

Abstract
Human blood plasma proteome reflects physiological changes associated with a child’s development as well as development of disease states. While age-specific normative values are available for proteins routinely measured in clinical practice, there is paucity of comprehensive longitudinal data regarding changes in human plasma proteome during childhood. We applied TMT-10plex isobaric labeling-based quantitative proteomics to longitudinally profile the plasma proteome in 10 healthy children during their development, each with 9 serial time points from 9 months to 15 years of age. In total, 1828 protein groups were identified at peptide and protein level false discovery rate of 1% and with at least two razor and unique peptides. The longitudinal expression profiles of 1747 protein groups were statistically modeled and their temporal changes were categorized into 7 different patterns. The patterns and relative abundance of proteins obtained by LC-MS were also verified with ELISA. To our knowledge, this study represents the most comprehensive longitudinal profiling of human plasma proteome to date. The temporal profiles of plasma proteome obtained in this study provide a comprehensive resource and reference for biomarker studies in childhood diseases.
Keywords: human plasma proteome, childhood development, pediatric proteome, temporal proteome profiling, tandem mass tags, TMT10
Graphical Abstract
INTRODUCTION
Human blood plasma communicates with almost all of the cells in the body and the plasma proteome contains not only classical plasma proteins, but also tissue leakage proteins, aberrant secretions from tumor cells and foreign proteins of infectious organisms and parasites [1–3]. Because of its accessibility, human blood plasma is a widely used biospecimen for clinical diagnosis of diseases, it also holds great potential in proteomic biomarker research where novel and more specific biomarkers are desired for more accurate disease diagnosis and assessment of therapeutic responses. The majority of the plasma biomarker studies up to date have focused on the adult population, for which the plasma proteome remains relatively stable on an individual basis. The childhood development from neonates to puberty is characterized by rapid physical, physiological and psychological changes. However, the proteome at this phase of life has been less explored [4]. Understanding of plasma proteome changes throughout childhood could help prediction and early diagnosis of childhood diseases [5]. In addition, longitudinal changes of a person’s proteome reflect the influence of genetic factor in the native abundance of proteins - the cornerstone of personalized medicine [6].
Significant technical challenges exist in plasma proteome research in that plasma proteome has over 10 orders of magnitude in protein abundances, which greatly exceeds the dynamic range of any proteomics platform [3, 7, 8]. The relative low abundance of disease-specific biomarkers can be easily masked by the high and medium abundant proteins in the extremely complex human plasma proteome [2]. To this end, 2D gel electrophoresis, immunoaffnity-based depletion of high and medium abundant proteins and peptide level fractionation to reduce sample complexity in LC-MS/MS analysis have been used to broaden the coverage of plasma proteome [8]. In addition, isobaric labeling-based quantitative proteomics, such as using iTRAQ and tandem mass tag (TMT) for peptide level labeling has been widely used for identification and more accurate relative quantification of proteome-wide changes in biological samples [9]. In particular, the multiplexing ability of TMT reagent and the resulted high throughput analysis enable the analysis of 1,000 plasma samples in a realistic time frame with comprehensive proteome coverage [10], and up to ten different experimental conditions can be analyzed simultaneously using the TMT10 reagent [11–13].
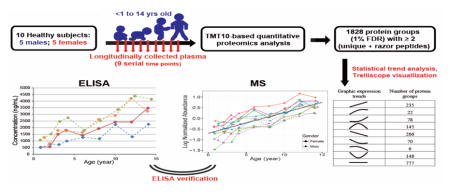
Here in this work, we aim to establish a pediatric plasma proteome database with longitudinal expression patterns/trends of plasma proteins. Toward this goal, 9 time point series of plasma samples longitudinally collected from 10 healthy subjects from birth to puberty were analyzed. Immune-depletion for high abundant plasma protein removal, extensive peptide level fractionation to reduce sample complexity, and high-performance nanoUHPLC-MS/MS for peptide separation and detection were used to broaden the proteome coverage. For accurate relative quantification, TMT10-based isobaric labeling approach was applied to maximize the number of time point samples that can be quantified in parallel. Statistical modeling was applied to fit the temporal trend in abundance of each protein within the same subject. To confirm the fidelity of these mass spectrometry derived data, certain proteins were selected for independent verification by ELISA. To our knowledge, this study represents the most comprehensive longitudinal profiling of childhood plasma proteome.
EXPERIMENTAL SECTION
Blood plasma samples from healthy subjects
Participants were selected from the Diabetes Autoimmunity Study in the Young (DAISY) prospective cohort, and the details of follow-up have been published previously [14]. Informed consent was obtained from the parents of each study subject. The Colorado Multiple Institutional Review Board approved all study protocols. Healthy children included in this study have been negative for islet autoantibodies during the entire follow-up period. Venous non-fasting blood samples were collected at each study visit and plasma was separated and stored at −80 °C. Archived plasma samples from 10 healthy subjects were selected for this study. Frozen plasma samples were transferred to proteomics measurement laboratory for sample processing and measurement. In total, 90 plasma samples from the healthy children were used in this MS-based proteomics profiling study and they were processed according to the experimental strategy (Figure 1).
Figure 1.

Schematic representation of experimental workflow in this study.
Proteomic sample preparation
Unless otherwise specified, all reagents and chemicals used in this study were purchased from Sigma Aldrich (St. Louis, MO). The kit for BCA protein assay was obtained from ThermoFisher Scientific (Rockford, IL). Sequencing-grade trypsin was purchased from Promega (Madison WI). All solvents used were HPLC-grade.
All samples were prepared in the same batch by the same researcher to ensure minimal variations in sample preparation. MARS Human-14 column (Agilent Technologies) was used to deplete the top 14 most abundant proteins according to manufacturer’s instructions. The flow-through fractions (depleted plasma) were collected and concentrated using 3kDa molecular weight cutoff filters (Millipore). The depleted plasma proteins were denatured, reduced, alkylated, and digested as previously described [15]. The digestion mixture was desalted using C18 SPE cartridges and the eluted peptide solution was completely dried before isobaric labeling. Aliquots of each individual sample from all healthy subjects were equally pooled to create the common reference sample, to be used in subsequent TMT labeling as reference channel (Supplementary material Table S1). The 9 serial samples from each individual and the common reference sample were included in one labeling experiment set, instead of randomly assigning the time points of the same subject into different experiments in order to avoid the potential missing value issues resulted from data-dependent MS/MS acquisitions [16]. TMT-10plex labeling on peptides was performed according to the manufacturer’s instructions (ThermoFisher Scientific). After labeling, the 10 labeled samples in each experiment set were pooled, concentrated and fractionated using high-pH RPLC on an XBridge C18 analytical column (particle size of 5μm, 250 × 4.6mm, Waters) at a flow rate of 0.5 mL min−1. Mobile phases A and B consisted of 10 mM ammonium formate in water (pH 10) and 90% ACN (pH 10), respectively. Ninety six fractions were collected and concatenated into 24 fractions [17, 18]. Peptide solutions were dried, stored at −80 °C, and reconstituted in 0.1% FA until LC-MS/MS analysis.
Quantitative LC-MS/MS analysis
LC-MS/MS analysis was conducted using an UltiMate 3000 RSLCnano system coupled to a Q Exactive HF mass spectrometer through an EASY-Spray ion source (ThermoFisher Scientific). Peptides were separated on a PepMap C18 analytical column (2 μm particle, 50 cm × 75 μm i.d., ThermoFisher Scientific). A binary solvent system consisting of 0.1% FA in ddH2O (solvent A) and 0.1% FA in ACN (solvent B) was used to separate peptides at a flow rate of 250 nL min−1. LC separation was performed using the following gradient setting: held at 4% B for 3 min, from 4% to 8% B in 0.1 min, 8% to 40% B in 90 min, 40% to 90% B in 0.1% min, held at 90% B for 10 min, 90% to 4% B in 0.1 min, and held at 4% B for 17 min for re-equilibrating column.
MS data was acquired in profile mode using a data-dependent top 15 method and resolution for full scans (400 to 1950 m/z) was set to 120,000 at m/z 200. Precursors were isolated with a window of 1.4 m/z [19] and fragmented with HCD fragmentation (higher-energy collisional dissociation, normalized collision energy of 32). Resolution for MS/MS spectrum was set to 60,000 at m/z 200, which ensured 6 mDa difference between reporter ions can be clearly resolved. Automatic gain control targets for full MS and MS/MS scans were 3e6 and 1e5, with maximum ion injection time of 50 and 100 ms, respectively. Precursor ions with unassigned, single, seven and higher charge states were excluded, and dynamic exclusion time was set to 20 s.
Database search
All raw files obtained from LC-MS/MS analyses were analyzed using MaxQuant software version 1.5.3.30, and searched against Swiss-Prot human protein database (91,960 protein entries, 02/17/2016 release) using the built-in Andromeda search engine. Semispecific Trypsin/P was selected as the enzyme. Cysteine carbamidomethylation and TMT-10plex labeled N-terminus and lysine were set as fixed modifications. Methionine oxidation was set as variable modifications. FDR was set at 1% for protein and peptide level identification using decoy database, and precursor intensity fraction was set to >0.75 [20]. Other parameters were used as default settings for Orbitrap-type data. The search results in proteinGroups.txt generated by MaxQuant were processed in Perseus software version 1.5.1.6 [21]. Identified protein is represented as protein group in the database search output when the search algorithm identifies a cluster of proteins with high sequence similarity or protein isoforms, which cannot be further differentiated on the basis of shared peptides. The potential contaminants (manually selected for contaminants with no protein names), reverse hits and proteins only identified by modification site were excluded from the identification list. Further, a filtering criterion was set to keep the identified proteins with the quantified values of all ten reporter ions (no missing value) in at least one individual. The longitudinal expression data of this confidently identified protein list was used for statistical trend analysis.
Statistical trend analysis
Quantified protein data based on the reporter ion intensity was normalized to the reference sample, log2 transformed, and median centered [22], i.e. bring the median intensity values of each sample to the same level to correct for the slight variations in the amount of sample used for each labeling channel. Two mixed effect linear models were fit to the log normalized abundance data: a quadratic model with random quadratic, linear, and intercept terms (Equation 1), and a linear model with random linear and intercept terms (Equation 2). Age was used as the fixed effect in these models to examine trends in abundance over time, while controlling for age difference between subjects and random effects for each subject were included to account for the subject variability and non-independent nature of the data over time.
| (Eq. 1) |
| (Eq. 2) |
Likelihood ratio tests were conducted to test for significant quadratic and linear fixed effects and p-values were recorded. An α =0.05 level of significance was used to determine which models were significant. All models were fit in R using the lme4 package [23].
Gene ontology (GO) functional annotation and pathway enrichment
For GO functional annotation analysis, the UniProt accession IDs from protein groups identified in the study were analyzed by the PANTHER classification system [24]. PANTHER version 10.0 (release date May 15, 2015) was used, which includes 11,929 protein families with 83,190 functionally distinct protein subfamilies. Homo Sapiens was chosen as the organism for GO annotations. KEGG pathway enrichment was conducted using DAVID Bioinformatics Resource 6.7, and Homo sapiens was used as “background” for enrichment calculation [25]. GO term biological process enrichment was performed using 1828 plasma protein groups identified in this study as “background”.
Verification of expression patterns using ELISA
ELISA kits for Insulin-like growth factor I (IGF1, #ab211651, intra-assay coefficient of variation (CV) 2.3%) and Insulin-like growth factor-binding protein 3 (IGFBP3, #ab211652, intra-assay CV 3.1%) were purchased from Abcam (Cambridge, MA), and ELISA kit for Insulin-like growth factor-binding protein 2 (IGFBP2, #DGB200, intra-assay CV 5%) was from R&D Systems (Minneapolis, MN), and ELISA kit for Anti-mullerian hormone (AMH, #AL-124, intra-assay CV 3.7%) was purchased from AnshLabs (Webster, TX). The plasma samples from each time point of selected subjects were diluted with dilution factors of 1:80, 1:100, and 1:2000 for IGF1, IGFBP2, and IGFBP3 detection, respectively; and for AMH measurement, dilution factors of 1:10 and 1:500 were used for samples from females and males, respectively. All ELISA measurements were conducted according to manufacturer’s protocol. Duplicated wells were used to average the absorbance values and the concentration of target proteins in the plasma were calculated based on the calibration curve (R2 = 0.9848, 0.9999, 0.9936 and 0.9971 for IGF1, IGFBP2, IGFBP3, and AMH, respectively.) and dilution factor after subtraction of background.
RESULTS AND DISCUSSION
Experimental design to longitudinally profile changes of plasma proteome
Temporal expression trends of human plasma proteins in healthy children, over the age from infant to puberty were measured in this quantitative proteomics study. Five boys and girls were selected, and 9 out of the 10 subjects were Caucasian (Detailed clinical data is shown in Supplementary material Table S1). The 9 time points selected from each of the 10 subjects were at almost identical sampling ages (Figure 2). TMT-10plex isobaric labeling enables multiplexed analysis of up to 10 samples in one experiment, and it was applied in this study to simultaneously compare samples from 9 time points of the same subject in the same experiment, while the 10th channel is being used as common reference for between-subject comparison [26]. Assignment of 9 time points from each individual into one labeling experiment significantly prevented the potential issue of missing values that are commonly observed in data-dependent acquisition shotgun proteomics [16], which is detrimental to expression pattern analysis. Although we did not randomly label the samples of 9 time points from the same individual within and between TMT experiments, the data analysis showed that this labeling design did not result in systematic bias to samples of any individual time point, because the expression patterns of the >1,800 proteins were randomly distributed.
Figure 2.

The serial plasma samples from healthy subjects used in this study (NP01 to NP10, n = 10). The filled blue circles indicate the sampling time point.
Comprehensive identification of plasma proteins
Human plasma proteome has wide dynamic range (>109 concentration range) and complexity, with the top 22 high abundant proteins (HAPs) constitute 99% of the total protein content [1]. To significantly increase the depth of detection for plasma proteome, removal of the HAPs is needed to uncover the low abundant proteins (LAPs) that are of interest as disease markers [1, 7]. As shown in Figure 1, immunodepletion was performed using an Agilent MARS-Human 14 column to remove the HAPs prior to tryptic digestion [27, 28]. High-pH RPLC was further utilized to fractionate the TMT-labeled peptide mixtures before nanoLC-MS/MS analysis using a fast scanning Q Exactive HF benchtop Orbitrap mass spectrometer [8, 17–19]. As a result, 240 raw files were generated and processed using MaxQuant for protein identification and quantification [29]. Under rigorous peptide and protein level FDR cut off of 1%, 1828 protein groups were identified and relatively quantified with at least two razor + unique peptides and one unique peptide ( razor peptides are defined as non-unique peptides assigned to the protein group with the largest number of total peptides identified [30]). Among them, 1009 proteins were commonly identified in all 100 samples (Supplementary material Table S2).
Keshishian et al. reported identification of 5304 proteins using their optimized plasma analytical platform. While their study population - 4 adult patients studied at 4 time-points during planned myocardial infarction for the treatment of hypertrophic obstructive cardiomyopathy – was diametrically different from healthy children, some comparisons can be made. Among identified proteins, 3400 proteins were identified and quantified with at least 2 “distinct” peptides in all 16 samples using iTRAQ-4plex labeling [8]. It is of note that the numbers of proteins were obtained using Spectrum Mill software and under different search parameters than ours. When their 120 raw files were reanalyzed using MaxQuant under the same search parameters (except iTRAQ-4plex option used as quantification) and protein database as ours, 2558 protein groups (under the same criteria of at least two razor + unique peptides and one unique peptide) were identified in at least one sample, and 1526 protein groups were quantified in all 16 samples. The additional 30% protein groups identified in their study likely is a result of 1) depletion of medium abundant proteins using Supermix column [31]; 2) more extensive peptide level fractionations (30 vs 24 in our study); and 3) slightly higher efficiency of iTRAQ-4plex than TMT labeling in peptide identification [32]. However, this was achieved at the cost of doubling instrument time. Just for the LC-MS/MS effective gradient alone, 75 hours were devoted to analyze the 4 samples of one subject in their study, compared to the 36 hours for the 9 samples of one subject in our study. To explore higher multiplicity for large scale quantitative proteomics, Keshishian et al. also applied TMT-10plex to label the aliquots from a pooled healthy human plasma sample, and 2066 proteins with at least 2 peptides were reported [8]. For a fair comparison, their TMT-10plex data was reanalyzed using MaxQuant under our parameters, and 1504 protein groups (under the same filtering criteria) were identified in at least one sample/labeling channel, which is ~17% lower than the 1828 proteins that we identified.
Depth of proteome coverage
As plotted in Figure 3, the reporter ion intensities of each protein were averaged across the samples, and their distributions span over 5 orders of magnitude. To evaluate the sensitivity of our quantification platform, we looked at how LAPs can be identified and quantified by comparison with published data. Farrah et al. reported estimated concentrations of 1243 plasma proteins by applying spectral counting to reanalyze diverse tandem mass spectrometry data generated in laboratories around the world and deposited online [33]. For the 100 proteins reported in their list with the lowest concentration (0.5 – 2.0 ng mL−1), 70 proteins were identified in this study, and the distributions of those proteins were scattered over the middle of intensity range rather than aggregated on the left side as expected for the LAPs (Figure 3). Several well-known LAPs as summarized in Zhou et al. [34] were also identified in our study, which include Interleukin-6 receptor subunit alpha, Platelet factor 4, Metalloproteinase inhibitor 1, Intercellular adhesion molecule 3, Fructose-bisphosphate aldolase B, and Neutrophil defensin. This observation clearly showed that the TMT-10plex quantitative proteomics platform that we implemented in this study achieved very high detection sensitivity and depth of plasma proteome coverage.
Figure 3.

Histogram of the 1828 identified protein groups with average reporter ion intensity. The averaged intensities were observed to span over 5 orders of magnitude. The 70 out of the 100 lowest abundant proteins reported by Farrah et al. (2011) were marked with red color.[33]
Gene ontology and pathway analysis of plasma proteins
For an overall understanding of the human plasma proteome identified in the present study, PANTHER Gene Ontology (GO) classification system was utilized for functional classification of those 1828 proteins. As shown in the Supplementary material Figure S1, 3859, 1973, and 952 annotation hits were found in the categories of biological process (Supplementary material Figure S1A), molecular function (Supplementary material Figure S1B), and cellular component (Supplementary material Figure S1C), respectively. In the category of biological process, metabolic and cellular are the top two processes that the plasma proteome are associated with, and 15.3% of the proteins relate to the processes of response to stimulus and immune system (Supplementary material Figure S1). As the main subcategory, the metabolic process can be further classified into primary metabolic process, phosphate-containing compound metabolic process, nitrogen compound metabolic process, etc., with the majority (71.4%) of the metabolic process hits are primary metabolic process related (i.e., associated with protein, lipid, nucleobase-containing compound, and carbohydrate). For molecular function annotation, >75% of the annotation hits are involved in the functions of catalytic activity, binding, receptor activity, and enzyme regulator activity (Supplementary material Figure S1B). In addition, the activities of hydrolase, enzyme regulator and transferase are the top three ranks in the subcategory of catalytic activity. Cell part, extracellular region, and organelle are the top three categories in the cellular component annotation result (Supplementary material Figure S1C). Furthermore, we applied KEGG pathway enrichment analysis for those 1828 protein groups identified in this study using DAVID bioinformatics database. Under 1% FDR criterion, 11 pathways were enriched including complement and coagulation cascades, ECM-receptor interaction, focal adhesion, among others (Supplementary material Table S3).
We also compared the results of GO annotation for the proteins in this study and from the datasets acquired by Keshishian et al. [8] and re-searched using MaxQuant (Supplementary material Figure S2). Although around 40% of proteins are different between those protein lists, no obvious difference in GO annotations was observed among the three datasets. While this similarity can be largely attributed to the same type of specimen being analyzed, it also showed that different isobaric labeling technique did not introduce bias toward certain categories of proteins, regardless of the number of proteins identified.
Temporal expression patterns of plasma proteins in healthy children
Statistical modeling was applied to fit the temporal changes of each protein within the same subject, and statistical p-values were obtained to evaluate the confidence of fit to a linear or quadratic model and to observe whether the expression trend changed with time/age (Supplementary material Table S2). For this trend analysis, data was further filtered to include only protein groups that were identified in at least 2 individuals. The expression patterns/trends of those 1747 proteins were uploaded online and can be visualized using Trelliscope (https://ascm.shinyapps.io/Zhang_Control_Trend_Analysis/), and a brief introduction for accessing this database was provided in the Supplementary Material [35]. Overall, more than 50% of plasma proteins identified in this study had age-dependent expression trends, which demonstrates the importance of longitudinal profiling study to identify the potential biomarkers specific to childhood diseases, and the requirement of strictly age-matched clinical samples in a cross-sectional study in pediatric population. To our knowledge, the current study represents the largest longitudinal expression profiles of human plasma proteome to date.
The expression trends were categorized into seven major patterns including: increase, increase-then-decrease, curved increase, decrease, decrease-then-increase, curved decrease, and flat. Among those 1747 protein groups (Table 1), 777 protein groups had p-values higher than 0.05 in linear or quadratic models, which indicated that those patterns belong to the category of flat, and no significant expression change occurred from birth to puberty. There were 235 and 266 protein groups having the linear increase and decrease trends along with the age increase, respectively. Also, 145 and 148 protein groups had the opposite trends of increase-then-decrease and decrease-then-increase, with transition points observed at ages of six and eight. Furthermore, 100 and 76 protein groups had the trends of curved increase and curved decrease, respectively. More specifically, among those 100 protein groups with the trend of curved increase, 22 had the curved increase trend in the beginning of childhood then flat trends afterwards, while 78 had flat expressions then curved increase trend in the later phase of childhood. Similarly, 76 protein groups were observed with the trend of curved decrease, of which 70 had the curved decrease trend in the beginning, followed by flat trend later in life, and 6 had reverse patterns.
Table 1.
Temporal Expression Pattern Categories observed in this Study
| Trend category | Trend subcategory | Graphic trend | Number of protein groups | GO-biological process enrichmenta |
|---|---|---|---|---|
| Increase |
|
235 | Cellular protein/macromolecule catabolic process and corresponding regulations | |
| Curved increase | Curved-increase-then-flat |
|
22 | |
| Flat-then-curved-increase |
|
78 | ||
| Increase-then-decrease |
|
145 | ||
| Decrease |
|
266 | Cell adhesion | |
| Curved decrease | Curved-decrease-then-flat |
|
70 | |
| Flat-then-curved-decrease |
|
6 | ||
| Decrease-then-increase |
|
148 | ||
| Flat |
|
777 |
GO-biological process enrichment analysis of plasma proteins with trends of “increase or curved increase” and “decrease or curved decrease”, see Supporting Information Table S4 and S5 for details.
Moreover, biological process of GO term enrichment analysis was performed for those 335 and 342 protein groups with overall trends of increase (“increase” or “curved increase”) and decrease (“decrease” or “curved decrease”), respectively, and the 1828 protein groups identified in this study were used as “background” for enrichment calculation (Table 1 and Supplementary material Table S4 and S5). GO terms related to cellular protein/macromolecule catabolic process and corresponding regulations were enriched (1% FDR) for those protein groups with overall trends of increase. This is expected because of the increasing need of anabolic reactions during the rapid growth phase of childhood development. On the other hand, GO term related to cell adhesion, which mediates the cell-cell interactions and molecule-cell interactions, was enriched for those protein groups with overall trends of decrease. The link between reduced expressions of cell adhesion process and increased age/growth has not yet been directly established, but could be an interesting topic for further study.
Verification of temporal expression patterns using ELISA
As shown in Figure 1, multiple sample preparation, data acquisition, data processing and statistical analysis steps were employed in this MS-based plasma proteomic profiling study. To confirm the temporal expression trends reported in our study, we used ELISA to validate several protein candidates with unique expression trends. It needs to point out that ELISA-based targeted protein measurement only involves minimal sample manipulation, i.e. dilution of the original plasma sample and relative simple data processing.
Insulin-like growth factor I (IGF1) is one of the IGF protein families which have significant growth-promoting activity [36], and IGFs are also important in regulating cartilage and bone development, normal growth and ageing [37, 38]. Based on the primary function of IGF1, the expression trend of linear increase with age is expected and has been confirmed in our temporal MS and ELISA data (Figure 4A). It is of note that 1) levels of the first two time point samples from subject NP04 were lower than the detection sensitivity (51 pg/mL) of IGF1 ELISA utilized herein (using 1:80 diluted plasma); 2) In MS data, the abundance of IGF1 in subject NP10 after the third time point was significantly higher than those in other three subjects, which was also unambiguously confirmed by ELISA data. Moreover, IGF binding proteins (IGFBPs) are key modulators of functions and half-life of circulating IGFs. Recent study by Diener et al. demonstrated a positive association between IGF1 and IGFBP3 based on the serum data from 6,061 adult participants [39]. Consistently, the expression trend of IGFBP3 had similar linear increase trend as of IGF1 and also verified by ELISA (Figure 4B). On the other hand, IGFBP2 has been reported as a negative regulator for IGF-mediated growth, developmental rates, and bone growth [38]. As expected, the curved decrease trend was observed in IGFBP2 in both our MS and ELISA data (Figure 4C). In addition to the consistent patterns observed between MS and ELISA data, it is worth noting the accuracy in relative quantification for those three proteins. The average reporter ion intensities for IGF1, IGFBP3, and IGFBP2 were 89751, 457091 and 95362, respectively (Supplementary material Table S2). The intensity ratio of IGF1:IGFBP3 was 1/5, which is similar to the molar ratio of IGF1:IGFBP3 reported by Diener et al. [39] ELISA data for those three proteins also confirmed that IGFBP3 concentration was much higher than IGF1 and IGFBP2 (Figure 4). Therefore, the relative quantitation accuracy of our LC-MS/MS based quantitative proteomics platform has been verified by ELISA, which is also consistent with the biological functions and findings reported by others.
Figure 4.

The temporal expression patterns of protein IGF1 (A), IGFBP3 (B), and IGFBP2 (C) observed by LC-MS/MS analysis (Left panels) and their corresponding verification trends by ELISA (Right panels). The dots and triangles indicate the sampling time points from female and male, respectively, and the lines with different colors indicate each individual subject. X- and Y-axis are the real age and log2 scales of normalized protein relative abundance (MS data) and protein concentration in ng/mL (ELISA data), respectively. IGF1: Insulin-like growth factor I; IGFBP3: Insulin-like growth factor-binding protein 3; IGFBP2: Insulin-like growth factor-binding protein 2.
Proteins with differential sex-dependent expression
Sex-dependent expression of AMH (anti-mullerian hormone) has been well documented, which is a protein produced by the Sertoli cells of the testes in male during embryonal development to cause a regression of the Muellerian duct [40, 41]. In female, AMH is secreted by the granulosa cells in ovary and can be measurable in serum. It is of note that the levels of AMH circulating in blood of a female are significantly lower than those in male, at least one order of magnitude [42, 43]. In our MS data (Figure 5), AMH was detected in all 5 males without any missing value, however, AMH was only observed in the samples from 3 females. The relative abundance of AMH in female was also at least ten folds lower than those in male, consistent with ELISA data reported by others [42, 43]. Importantly, the decreasing trend in samples from male subjects was also consistent with the findings from Hero et al., in which the decrease trend was observed since the age of 9 to puberty [44].
Figure 5.

The temporal expression patterns of protein AMH observed by LC-MS/MS analysis (Left panel) and their corresponding verification trends by ELISA (Right panel). The dots and triangles indicate the sampling time points from female and male, respectively, and the lines with different colors indicate each individual subject. X- and Y-axis are the real age and log2 scales of normalized protein relative abundance (MS data) and protein concentration in pg/mL (ELISA data)respectively. AMH: Anti-mullerian hormone.
Consistent with previous reports [45], abundances of IGF1 and IGFBP3 in female subjects were relatively higher than those in male subjects (Figure 4A and B); in contrast, IGFBP2 had relatively higher expressions in males than females (Figure 4C).
CONCLUSION
Using a TMT-10plex-based isobaric labeling quantitative LC-MS/MS platform, we performed a very comprehensive longitudinal profiling of human plasma proteome for healthy children during their development, with 9 serial time points staring from birth to adolescence. Statistical modeling analysis was applied to fit the temporal changes of each protein within the same subject. In total, we reported the expression trends of 1747 protein groups, which were provided to the research community as a freely accessible interactive database. The expression trends of selected proteins, IGF1, IGFBP2, and IGFBP3 were further verified by ELISA, which demonstrates the consistency in both the expression patterns and relative abundance of these proteins as expected from their biological functions. Taken together, this longitudinal profiling of human plasma proteome provided a comprehensive resource for the changes of plasma proteins during childhood development, which could serve as a reference for proteomic biomarker studies of childhood diseases.
Supplementary Material
SIGNIFICANCE.
A pediatric plasma proteome database with longitudinal expression patterns of 1747 proteins from neonate to adolescence was provided to the research community. 970 plasma proteins had age-dependent expression trends, which demonstrated the importance of longitudinal profiling study to identify the potential biomarkers specific to childhood diseases, and the requirement of strictly age-matched clinical samples in a cross-sectional study in pediatric population.
Highlights.
Comprehensive longitudinal profiling of human plasma proteome during childhood.
TMT-10plex quantitative proteomics to improve proteome coverage and data quality.
A pediatric plasma proteome database with longitudinal expression patterns.
Temporal changes of plasma proteome as reference to childhood disease research.
Acknowledgments
The authors gratefully thank Athena Schepmoes for preparation of proteomic samples. The work was supported by National Institutes of Health grants DK099174, DK32493, DK32083, DK050979, DK57516, and by the Juvenile Diabetes Research Foundation grant 17-2013-535.
ABBREVIATIONS
- TMT
tandem mass tag
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- GO
gene ontology
- ELISA
enzyme-linked immunosorbent assay
- IGF1
insulin-like growth factor I
- IGFBPs
insulin-like growth factor-binding proteins
- AMH
anti-mullerian hormone
- iTRAQ
isobaric tags for relative and absolute quantitation
- FDR
false discovery rate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1(11):845–67. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–83. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 3.Surinova S, Schiess R, Huttenhain R, Cerciello F, Wollscheid B, Aebersold R. On the development of plasma protein biomarkers. J Proteome Res. 2011;10(1):5–16. doi: 10.1021/pr1008515. [DOI] [PubMed] [Google Scholar]
- 4.Ignjatovic V, Lai C, Summerhayes R, Mathesius U, Tawfilis S, Perugini MA, Monagle P. Age-related differences in plasma proteins: how plasma proteins change from neonates to adults. PLoS One. 2011;6(2):e17213. doi: 10.1371/journal.pone.0017213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moulder R, Bhosale SD, Erkkila T, Laajala E, Salmi J, Nguyen EV, Kallionpaa H, Mykkanen J, Vaha-Makila M, Hyoty H, Veijola R, Ilonen J, Simell T, Toppari J, Knip M, Goodlett DR, Lahdesmaki H, Simell O, Lahesmaa R. Serum Proteomes Distinguish Children Developing Type 1 Diabetes in a Cohort With HLA-Conferred Susceptibility. Diabetes. 2015;64(6):2265–78. doi: 10.2337/db14-0983. [DOI] [PubMed] [Google Scholar]
- 6.Hennig R, Cajic S, Borowiak M, Hoffmann M, Kottler R, Reichl U, Rapp E. Towards personalized diagnostics via longitudinal study of the human plasma N-glycome. Biochim Biophys Acta. 2016;1860(8):1728–38. doi: 10.1016/j.bbagen.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 7.Pernemalm M, Lehtio J. Mass spectrometry-based plasma proteomics: state of the art and future outlook. Expert Rev Proteomics. 2014;11(4):431–48. doi: 10.1586/14789450.2014.901157. [DOI] [PubMed] [Google Scholar]
- 8.Keshishian H, Burgess MW, Gillette MA, Mertins P, Clauser KR, Mani DR, Kuhn EW, Farrell LA, Gerszten RE, Carr SA. Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol Cell Proteomics. 2015;14(9):2375–93. doi: 10.1074/mcp.M114.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical chemistry. 2003;75(8):1895–904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 10.Cominetti O, Nunez Galindo A, Corthesy J, Oller Moreno S, Irincheeva I, Valsesia A, Astrup A, Saris WH, Hager J, Kussmann M, Dayon L. Proteomic Biomarker Discovery in 1000 Human Plasma Samples with Mass Spectrometry. J Proteome Res. 2016;15(2):389–99. doi: 10.1021/acs.jproteome.5b00901. [DOI] [PubMed] [Google Scholar]
- 11.McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, Gygi SP. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84(17):7469–78. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erickson BK, Jedrychowski MP, McAlister GC, Everley RA, Kunz R, Gygi SP. Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Analytical chemistry. 2015;87(2):1241–9. doi: 10.1021/ac503934f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy JP, Stepanova E, Everley RA, Paulo JA, Gygi SP. Comprehensive Temporal Protein Dynamics during the Diauxic Shift in Saccharomyces cerevisiae. Mol Cell Proteomics. 2015;14(9):2454–65. doi: 10.1074/mcp.M114.045849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norris JM, Yin X, Lamb MM, Barriga K, Seifert J, Hoffman M, Orton HD, Baron AE, Clare-Salzler M, Chase HP, Szabo NJ, Erlich H, Eisenbarth GS, Rewers M. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. Jama. 2007;298(12):1420–8. doi: 10.1001/jama.298.12.1420. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q, Fillmore TL, Schepmoes AA, Clauss TR, Gritsenko MA, Mueller PW, Rewers M, Atkinson MA, Smith RD, Metz TO. Serum proteomics reveals systemic dysregulation of innate immunity in type 1 diabetes. J Exp Med. 2013;210(1):191–203. doi: 10.1084/jem.20111843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmer JS, Monroe ME, Qian WJ, Smith RD. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom Rev. 2006;25(3):450–82. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T, Moore RJ, Klemke RL, Camp DG, 2nd, Smith RD. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11(10):2019–26. doi: 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu CW, Atkinson MA, Zhang Q. Type 1 diabetes cadaveric human pancreata exhibit a unique exocrine tissue proteomic profile. Proteomics. 2016;16(9):1432–46. doi: 10.1002/pmic.201500333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A, Mann M. The Q Exactive HF, a Benchtop Mass Spectrometer with a Pre-filter, High-performance Quadrupole and an Ultra-high-field Orbitrap Analyzer. Mol Cell Proteomics. 2014;13(12):3698–708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michalski A, Cox J, Mann M. More than 100, 000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J Proteome Res. 2011;10(4):1785–93. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 21.Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics. 2012;13(Suppl 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callister SJ, Barry RC, Adkins JN, Johnson ET, Qian WJ, Webb-Robertson BJ, Smith RD, Lipton MS. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J Proteome Res. 2006;5(2):277–86. doi: 10.1021/pr050300l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bates D, Maechler M, Bolker B, Walker S. lme4: Linear mixed-effects models using Eigen and S4. R package version 1.1–7. 2014 [Google Scholar]
- 24.Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ, Kitano H, Thomas PD. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic acids research. 2005;33(Database issue):D284–8. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 26.Rauniyar N, Yates JR., 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res. 2014;13(12):5293–309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith MP, Wood SL, Zougman A, Ho JT, Peng J, Jackson D, Cairns DA, Lewington AJ, Selby PJ, Banks RE. A systematic analysis of the effects of increasing degrees of serum immunodepletion in terms of depth of coverage and other key aspects in top-down and bottom-up proteomic analyses. Proteomics. 2011;11(11):2222–35. doi: 10.1002/pmic.201100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu C, Rudnick PA, Martinez MY, Cheek KL, Stein SE, Slebos RJ, Liebler DC. Depletion of abundant plasma proteins and limitations of plasma proteomics. J Proteome Res. 2010;9(10):4982–91. doi: 10.1021/pr100646w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 30.Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nature protocols. 2009;4(5):698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- 31.Shi T, Zhou JY, Gritsenko MA, Hossain M, Camp DG, 2nd, Smith RD, Qian WJ. IgY14 and SuperMix immunoaffinity separations coupled with liquid chromatography-mass spectrometry for human plasma proteomics biomarker discovery. Methods. 2012;56(2):246–53. doi: 10.1016/j.ymeth.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pichler P, Kocher T, Holzmann J, Mazanek M, Taus T, Ammerer G, Mechtler K. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal Chem. 2010;82(15):6549–58. doi: 10.1021/ac100890k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmstrom J, Ossola R, Watts JD, Lin B, Zhang H, Moritz RL, Aebersold R. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics. 2011;10(9):M110 006353. doi: 10.1074/mcp.M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou C, Simpson KL, Lancashire LJ, Walker MJ, Dawson MJ, Unwin RD, Rembielak A, Price P, West C, Dive C, Whetton AD. Statistical considerations of optimal study design for human plasma proteomics and biomarker discovery. J Proteome Res. 2012;11(4):2103–13. doi: 10.1021/pr200636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hafen R, Gosink L, McDermott J, Rodland K, Dam KK-V, Cleveland WS. Trelliscope: A system for detailed visualization in the deep analysis of large complex data, Large-Scale Data Analysis and Visualization (LDAV). 2013 IEEE Symposium on, IEEE; Atlanta, Georgia. 2013; pp. 105–112. [Google Scholar]
- 36.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocrine reviews. 1995;16(1):3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 37.Zofkova I. Pathophysiological and clinical importance of insulin-like growth factor-I with respect to bone metabolism. Physiological research/Academia Scientiarum Bohemoslovaca. 2003;52(6):657–79. [PubMed] [Google Scholar]
- 38.Fisher MC, Meyer C, Garber G, Dealy CN. Role of IGFBP2, IGF-I and IGF-II in regulating long bone growth. Bone. 2005;37(6):741–50. doi: 10.1016/j.bone.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 39.Diener A, Rohrmann S. Associations of serum carotenoid concentrations and fruit or vegetable consumption with serum insulin-like growth factor (IGF)-1 and IGF binding protein-3 concentrations in the Third National Health and Nutrition Examination Survey (NHANES III) Journal of nutritional science. 2016;5:e13. doi: 10.1017/jns.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dewailly D, Andersen CY, Balen A, Broekmans F, Dilaver N, Fanchin R, Griesinger G, Kelsey TW, La Marca A, Lambalk C, Mason H, Nelson SM, Visser JA, Wallace WH, Anderson RA. The physiology and clinical utility of anti-Mullerian hormone in women. Human reproduction update. 2014;20(3):370–85. doi: 10.1093/humupd/dmt062. [DOI] [PubMed] [Google Scholar]
- 41.Broer SL, Broekmans FJ, Laven JS, Fauser BC. Anti-Mullerian hormone: ovarian reserve testing and its potential clinical implications. Human reproduction update. 2014;20(5):688–701. doi: 10.1093/humupd/dmu020. [DOI] [PubMed] [Google Scholar]
- 42.Grinspon RP, Rey RA. Anti-mullerian hormone and sertoli cell function in paediatric male hypogonadism. Hormone research in paediatrics. 2010;73(2):81–92. doi: 10.1159/000277140. [DOI] [PubMed] [Google Scholar]
- 43.Lie Fong S, Visser JA, Welt CK, de Rijke YB, Eijkemans MJ, Broekmans FJ, Roes EM, Peters WH, Hokken-Koelega AC, Fauser BC, Themmen AP, de Jong FH, Schipper I, Laven JS. Serum anti-mullerian hormone levels in healthy females: a nomogram ranging from infancy to adulthood. The Journal of clinical endocrinology and metabolism. 2012;97(12):4650–5. doi: 10.1210/jc.2012-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hero M, Tommiska J, Vaaralahti K, Laitinen EM, Sipila I, Puhakka L, Dunkel L, Raivio T. Circulating antimullerian hormone levels in boys decline during early puberty and correlate with inhibin B. Fertility and sterility. 2012;97(5):1242–7. doi: 10.1016/j.fertnstert.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 45.Lofqvist C, Andersson E, Gelander L, Rosberg S, Hulthen L, Blum WF, Wikland KA. Reference values for insulin-like growth factor-binding protein-3 (IGFBP-3) and the ratio of insulin-like growth factor-I to IGFBP-3 throughout childhood and adolescence. J Clin Endocrinol Metab. 2005;90(3):1420–7. doi: 10.1210/jc.2004-0812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.