

Abstract
Lutibacter profundi LP1T within the family Flavobacteriaceae was isolated from a biofilm growing on the surface of a black smoker chimney at the Loki’s Castle vent field, located on the Arctic Mid-Ocean Ridge. The complete genome of L. profundi LP1T is the first genome to be published within the genus Lutibacter. L. profundi LP1T consists of a single 2,966,978 bp circular chromosome with a GC content of 29.8%. The genome comprises 2,537 protein-coding genes, 40 tRNA species and 2 rRNA operons. The microaerophilic, organotrophic isolate contains genes for all central carbohydrate metabolic pathways. However, genes for the oxidative branch of the pentose-phosphate-pathway, the glyoxylate shunt of the tricarboxylic acid cycle and the ATP citrate lyase for reverse TCA are not present. L. profundi LP1T utilizes starch, sucrose and diverse proteinous carbon sources. In accordance, the genome harbours 130 proteases and 104 carbohydrate-active enzymes, indicating a specialization in degrading organic matter. Among a small arsenal of 24 glycosyl hydrolases, which offer the possibility to hydrolyse diverse poly- and oligosaccharides, a starch utilization cluster was identified. Furthermore, a variety of enzymes may be secreted via T9SS and contribute to the hydrolytic variety of the microorganism. Genes for gliding motility are present, which may enable the bacteria to move within the biofilm. A substantial number of genes encoding for extracellular polysaccharide synthesis pathways, curli fibres and attachment to surfaces could mediate adhesion in the biofilm and may contribute to the biofilm formation. In addition to aerobic respiration, the complete denitrification pathway and genes for sulphide oxidation e.g. sulphide:quinone reductase are present in the genome. sulphide:quinone reductase and denitrification may serve as detoxification systems allowing L. profundi LP1T to thrive in a sulphide and nitrate enriched environment. The information gained from the genome gives a greater insight in the functional role of L. profundi LP1T in the biofilm and its adaption strategy in an extreme environment.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-016-0219-x) contains supplementary material, which is available to authorized users.
Keywords: Lutibacter, Flavobacteriaceae, Loki’s castle, Biofilm, Deep-sea hydrothermal vent
Introduction
The type strain Lutibacter profundi LP1T (=DSM 100437 T =JCM 30585 T) belongs to the family Flavobacteriaceae within the phylum Bacteroidetes [1]. Members of this family are abundant in marine and freshwater habitats and have been isolated from seawater [2, 3], sea ice [4], fresh water [5], glaciers [6, 7], marine plants and animals [4, 8]. In addition, metagenomic studies have shown the presence of Bacteroidetes in marine sediments [9, 10]. Members of the Flavobacteriaceae are also found in the human microbiota [11, 12], soil [13], insects [14], food and dairy products [15]. The family Flavobacteriaceae has been proposed to play an important role in the degradation of organic matter and nutrition turnover in the oceans [16]. They have been identified either as free-living or attached to organic detritus particles and phytoplankton in marine surfaces [17, 18] and in deep-sea planktonic communities [19]. Biopolymers, such as cellulose, chitin and proteins are part of the high molecular mass fraction of (dissolved) organic material in aquatic habitats. The ability to degrade such polymers has been shown for Flavobacteriaceae in both culture-dependent and independent studies [16, 20]. A multiplicity of strains has been isolated and several genomes sequenced [21–23]. Genomic analyses of marine isolates have revealed a large number of GHs, GTs, peptidases and adhesion proteins, as well as genes for gliding motility, supporting an organotrophic life style as HMW organic matter degraders [21–24].
In 2006 the first Lutibacter strain, L. litoralis CL-TF09T, was isolated and introduced as a new organotrophic genus of the Flavobacteriaceae family [25]. Until now, all published strains have been isolated from Korean coastal waters or the Sea of Japan, and found either as free-living or in association with invertebrates [3, 25–31]. In contrast, L. profundi LP1T was isolated from a biofilm attached to the outer surface of a black smoker chimney from the LCVF located at the AMOR [1]. In the biofilm, the Bacteroidetes population was attached as ectobionts on the outer surface of filamentous Epsilonproteobacteria [32]. Here we present the complete genome of Lutibacter profundi LP1T, the first genome to be published from the genus Lutibacter. The genomic features of L. profundi are presented and its possible role in the biofilm community and its biotechnological potential is discussed.
Organism Information
The isolation and characterization of L. profundi LP1T has previously been described [1]. Thus, the organism information will be given as a short summary supplemented with additional information.
Classification and features
L. profundi LP1T was isolated from a biofilm attached to the surface of a black smoker chimney wall at the LCVF, on the AMOR [32–34]. A steep temperature gradient between the up to 320°C hydrothermal fluids and the −0.7°C cold surrounding seawater places the biofilm in a mesophilic temperature range [33, 35]. Artificial seawater medium [36] supplemented with modified Wolfe’s mineral solution without NaCl or CaCl2 (0.001%), Wolfe’s vitamin solution (0.5%), 10mM Na2S and yeast extract (0.01%) under microaerobic conditions was used for primary enrichments and isolation of L. profundi LP1T [1].
The genus Lutibacter , including L. profundi LP1T, thus far comprise nine strains which are proposed to represent novel species: L. litoralis CL-TF09T [25], L. maritimus S7-2T [26], L. aestuarii MA-My1T [27], L. flavus T [29], L. agarilyticus KYW566T [28], L. oricola UDC377T [3], L. crassostreae TYO-8T [30] and L. holmesii KMM 6277 T [31]. The strain L. crassostreae TYO-8T was isolated from an oyster collected from the South Sea, South Korea [30], whereas L. holmesii KMM 6277 T was isolated from an sea urchin collected from Troitas Bay, Sea of Japan [31]. The other species were isolated from shallow coastal waters or tidal areas around the coast of South Korea [3, 25–29]. So far, L. profundi LP1T is the only Lutibacter strain isolated outside of South Korean Territory. L. profundi LP1T shared between 94.7% (L. maritimus S7-2T) and 97.5% (L. holmesii KMM 6277 T) 16S rRNA gene identity with the other Lutibacter strains. 16S rRNA phylogenetic analysis placed strain LP1T closest to L. agarilyticus KYW566T and L. holmesii KMM 6277 T within the Lutibacter group, as previously described (Fig. 1) [1].
Fig. 1.
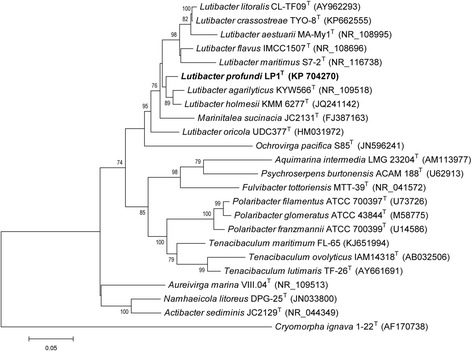
Phylogenetic tree displays the position of Lutibacter profundi LP1T (shown in bold) relative to the other type strains of Lutibacter based on 16S rRNA. The phylogenetic tree was generated after trimming the alignment using MUSCLE [82, 83] to 1323 aligned positions, using maximum likelihood method with general time reversible model as preferred model incorporated in MEGA v. 6.06 [84]. At the branch points bootstrap values above 70, expressed as percentage of 1000 replicates, are shown. Bar: 0.05 substitutions per nucleotide position. Cryomorpha ignava 1-22T (AF170738) was used as outgroup
L. profundi LP1T was described as Gram-negative, microaerophilic, non-motile rods [1] (Fig. 2a). L. profundi LP1T grew in a temperature range between 13 and 34 °C with an optimum of 23 °C, a pH range between 5.5 and 7.5 with pH 6–6.5 as optimum [1]. L. profundi LP1T grew in NaCl concentrations ranging from 1 to 3%, with an optimal concentration of 2% (Table 1). However, the strain was not able to grow with NaCl as the sole source of salt. No growth was observed under fermentative or anaerobic conditions using NO3 − and S2O3 2− as electron acceptors. Nevertheless, nitrate was reduced to nitrite under anaerobic and microaerophilic conditions. L. profundi LP1T was tested positive for oxidase and catalase activity [1]. Using the API ZYM system (BioMérieux, France), L. profundi LP1T showed strong activity for alkaline phosphatase, leucine arylamidase, valine arylamidase, trypsin, acid phosphatase, naphtol-AS-BI-phosphohydrolase and N-acetyl-Beta-glucosaminidase, as well as weak activity for esterase lipase and alpha-glucosidase. Following carbon sources were utilized in an AN microplate™ (Biolog, USA) test: pyruvic acid, L-alanyl-L-glutamine, L-alanyl-L-threonine, L-glutamic acid, glycyl-L-proline and L-threonine, in addition to L-proline, L-glutamate and pyruvate with 0.01% extra YE [1]. Furthermore, L. profundi LP1T was able to grow on D-sucrose supplemented with 0.01% yeast extract, but not on D-glucose, D-fructose, D-cellobiose and D-galactose. L. profundi LP1T did not utilize glycerol, citrate, succinate, L-leucine and tartrate supplemented with yeast extract [1]. L. profundi LP1T was able to hydrolyse gelatin, casein, starch and indoxyl acetate, but not agar, cellulose, urea, esculine, lecithin, tween 80 or tween 20. Cells were resistant to streptomycin, however susceptible to ampicillin, penicillin, erythromycin, tetracyclin and chloramphenicol.
Fig. 2.

Scanning electron microscopy of Lutibacter profundi LP1T. a Normal cultivation conditions, b Oxygen stress (atmospheric oxygen level)
Table 1.
Classification and general features of Lutibacter profundi LP1T according to MIGS standards [39]
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [75] | |
| Phylum Bacteroidetes | TAS [76, 77] | ||
| Class Flavobacteriia | TAS [78] | ||
| Order Flavobacteriales | TAS [77, 79] | ||
| Family Flavobacteriaceae | TAS [80] | ||
| Genus Lutibacter | TAS [25, 27] | ||
| Species Lutibacter profundi | TAS [1] | ||
| Type strain: LP1 (DSMZ 100437T = T) | TAS [1] | ||
| Gram stain | Gram-negative | TAS [1] | |
| Cell shape | Rod | TAS [1] | |
| Motility | Non-motile | TAS [1] | |
| Sporulation | no | TAS [1] | |
| Temperature range | 13–34°C | TAS [1] | |
| Optimum Temperature | 23°C | TAS [1] | |
| pH range; optimum | 5.2–7.5; 6.2 | TAS [1] | |
| Carbon sources | tryptone | TAS [1] | |
| MIGS-6 | Habitat | Marine, biofilm attached to black smoker chimney | TAS [1] |
| MIGS-6.3 | Salinity | 1–3% | TAS [1] |
| MIGS-22 | Oxygen requirement | Microaerobic, aerobic | TAS [1] |
| MIGS-15 | Biotic relationship | Free-living | TAS [1] |
| MIGS-14 | Pathogenicity | Non-pathogen | NAS |
| MIGS-4 | Geographic location | Loki’s Castle, Arctic mid-Ocean ridge | TAS [33] |
| MIGS-5 | Sample collection | Summer 2009 | TAS [32, 34] |
| MIGS-4.1 | Latitude | 73.33.97N, | TAS [1, 32, 34] |
| MIGS-4.2 | Longitude | 08.09.51E | TAS [1, 32, 34] |
| MIGS-4.4 | Altitude | −2350m | TAS [1, 32, 34] |
Evidence codes – IDA inferred from direct assay, TAS traceable author statement (i.e., a direct report exists in the literature), NAS non-traceable author statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [81]
In the current study, L. profundi LP1T tested negative for the utilization of the following additional carbohydrates; D-maltose, D-mannose, L-arabinose, D-trehalose, D-xylose, D-cellulose and chitin.
Chemotaxonomic data
The composition of the major cellular fatty acids in L. profundi LP1T varies depending on the used media and growth condition [1]. After growth on marine broth 2216 agar plates the major cellular fatty acids are iso-C15:0 (25.2%), iso-C15:0 3-OH (14.5%), iso-C17:0 3-OH (9.6%), iso-C15:1 (G) (9.0%), anteiso-C15:0 (8.2%), iso-C16:0 3-OH (5.4%) and summed feature I iso-C15:1 (H)/C13:0 3OH (7.4%) [1]. The major cellular fatty acid composition varied between the different Lutibacter type strains [1]. The major polar lipids of L. profundi LP1T are DPG, PE, one unidentified aminolipid and two unidentified lipids, where PE is the main polar lipid. In accordance with the genus, menaquinone-6 (MK-6) is the only respiratory quinone [1].
Genome sequencing information
Genome project history
L. profundi LP1T as the type strain is the first Lutibacter isolate from a deep-sea hydrothermal vent system. The bacterium was chosen for sequencing to study its genomic features in relation to the environmental system it originated from and its biotechnological potential.
Sequencing was conducted at NSC, Norway [37]. Assembly, finishing and polishing steps were performed at the Centre for Geobiology, University of Bergen, Norway. To fulfil NCBI standards the annotation of the genome was performed using the automatic NCBI PGAAP [38]. The complete genome sequence and annotation data of L. profundi LP1T is accessible in GenBank under the accession number CP013355. The project information and its association with MIGS version 2.0 compliance [39] have been summarized in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Pacific Biosciences 10 kb library |
| MIGS-29 | Sequencing platform | PacBio |
| MIGS-31.2 | Fold coverage | 76x |
| MIGS-30 | Assemblers | Hierarchical Genome Assembly Process (HGAP) v2 |
| MIGS-32 | Gene calling method | Prodigal |
| Locus Tag | Lupro | |
| Genbank ID | CP013355 | |
| Genbank Date of Release | February 1., 2016 | |
| GOLD ID | Gp0134121 | |
| BIOPROJECT | PRJNA304382 | |
| MIGS-13 | Source Material Identifier | DSMZ 100437T =JCM 30585T |
| Project relevance | Environmental |
Growth conditions and genomic DNA preparation
L. profundi LP1T was grown by gently shaking in M1 broth medium at microaerophilic conditions and 23 °C. The high molecular DNA of a 60 ml culture was isolated using a modified method of Marmur [40, 41].
Genome sequencing and assembly
A 10 kb library was prepared using Pacific Bioscience 10 kb library preparation protocol and BluePippin (Sage Science) for the final size selection. Two SMRT cells were used for sequencing the library on a Pacific Bioscience RS II instrument in combination with the P4-C2 chemistry. In total, 63,994 reads with an average length of 5671 bp were obtained generating a total number of 362.9 Mbp. The raw reads were filtered prior de novo assembly using HGAP v2 (Pacific Bioscience) [42], which resulted in one 2,978,418 bp contig with an average coverage of 76.29. Using the Gepard dotplot [43], verified a single highly accurate self-overlapping contig. Minimus2 from the AMOS software package [44] was used to perform the circularization and trimming of the chromosomal contig. Final polishing steps using the RS_Resequencing protocol implemented in the SMRT Analysis software (Pacific Biosciences), resulted in a 2,966,978 bp circular chromosome with a consensus concordance of 99.9%. The location of the dnaA gene was manually relocated and used as start of the chromosome.
Genome annotation
In order to comply to NCBI standards, the annotation of the genome was performed using the automatic NCBI PGAAP [38]. In addition, SignalP and TMHH-plugins in CLC Genomics Workbench (Qiagen, version 9) was used for the identification of genes with signal peptides and transmembrane helices, respectively.
Genome properties
The circular genome of L. profundi LP1T consists of 2,966,978 bp with a GC content of 29.8%. The chromosome comprises 6 rRNAs located in two operons, 40 tRNAs and one ncRNA (Table 3). The two 16S rRNA genes are identical in DNA sequence. Of 2537 predicted protein-coding genes 1531 were assigned to a putative function and 1006 as hypothetical proteins. In total 96.3% of protein-coding genes were assigned to COG functional categories summarized in Table 4. A Circos [45] genome atlas is presented in Fig. 3. The MEROPS peptidase database [46] and dbCAN [47] were used for identification of peptidases and carbohydrate-degrading enzymes. Identification of conserved domains using the NCBI Batch web CD-Search Tool [48] complemented the analysis. A putative episome of 89 genes is located inside the genome (10.15 kb: 608003-709593) including several plasmid stabilization genes and hypothetical genes.
Table 3.
Genome statistics
| Attribute | Value | Percent of total |
|---|---|---|
| Genome size (bp) | 2,966,978 | 100.00 |
| DNA coding (bp) | 2,681,332 | 90.4 |
| DNA G + C (bp) | 815,201 | 27.5 |
| DNA scaffolds | 1 | |
| Total genes | 2,611 | 100 |
| Protein coding genes | 2,537 | 97.2 |
| RNA genes | 47 | 1.8 |
| Pseudo genes | 27 | 1 |
| Genes in internal clusters | ND | |
| Genes with function prediction | 1,531 | 58.6 |
| Genes assigned to COGs | 2,447 | 96.3 |
| Genes with Pfam domains | 2,092 | 82.2 |
| Genes with signal peptides | 219 | 8.4 |
| Genes with transmembrane helices | 617 | 23.6 |
| CRISPR repeats | 2 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | Percent agea | Description |
|---|---|---|---|
| J | 131 | 5,2 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0 | RNA processing and modification |
| K | 88 | 3,5 | Transcription |
| L | 105 | 4,1 | Replication, recombination and repair |
| B | 0 | 0 | Chromatin structure and dynamics |
| D | 17 | 0,7 | Cell cycle control, cell division, chromosome partitioning |
| V | 35 | 1,4 | Defence mechanisms |
| T | 67 | 2,6 | Signal transduction mechanisms |
| M | 160 | 6,3 | Cell wall/membrane/envelope biogenesis |
| N | 3 | 0,1 | Cell motility |
| U | 21 | 0,8 | Intracellular trafficking, secretion, and vesicular transport |
| O | 92 | 3,6 | Posttranslational modification, protein turnover, chaperones |
| C | 161 | 6,3 | Energy production and conversion |
| G | 55 | 2,2 | Carbohydrate transport and metabolism |
| E | 179 | 7,1 | Amino acid transport and metabolism |
| F | 63 | 2,5 | Nucleotide transport and metabolism |
| H | 69 | 2,7 | Coenzyme transport and metabolism |
| I | 57 | 2,2 | Lipid transport and metabolism |
| P | 107 | 4,2 | Inorganic ion transport and metabolism |
| Q | 12 | 0,5 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 0 | 0 | General function prediction only |
| S | 1025 | 40,4 | Function unknown |
| - | 94 | 3,7 | Not in COGs |
athe total is based on the number of protein coding genes in the annotated genome
Fig. 3.
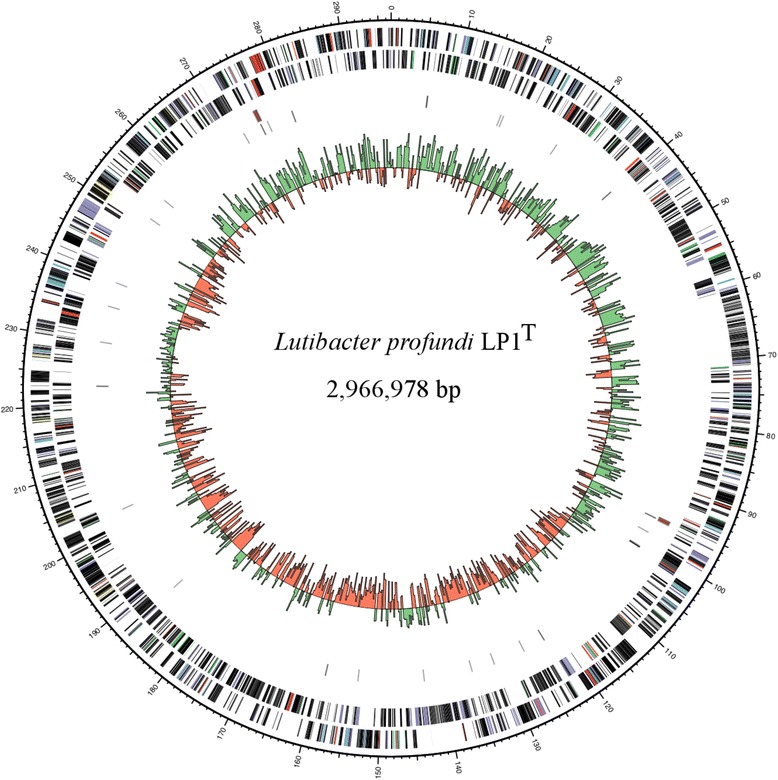
Circular representation of the Lutibacter profundi LP1T genome displaying relevant genome features. Circles representing the following (from centre to outside): 1, G + C skew [(G – C)/(G + C) using a 2-kbp sliding window] (green, positive G + C skew; red, negative G + C skew); 2, tRNAs (black); 3, rRNA operons (red); 4, Coding DNA sequence on the reverse strand; 6, CDS on the forward strand. Colour coding of CDS was based on COG categories. The figure was build using Circos version. 0.67–6 [45]
Insights from the genome sequence
In addition to the automatic genome annotation by PGAAP, KAAS [49] was used to analyse metabolic features of the strain LP1T. The L. profundi LP1T genome encodes for all central carbohydrate metabolic pathways (Additional file 1: Table S1); Embden-Meyerhof-Parnas pathway, gluconeogenesis and the TCA cycle. The genome contains genes for the non-oxidative branch of the pentose-phosphate-pathway, however misses the genes for the oxidative branch. Genes for the glyoxylate shunt of the TCA cycle are not present. The key enzyme ATP citrate lyase (EC 2.3.3.8) of the rTCA was not found. Besides the pyruvate dehydrogenase complex, a pyruvate:ferredoxin oxidoreductase (Lupro_00440) was identified, which may also catalyse the reverse reaction from acetyl-CoA to pyruvate. L. profundi LP1T harbours the gene for phosphoenolpyruvate carboxylase (Lupro_02180), which may convert phosphoenolpyruvate into oxaloacetate, fixing CO2 in an anaplerotic reaction [22, 23]. Genes for energy generation via oxidative phosphorylation were identified (Additional file 1: Table S1). The major components comprise the NADH-dehydrogenase complex I, the succinate dehydrogenase/fumarate reductase complex II, a variety of quinone, and cytochrome c terminal oxidoreductases. Energy generation in form of ATP could be provided by the encoded F0F1-type ATP synthase. In addition to a H(+)-translocating NADH-dehydrogenase complex, a Na(+)-translocating NADH-quinone reductase is encoded in the genome, a feature common in marine bacteria [50]. Different aerobic terminal oxidoreductases could be identified, such as cytochrome c oxidases, cytochrome bo3 ubiquinol oxidase, cbb3-type cytochrome c oxidase and quinol oxidizing cytochrome bd-I terminal oxidase.
All genes for the complete denitrification pathway, from nitrate to nitrogen (NapAB, NirS, NorBC, NosZ), were identified in the strain LP1T (Additional file 1: Table S1). Nitrate reduction to nitrite was confirmed in growth experiments under aerobic and microaerophilic conditions, while anaerobic growth using nitrate as the sole electron acceptor was not observed [1]. One ammonium transporter (Lupro_05500) was detected for ammonia assimilation. Ammonia can be fixed indirectly by glutamine synthetase and GOGAT, or directly by NADP-dependent glutamate dehydrogenase forming glutamate. Two different forms of GOGAT were identified, a NADPH dependent and a ferredoxin-dependent. The absence of genes encoding for urease is in concordance with the phenotypic characterization [1]. Genes for oxidation of sulphide, SQR, polysulfide reductase and sulphate permease, were identified in the genome of L. profundi LP1T (Additional file 1: Table S1). However, growth of L. profundi LP1T was not stimulated in the presence of thiosulfate under microaerobic or anaerobic conditions [1]. The presence of a SQR could also be an adaptation to the elevated concentration of sulphide emitted from the vent fluids at LCVF, rather than growth.
Potential role of Lutibacter profundi LP1T as complex organic compound degrader in the deep-sea biofilm
The organotroph L. profundi LP1T was isolated from a microbial biofilm where a Bacteroidetes population was found attached to filamentous Epsilonproteobacteria producing a sugar biopolymer resembling chitin or cellulose [32]. The dbCAN analysis detected 101 proteins exhibiting one or more functional activities within CAZy [51, 52]. GTs (45) are mainly represented, followed by GHs (24), CEs (24), PLs (1) and CBMs (7). Ten GH families (Additional file 2: Table S2) are found in the genome, whereof GH13 and GH74 represent half of the enzymes. Diverse GH13 hydrolases, partially located in a Sus, cluster enable the bacterium to utilize starch. Characterization of L. profundi LP1T has shown its ability to grow on starch and sucrose as the single C-source [1]. The strain also has the ability to catabolise monosaccharides such as mannose-6P, fructose-6P and glucose, as well as the disaccharides maltose, sucrose and trehalose. A sugar kinase (Lupro_07775) could activate monosaccharides such as mannose and fructose by phosphorylation [53]. The ability of the strain LP1T to degrade starch [1] was supported by the presence of a Sus (Lupro_12175-Lupro_12250). Additional, two other SusD proteins (Lupro_05305 and Lupro_02600) and three signal peptide containing proteins (Lupro_10330, Lurpo_05115 and Lurpo_05135), described as ‘Starch-binding associating with outer membrane’, were found in the genome adjacent to TonB-linked outer membrane transporter proteins. These proteins harbour a SusD-like_2 domain and facilitate extracellular starch-binding, while being associated to the outer membrane with an N-terminal lipid tail [54]. For the polysaccharide degradation specialist Bacteroides thetaiotaomicron, SusC and SusD alone account for ~60% of the polysaccharide-degrading ability [55]. Furthermore, a gene for a bacterial glycogen synthase (Lupro_08100) was found in the L. profundi LP1T genome that would allow energy conservation in form of glycogen.
Conserved domain [48] prediction revealed a possible neuraminidase/sialidase function for the GH74 hydrolases, alongside with a general function for β-1,4-linked glucan hydrolase activity for this family based on CAZypedia [56]. Bacterial sialidases are involved in the removal of sialic acid from various glycoconjugates [57] and are so far classified in the GH families 33 and 58 [58]. However, most GH74 hydrolases exhibit specificities towards xyloglucans and/or xyloglucan-oligosaccharides found in plant cell walls [59]. Either way, these predicted enzymes might be involved in the degradation of oligosaccharides. GHs, belonging to GH3, GH20, GH23, GH73 and GH109, can be linked to modification/degradation of cell wall components such as peptidoglycan, glycoproteins and lipopolysaccharide. Two peptidoglycan-modifying enzymes, Lupro_08335 (GH23) and Lupro_11420 (GH73), are supplemented with a CBM family 50 mediating the binding to N-acetylglucosamine residues [60]. Various outer membrane proteins containing SusC domains and TonB-dependent receptors enable oligosaccharide import into the periplasm and from there through sodium/glucose co-transporter and L-fucose-proton symporter to the cytosol. Compared to carbohydrate active enzymes, L. profundi LP1T harbours a larger number of proteases. Positive degradation of gelatine and casein on agar plates was observed for L. profundi LP1T [1]. 131 gene-encoding sequences were assigned to 51 MEROPS peptidase families, mostly metallo- and serine proteases (Additional file 3: Table S3), whereof 27 contained a signal peptide. From marine sedimentary bacteria the majority of extracellular peptidases have been identified as serine- and metalloproteases [61, 62]. The peptidase families C26, M01, M14, M20, M23, S09, S12, S33, and S41 were found more frequently than others. The amount of M01 and S09 peptidases are similar to the deep-sea Bacteroidetes Zunongwangia profunda SM-A87, as well as the high number of peptidase genes from the families M01, M23, S09, and S41 [62]. Secreted M01 aminopeptidase in Z. profunda SM-A87 has been proposed as a response to HMW dissolved organic nitrogen degradation, whereas the prolyl oligopeptidases of family S09 specifically hydrolyse oligopeptides shorter than 30 residues [62].
For the accessibility of nutrition deriving from HMW organic matter, hydrolytic enzymes need to be exported across the cell envelope into the extracellular environment. In total, 71 genes encoding for proteins of the double-membrane-spanning secretion systems type I (T1SS), and efflux pumps are incorporated in the L. profundi LP1T genome (Additional file 4: Table S4). Both systems are often associated with nutrition acquisition and antimicrobial resistance mechanisms [63]. The T1SS use ABC transporters for substrate translocation across the cytoplasma membrane, whereas efflux pumps use Na+/H+ drug antiports or the proton-motive force [64]. 32 proteins were associated with ABC transport across the inner membrane. Whereas 6 RND transporters, 13 major facility transporters and 7 multidrug and toxic compound extrusion family proteins was identified as efflux pumps. In total, 10 outer membrane channel proteins TolC were identified, transporting substrate from the periplasm across the outer membrane in both systems [64].
Six genes related to the curli biogenesis system (Lupro_11990-Lupro_12015) were found. Curli fibers produced by the curli biogenesis system have shown to be involved in adhesion to surfaces, cell aggregation and biofilm formation [65]. Cell morphology changes were observed in L. profundi LP1T into filamentous rods and cell aggregation under sub-optimal cultivation condition, such as the presence of ampicillin, non-optimal temperatures, unfavourable carbon source or extended growth periods above one week (Fig. 2b) [1]. The abilities to aggregate or produce biofilms are also beneficial, and perhaps vital for L. profundi LP1T to survive the fluctuating chemical and physical conditions of the deep-sea hydrothermal vent system. A variety of protein domains involved in adhesion was identified using NCBI Batch web CD-Search Tool [48]. In total 60 ORFs were revealed from the genome, containing adhesion domains such as FN3, TSP_3, vWA, CBM’s, LamG, PKD, among others (Additional file 5: Table S5). Many bacterial species also produce extracellular polysaccharides that are able to promote adhesion [66]. In the genome of L. profundi LP1T three genes encoding for poly-β-1,6-N-acetyl-D-glucosamine synthase/GT family 2 (Lupro_00610, Lupro_00765, Lupro_09885) and a potential polysaccharide deacetylase gene (Lupro_10410) were found, which may enable the bacteria to produce poly-β-1,6-N-acetyl-D-glucosamine (PGA). The homopolymer PGA mediates cell-to-cell and cell-to-surface adhesion in biofilms in E. coli and has effects on diverse host-microbe interactions [67]. The O-antigen of lipopolysaccharides can mediate attachment to host surfaces and biofilm formation [68, 69]. The strain LP1T comprises extracellular polysaccharide gene clusters containing several glycosyl transferases, besides genes encoding for lipid A synthesis, which may also attribute towards cell adhesion and biofilm formation.
Many members of the Bacteroidetes are able to glide along surfaces in search for nutrition or as response to environmental stimuli [21, 70]. Blast analysis of the L. profundi LP1T genome revealed 17 protein-encoding genes involved in gliding motility (Additional file 6: Table S6). However, no gliding motility has been observed for L. profundi LP1T [1]. Bacteroidetes strains, such as the non-motile oral pathogen Porphyromonas gingivalis or F. johnsoniae use the gliding motility apparatus in addition for secretion of extracellular enzymes participating in accessing nutrition or serve as virulence factors [71, 72]. The gliding motility apparatus has been suggested to refer to PorSS as the type IX secretion system (T9SSs) [70]. In the genome of strain LP1T, 17 proteins were found containing a Por_Secre_tail domain, which is responsible for translocation of proteins across the outer membrane via PorSS [73]. Amongst these proteins are adhesins, proteases, an endonuclease, an α-amylase and a putative sialidase. Therefore the PorSS may not only add to the transportation system of L. profundi LP1T, but also enhances its hydrolytic capacity.
Conclusions
The genome of Lutibacter profundi LP1T comprises a single chromosome of 2,966 Mbp, smaller compared to other marine Bacteroidetes [21, 62, 74]. A reduced genome, a range of transporter systems and metabolic features indicate a highly specialized organism toward a life in a deep-sea hydrothermal vent biofilm.
L. profundi LP1T originated from a biofilm attached to the outer surface of a deep-sea hydrothermal chimney. The mat consisted of long recalcitrant sugar polymers produced by the Epsilonproteobacteria Sulfurovum with Bacteroidetes attached along the filament surface [32]. As organotrophs, Flavobacteriaceae have been linked to HMW organic matter degradation such as polysaccharides and proteins. L. profundi LP1T features a small selected arsenal of 24 GHs, which is rather a minor amount compared to other members of the family [21, 74], nevertheless it offers the possibility to hydrolyse α-glucosidic poly- and oligosaccharides, peptidoglycans and β-glycans. The utilisation of starch and sucrose was confirmed by the presence of a Sus cluster. Together with the large number of proteases strain LP1T seems predestined to utilize complex organic matter efficiently, derived from a microbial biofilm. Diverse TonB-dependent receptors located close to glycoside hydrolases and proteases, as well as sodium/glucose cotransporter, amino acid permeases and transporter confirm the organotrophic life style. L. profundi LP1T contains a set of genes for gliding motility, which is common in Bacteroidetes [70], and may allow the strain to move in the biofilm. Furthermore, the gliding motility apparatus seems to add to the transportation system of L. profundi LP1T, by exporting Por secretion signal containing proteins such as protease, endonuclease, amylase, putative sialidase or proteins with adhesive properties, which contributes to accessibility of nutrition’s for the bacteria. L. profundi LP1T can mediate attachment to surfaces via a multitude of adhesins and extracellular polysaccharides and thereby may contribute to the biofilm generation. The presence of various cytochrome c oxidases with different oxygen affinities enables the bacteria to thrive in microaerophilic to aereophilic conditions, like they are present in biofilms or hydrothermal environments influenced by fluctuation of hydrothermal fluids mixed with sea water. The microaerobic life style is further indicated by diverse ferredoxin utilizing enzymes. The complete pathway for denitrification is present in L. profundi LP1T in addition to oxygen respiration and the activity of nitrate reduction to nitrite has been confirmed under microaerobic conditions, although it did not enhance the growth [1]. Furthermore, SQR involved in the sulphur metabolisms may play an important role in sulphide detoxification in an environment with high sulphide concentration.
Acknowledgements
This work was funded by the Norwegian Research Council via the following projects; Mining of a Norwegian Biogoldmine through metagenomics, project nr. 208491, NorZymeD project nr. 221568 and Centre for Geobiology project nr. 179560.
Authors’ contributions
Conceived and designed the experiments: IHS and RS. Performed bioinformatics analysis and assembly refinement: RS. Analysed the data: JW, SLMB, IHS and RS. Wrote the paper: JW, SLMB, IHS and RS. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- AMOR
Arctic mid-Ocean ridge
- CAZy
Carbohydrate active enzyme families
- CBMs
Carbohydrate binding modules
- CEs
Carbohydrate esterases
- DPG
Diphosphatidylglycerol
- GHs
Glycoside hydrolases
- GOGAT
Glutamate-oxoglutarate aminotransferase
- GTs
Glycosyl transferases
- HMW
High-molecular weight
- KAAS
KEGG automatic annotation server
- LCVF
Loki’s castle vent field
- NSC
Norwegian sequencing centre
- PE
Phosphatidylethanolamine
- PGAAP
Prokaryotic Genome annotation pipeline
- PLs
Carbohydrate esterases
- RND
Resistance, Nodulation and cell Division
- SQR
Sulphide:quinone reductase
- Sus
Starch utilization cluster
Additional files
Central metabolism of Lutibacter profundi LP1T. (XLSX 37 kb)
Glycoside hydrolases of Lutibacter profundi LP1T revealed by dbCAN analyses. different export signals are SP: signal peptid, PorSS: Por_Secre_tail from type IX secretion system. (XLSX 47 kb)
Proteases of Lutibacter profundi LP1T revealed by MEROPS analyses, different export signals are SP: signal peptid, PorSS: Por_Secre_tail from type IX secretion system. (XLSX 44 kb)
Secretion systems of Lutibacter profundi LP1T. (XLSX 47 kb)
Genes and domains with a potential role in adhesion found in the L. profundi LP1T genome. (XLSX 38 kb)
CDS for gliding motility. (XLSX 38 kb)
References
- 1.Le Moine Bauer S, Roalkvam I, Steen IH, Dahle H. Lutibacter profundi sp. nov., isolated from a deep-sea hydrothermal system on the Arctic Mid-Ocean Ridge and emended description of the genus Lutibacter. Int J Syst Evol Microbiol. 2016. doi:10.1099/ijsem.0.001105. [DOI] [PubMed]
- 2.Chen Y, Zhang Z, Fu Y, Wang Y, Wang Y, Jiao N. Altuibacter lentus gen. nov., sp. nov., a novel member of family Flavobacteriaceae isolated from deep seawater of the South China Sea. Antonie Van Leeuwenhoek. 2013;104(6):1151–7. doi: 10.1007/s10482-013-0037-8. [DOI] [PubMed] [Google Scholar]
- 3.Sung H-RR, Shin K-SS, Ghim S-YY. Lutibacter oricola sp. nov., a marine bacterium isolated from seawater. Int J Syst Evol Microbiol. 2015;65(Pt 2):485–90. doi: 10.1099/ijs.0.067132-0. [DOI] [PubMed] [Google Scholar]
- 4.Bowman JP, Nichols DS. Novel members of the family Flavobacteriaceae from Antarctic maritime habitats including Subsaximicrobium wynnwilliamsii gen. nov., sp. nov., Subsaximicrobium saxinquilinus sp. nov., Subsaxibacter broadyi gen. nov., sp. nov., Lacinutrix copepodicola gen. nov., sp. nov., and novel species of the genera Bizionia, Gelidibacter and Gillisia. Int J Syst Evol Microbiol. 2005;55(Pt 4):1471–86. doi: 10.1099/ijs.0.63527-0. [DOI] [PubMed] [Google Scholar]
- 5.Zeder M, Peter S, Shabarova T, Pernthaler J. A small population of planktonic Flavobacteria with disproportionally high growth during the spring phytoplankton bloom in a prealpine lake. Environ Microbiol. 2009;11(10):2676–86. doi: 10.1111/j.1462-2920.2009.01994.x. [DOI] [PubMed] [Google Scholar]
- 6.Kwon YM, Yang SH, Kwon KK, Kim SJ. Nonlabens antarcticus sp. nov., a psychrophilic bacterium isolated from glacier ice, and emended descriptions of Nonlabens marinus Park et al. 2012 and Nonlabens agnitus Yi and Chun 2012. Int J Syst Evol Microbiol. 2014;64(Pt 2):400–5. doi: 10.1099/ijs.0.056606-0. [DOI] [PubMed] [Google Scholar]
- 7.Dong K, Liu H, Zhang J, Zhou Y, Xin Y. Flavobacterium xueshanense sp. nov. and Flavobacterium urumqiense sp. nov., two psychrophilic bacteria isolated from glacier ice. Int J Syst Evol Microbiol. 2012;62(Pt 5):1151–7. doi: 10.1099/ijs.0.030049-0. [DOI] [PubMed] [Google Scholar]
- 8.Lau SC, Tsoi MM, Li X, Plakhotnikova I, Dobretsov S, Wu M, et al. Stenothermobacter spongiae gen. nov., sp. nov., a novel member of the family Flavobacteriaceae isolated from a marine sponge in the Bahamas, and emended description of Nonlabens tegetincola. Int J Syst Evol Microbiol. 2006;56(Pt 1):181–5. doi: 10.1099/ijs.0.63908-0. [DOI] [PubMed] [Google Scholar]
- 9.Klippel B, Sahm K, Basner A, Wiebusch S, John P, Lorenz U, et al. Carbohydrate-active enzymes identified by metagenomic analysis of deep-sea sediment bacteria. Extremophiles. 2014;18(5):853–63. doi: 10.1007/s00792-014-0676-3. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Yu Y, Luo W, Zeng Y, Chen B. Bacterial diversity in surface sediments from the Pacific Arctic Ocean. Extremophiles. 2009;13(2):233–46. doi: 10.1007/s00792-009-0225-7. [DOI] [PubMed] [Google Scholar]
- 11.Kibe R, Sakamoto M, Yokota H, Benno Y. Characterization of the Inhabitancy of Mouse Intestinal Bacteria (MIB) in Rodents and Humans by Real-Time PCR with Group-Specific Primers. Microbiol Immunol. 2007;51(4):349–57. doi: 10.1111/j.1348-0421.2007.tb03916.x. [DOI] [PubMed] [Google Scholar]
- 12.Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, et al. Microbial Co-occurrence Relationships in the Human Microbiome. PLoS Comput Biol. 2012;8(7):e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowman JP, Nichols DS. Aequorivita gen. nov., a member of the family Flavobacteriaceae isolated from terrestrial and marine Antarctic habitats. Int J Syst Evol Microbiol. 2002;52(Pt 5):1533–41. doi: 10.1099/00207713-52-5-1533. [DOI] [PubMed] [Google Scholar]
- 14.Campbell CL, Mummey DL, Schmidtmann ET, Wilson WC. Culture-independent analysis of midgut microbiota in the arbovirus vector Culicoides sonorensis (Diptera: Ceratopogonidae) J Med Entomol. 2004;41(3):340–8. doi: 10.1603/0022-2585-41.3.340. [DOI] [PubMed] [Google Scholar]
- 15.Hugo CJ, Segers P, Hoste B, Vancanneyt M, Kersters K. Chryseobacterium joostei sp. nov., isolated from the dairy environment. Int J Syst Evol Microbiol. 2003;53(Pt 3):771–7. doi: 10.1099/ijs.0.02232-0. [DOI] [PubMed] [Google Scholar]
- 16.Kirchman DL. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39:91–100. doi: 10.1111/j.1574-6941.2002.tb00910.x. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Pereira PR, Fuchs BM, Alonso C, Oliver MJ, van Beusekom JE, Amann R. Distinct flavobacterial communities in contrasting water masses of the north Atlantic Ocean. ISME J. 2010;4(4):472–87. doi: 10.1038/ismej.2009.142. [DOI] [PubMed] [Google Scholar]
- 18.Delong EF, Franks DG, Alldredge AL. Phylogenetic Diversity of Aggregate-Attached Vs Free-Living Marine Bacterial Assemblages. Limnol Oceanogr. 1993;38(5):924–34. doi: 10.4319/lo.1993.38.5.0924. [DOI] [Google Scholar]
- 19.Quaiser A, Zivanovic Y, Moreira D, Lopez-Garcia P. Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J. 2011;5(2):285–304. doi: 10.1038/ismej.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cottrell MT, Kirchman DL. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol. 2000;66(4):1692–7. doi: 10.1128/AEM.66.4.1692-1697.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E, et al. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ Microbiol. 2006;8(12):2201–13. doi: 10.1111/j.1462-2920.2006.01152.x. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez JM, Fernandez-Gomez B, Fernandez-Guerra A, Gomez-Consarnau L, Sanchez O, Coll-Llado M, et al. Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria) Proc Natl Acad Sci U S A. 2008;105(25):8724–9. doi: 10.1073/pnas.0712027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez JM, Pinhassi J, Fernandez-Gomez B, Coll-Llado M, Gonzalez-Velazquez M, Puigbo P, et al. Genomics of the proteorhodopsin-containing marine flavobacterium Dokdonia sp. strain MED134. Appl Environ Microbiol. 2011;77(24):8676–86. doi: 10.1128/AEM.06152-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez-Gomez B, Richter M, Schuler M, Pinhassi J, Acinas SG, Gonzalez JM, et al. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 2013;7(5):1026–37. doi: 10.1038/ismej.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi DH, Cho BC. Lutibacter litoralis gen. nov., sp. nov., a marine bacterium of the family Flavobacteriaceae isolated from tidal flat sediment. Int J Syst Evol Microbiol. 2006;56(Pt 4):771–6. doi: 10.1099/ijs.0.64146-0. [DOI] [PubMed] [Google Scholar]
- 26.Park S, Kang S-JJ OT-KK, Yoon J-HH. Lutibacter maritimus sp. nov., isolated from a tidal flat sediment. Int J Syst Evol Microbiol. 2010;60(Pt 3):610–4. doi: 10.1099/ijs.0.012401-0. [DOI] [PubMed] [Google Scholar]
- 27.Lee S-YY, Lee M-HH OT-KK, Yoon J-HH. Lutibacter aestuarii sp. nov., isolated from a tidal flat sediment, and emended description of the genus Lutibacter Choi and Cho 2006. Int J Syst Evol Microbiol. 2012;62(Pt 2):420–4. doi: 10.1099/ijs.0.030320-0. [DOI] [PubMed] [Google Scholar]
- 28.Park SC, Choe HN, Hwang YM, Baik KS, Seong CN. Lutibacter agarilyticus sp. nov., a marine bacterium isolated from shallow coastal seawater. Int J Syst Evol Microbiol. 2013;63(Pt 7):2678–83. doi: 10.1099/ijs.0.047670-0. [DOI] [PubMed] [Google Scholar]
- 29.Choi A, Yang S-JJ, Cho J-CC. Lutibacter flavus sp. nov., a marine bacterium isolated from a tidal flat sediment. Int J Syst Evol Microbiol. 2013;63(Pt 3):946–51. doi: 10.1099/ijs.0.043471-0. [DOI] [PubMed] [Google Scholar]
- 30.Park S, Park J-MM, Won S-MM, Park D-SS, Yoon J-HH. Lutibacter crassostreae sp. nov., isolated from oyster. Int J Syst Evol Microbiol. 2015. doi:10.1099/ijs.0.000324. [DOI] [PubMed]
- 31.Nedashkovskaya OI, Van Trappen S, Zhukova NV, De Vos P. Lutibacter holmesii sp. nov., a novel marine bacterium of the family Flavobacteriaceae isolated from the sea urchin Strongylocentrotus intermedius and emended description of the genus Lutibacter. Int J Syst Evol Microbiol. 2015. doi:10.1099/ijsem.0.000525. [DOI] [PubMed]
- 32.Stokke R, Dahle H, Roalkvam I, Wissuwa J, Daae FL, Tooming-Klunderud A, et al. Functional interactions among filamentous Epsilonproteobacteria and Bacteroidetes in a deep-sea hydrothermal vent biofilm. Environ Microbiol. 2015. doi:10.1111/1462-2920.12970. [DOI] [PubMed]
- 33.Pedersen RB, Rapp HT, Thorseth IH, Lilley MD, Barriga FJ, Baumberger T, et al. Discovery of a black smoker vent field and vent fauna at the Arctic Mid-Ocean Ridge. Nat Commun. 2010;1:126. doi: 10.1038/ncomms1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dahle H, Roalkvam I, Thorseth IH, Pedersen RB, Steen IH. The versatile in situ gene expression of an Epsilonproteobacteria-dominated biofilm from a hydrothermal chimney. Environ Microbiol Rep. 2013;5(2):282–90. doi: 10.1111/1758-2229.12016. [DOI] [PubMed] [Google Scholar]
- 35.Dahle H, Okland I, Thorseth IH, Pederesen RB, Steen IH. Energy landscapes shape microbial communities in hydrothermal systems on the Arctic Mid-Ocean Ridge. ISME J. 2015;9(7):1593–606. doi: 10.1038/ismej.2014.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emerson D, Floyd MM. Enrichment and isolation of iron-oxidizing bacteria at neutral pH. Methods Enzymol. 2005;397:112–23. doi: 10.1016/S0076-6879(05)97006-7. [DOI] [PubMed] [Google Scholar]
- 37.Norwegian Sequencing Centre. Oslo, Norway 2015. http://www.sequencing.no.
- 38.Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, Garrity GM, et al. Toward an Online Repository of Standard Operating Procedures (SOPs) for (Meta)genomic Annotation. OMICS. 2008;12(2):137–41. doi: 10.1089/omi.2008.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marmur J. Procedure for Isolation of Deoxyribonucleic Acid from Micro-Organisms. J Mol Biol. 1961;3(2):208–18. doi: 10.1016/S0022-2836(61)80047-8. [DOI] [Google Scholar]
- 41.Roalkvam I, Dronen K, Stokke R, Daae FL, Dahle H, Steen IH. Physiological and genomic characterization of Arcobacter anaerophilus IR-1 reveals new metabolic features in Epsilonproteobacteria. Front Microbiol. 2015;6:987. doi: 10.3389/fmicb.2015.00987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 43.Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23(8):1026–8. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- 44.Treangen TJ, Sommer DD, Angly FE, Koren S, Pop M. Next generation sequence assembly with AMOS. Curr Protoc Bioinformatics. 2011;Chapter 11:Unit 11 8. doi:10.1002/0471250953.bi1108s33. [DOI] [PMC free article] [PubMed]
- 45.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rawlings ND, Morton FR. The MEROPS batch BLAST: a tool to detect peptidases and their non-peptidase homologues in a genome. Biochimie. 2008;90(2):243–59. doi: 10.1016/j.biochi.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 47.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(Web Server issue):445–51. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43(Database issue):D222–6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barquera B. The sodium pumping NADH:quinone oxidoreductase (Na(+)-NQR), a unique redox-driven ion pump. J Bioenerg Biomembr. 2014;46(4):289–98. doi: 10.1007/s10863-014-9565-9. [DOI] [PubMed] [Google Scholar]
- 51.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–D5. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller BG, Raines RT. Identifying latent enzyme activities: substrate ambiguity within modern bacterial sugar kinases. Biochemistry. 2004;43(21):6387–92. doi: 10.1021/bi049424m. [DOI] [PubMed] [Google Scholar]
- 54.Shipman JA, Berleman JE, Salyers AA. Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J Bacteriol. 2000;182(19):5365–72. doi: 10.1128/JB.182.19.5365-5372.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem. 2009;284(37):24673–7. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yaoi K, Ishida T. Glycoside Hydrolase Family 74. http://www.cazypedia.org/. Accessed 4 Mar 2016. 16:34 UTC.
- 57.Taylor G. Sialidases: structures, biological significance and therapeutic potential. Curr Opin Struct Biol. 1996;6(6):830–7. doi: 10.1016/S0959-440X(96)80014-5. [DOI] [PubMed] [Google Scholar]
- 58.Juge N, Tailford L, Owen CD. Sialidases from gut bacteria: a mini-review. Biochem Soc Trans. 2016;44(1):166–75. doi: 10.1042/BST20150226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez-Fleites C, Guerreiro CIPD, Baumann MJ, Taylor EJ, Prates JAM, Ferreira LMA, et al. Crystal structures of Clostridium thermocellum xyloglucanase, XGH74A, reveal the structural basis for xyloglucan recognition and degradation. J Biol Chem. 2006;281(34):24922–33. doi: 10.1074/jbc.M603583200. [DOI] [PubMed] [Google Scholar]
- 60.Steen A, Buist G, Leenhouts KJ, El Khattabi M, Grijpstra F, Zomer AL, et al. Cell wall attachment of a widely distributed peptidoglycan binding domain is hindered by cell wall constituents. J Biol Chem. 2003;278(26):23874–81. doi: 10.1074/jbc.M211055200. [DOI] [PubMed] [Google Scholar]
- 61.Zhou MY, Chen XL, Zhao HL, Dang HY, Luan XW, Zhang XY, et al. Diversity of both the cultivable protease-producing bacteria and their extracellular proteases in the sediments of the South China sea. Microb Ecol. 2009;58(3):582–90. doi: 10.1007/s00248-009-9506-z. [DOI] [PubMed] [Google Scholar]
- 62.Qin QL, Zhang XY, Wang XM, Liu GM, Chen XL, Xie BB, et al. The complete genome of Zunongwangia profunda SM-A87 reveals its adaptation to the deep-sea environment and ecological role in sedimentary organic nitrogen degradation. BMC Genomics. 2010;11:247. doi: 10.1186/1471-2164-11-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anes J, McCusker MP, Fanning S, Martins M. The ins and outs of RND efflux pumps in Escherichia coli. Front Microbiol. 2015;6:587. doi: 10.3389/fmicb.2015.00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Costa TR, Felisberto-Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, et al. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat Rev Microbiol. 2015;13(6):343–59. doi: 10.1038/nrmicro3456. [DOI] [PubMed] [Google Scholar]
- 65.Barnhart MM, Chapman MR. Curli Biogenesis and Function. Annu Rev Microbiol. 2006;60:131–47. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berne C, Ducret A, Hardy GG, Brun YV. Adhesins Involved in Attachment to Abiotic Surfaces by Gram-Negative Bacteria. Microbiol Spectr. 2015;3(4). doi:10.1128/microbiolspec.MB-0018-2015. [DOI] [PMC free article] [PubMed]
- 67.Itoh Y, Rice JD, Goller C, Pannuri A, Taylor J, Meisner J, et al. Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-beta-1,6-N-acetyl-D-glucosamine. J Bacteriol. 2008;190(10):3670–80. doi: 10.1128/JB.01920-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hathroubi S, Hancock MA, Bosse JT, Langford PR, Tremblay YD, Labrie J et al. Surface polysaccharide mutants reveal that absence of O antigen reduces biofilm formation of Actinobacillus pleuropneumoniae. Infect Immun. 2015. doi:10.1128/IAI.00912-15. [DOI] [PMC free article] [PubMed]
- 69.Bogino PC, Oliva Mde L, Sorroche FG, Giordano W. The role of bacterial biofilms and surface components in plant-bacterial associations. Int J Mol Sci. 2013;14(8):15838–59. doi: 10.3390/ijms140815838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McBride MJ, Zhu Y. Gliding motility and Por secretion system genes are widespread among members of the phylum Bacteroidetes. J Bacteriol. 2013;195(2):270–8. doi: 10.1128/JB.01962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kharade SS, McBride MJ. Flavobacterium johnsoniae chitinase ChiA is required for chitin utilization and is secreted by the type IX secretion system. J Bacteriol. 2014;196(5):961–70. doi: 10.1128/JB.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sato K, Naito M, Yukitake H, Hirakawa H, Shoji M, McBride MJ, et al. A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc Natl Acad Sci U S A. 2010;107(1):276–81. doi: 10.1073/pnas.0912010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shoji M, Sato K, Yukitake H, Kondo Y, Narita Y, Kadowaki T, et al. Por secretion system-dependent secretion and glycosylation of Porphyromonas gingivalis hemin-binding protein 35. PLoS One. 2011;6(6):e21372. doi: 10.1371/journal.pone.0021372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mann AJ, Hahnke RL, Huang S, Werner J, Xing P, Barbeyron T, et al. The genome of the alga-associated marine flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl Environ Microbiol. 2013;79(21):6813–22. doi: 10.1128/AEM.01937-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krieg NR, Ludwig W, Euzeby J, Whitman WB. Phylum XIV. Bacteroidetes phyl.nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2010. p. 25. [Google Scholar]
- 77.Validation List No. 143. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2012;62(1):1–4. doi:10.1099/ijs.0.039487-0. [DOI] [PubMed]
- 78.Bernardet JF. Class II. Flavobacteriia class. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2010. p. 105. [Google Scholar]
- 79.Bernardet JF. Order I. Flavobacteriales ord. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2010. p. 105. [Google Scholar]
- 80.Bernardet JF. Family I. Flavobacteriaceae Reichenbach 1992b. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology. Second. New York: Springer; 2010. pp. 106–11. [Google Scholar]
- 81.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontology Consortium Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]