guo X, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 48.Guo Y, Long J, He J, Li CI, Cai Q, Shu XO, Zheng W, Li C. Exome sequencing generates high quality data in non-target regions. BMC Genomics. 2012;13:194. doi: 10.1186/1471-2164-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopez JP, Diallo A, Cruceanu C, Fiori LM, Laboissiere S, Guillet I, Fontaine J, Ragoussis J, Benes V, Turecki G, et al. Biomarker discovery: quantification of microRNAs and other small non-coding RNAs using next generation sequencing. BMC Med Genet. 2015;8. [DOI] [PMC free article] [PubMed]