

Abstract
Proteorhodopsin, a retinal-binding protein, represents a potentially significant source of light-driven energy production in the world's oceans. The distribution of photochemically divergent proteorhodopsins is stratified according to depth. Here, we present evidence that such photochemical diversity was tuned by Darwinian selection. By using a Bayesian method, we identified sites targeted by Darwinian selection and mapped them to three-dimensional models of proteorhodopsins. We suggest that spectral fine-tuning results from the combined effect of amino acids that directly interact with retinal and those that influence the confirmation of the retinal-binding pocket.
Proteorhodopsin (PR) is a retinal-binding membrane protein that functions as a light-driven proton pump (1–3). It belongs to a superfamily of microbial rhodopsins and only recently was discovered in an uncultivated marine bacterium by using environmental genomic methods (1). The discovery had wide-ranging significance, because it suggested that a globally important source of light-driven energy production was operating in oceanic surface waters that previously had been unknown to science (1, 4). It is not known whether bacteria that harbor PR can fix CO2; however, it is suggested that PR-mediated phototrophy could support a large fraction of a cell's energy requirements (4). It is well known that an ability to produce energy from light is expected to lower overall respiratory energy requirements (5). Moreover, PR-bearing bacteria appear to be ubiquitous in oceanic surface waters, having been detected in culture-independent surveys of coastal and oceanic regions of the Antarctic and Pacific oceans and the Mediterranean, Red, and Sargasso seas (4, 6–8).
Bacterial PRs embody considerable genetic and functional diversity. Different genetic variants of PR appear to be spectrally fine-tuned to different oceanic habitats; presumably, the wave-length of light absorption has been adapted to match different light intensities in the marine environment (4). Based on absorption maxima, there are several different pigment families of PRs, with variants spanning the range from blue (490 nm) to green (540 nm). In addition to the divergence of blue-absorbing PRs (B-PRs) and green-absorbing PRs (G-PRs), several smaller-scale changes in absorption maxima evolved among G-PRs (7). A major genetic component of this spectral fine tuning has been resolved recently; most of the observed difference in spectral tuning between B-PRs and G-PRs arises from changes at a single amino acid residue [position 105 in the amino acid sequence used by Man et al. (7) and hereafter referred to as position 105].
The role of natural selection in the evolution of PR photochemical diversity is unclear. Given the potential significance of PRs in bioenergetics of oceanic bacteria, it is important to understand whether selective forces acting on light-driven bacterial metabolism are responsible for divergence in PR function and their distribution in the marine photic zone. In this study, we begin to address these issues. We assembled a data set of 75 PR sequences representing a wide range of geographic localities. We inferred a phylogeny for this sample of gene sequences, which served as a framework for inferring the history of selection pressure on the PR family. Our analyses suggest that PRs were adapted to different light intensities in the marine environment by a process of Darwinian evolution that involved substitutions of major effect, as well as substitutions for fine-tuning of absorption maxima.
Materials and Methods
Sequence Data. For the phylogenetic study of the PR family of proteins, 75 PR sequences were assembled, representing samples from the central northern Pacific, eastern Pacific, and Southern oceans, the Mediterranean Sea, and the Red Sea. These PR sequences are available from GenBank (accession nos. AF349976–AF350001, AF350003, AF279106, AY250714, AY250716–AY250734, AY250736, AY250737, AY210898–AY210900, AY210902–AY210904, AY210906–AY210919, AY598757, AY598758, and AY601905). For the purpose of identifying the root, we compiled a second data set by adding five “deep-branching” PR gene sequences (GenBank accession nos. AY250738–AY250741 and AY728898) to the 75-PR data set above. The deep-branching PR sequences are excluded from the codon-based analyses because of excessive substitutional saturation at synonymous sites. We will hereafter refer to these five sequences as “outgroups” and the data set that included the outgroups as the “80-PR data set.” Sequences were aligned by using clustalx (9), followed by manual adjustments. Numbering of sites in our alignment yielded a slight difference from the numbering of sites in the reference sequence used by Man et al. (7). To facilitate comparison among studies, we adopted the numbering system of Man et al. (7) and related that system of numbering sites to our alignment in Fig. 3, which is published as supporting information on the PNAS web site. Alignment of the 75- and 80-PR data sets are listed in Data Set 1 and Data Set 2, which are published as supporting information on the PNAS web site.
Phylogenetic Analysis. To infer a location for the root of the tree, we conducted a phylogenetic analysis of the 80-PR data set. The deep-branching PR gene sequences are highly divergent; hence, phylogenetic analysis of the 80-PR data set was based on amino acid variation alone. Maximum likelihood (ML) was used to estimate pairwise distances under the Whelan and Goldman (WAG) substitution matrix (10), combined with empirical estimates of amino acid frequencies and a gamma model of among-sites rate variation (11). Genetic distances were computed under the above model as implemented in the codeml program of the paml package (12). A phylogenetic tree was inferred from the matrix of pairwise distance by using the minimum evolution criterion, as implemented in the dambe program (13). This phylogeny is shown in Fig. 4, which is published as supporting information on the PNAS web site.
Phylogenetic analysis was conducted on the nucleotide sequences of the 75-PR data set. ML was used as the optimality criterion, and tree searches were conducted under the HKY85 substitution matrix (14), combined with a gamma model of among-sites rate variation (11), as implemented in paup* 4.0b10 (15). The resulting topology was rooted according to the location inferred under the tree estimated from the 80-PR data set. Ancestral codon states at each node of the 75-PR phylogeny were reconstructed by using the ML approach (marginal method) developed by Yang et al. (16).
We tested for among-sites variation in phylogenetic signal by using the difference in the sum of squares (DSS) statistic (17) as implemented in the program topal 2 (18). We found no evidence for such variability in the PR alignment (see Fig. 5, which is published as supporting information on the PNAS web site).
Statistical Inference of Natural Selection Pressure. The phylogenetic approach of Goldman and Yang (19) was used to measure selection pressure. In this approach (reviewed in refs. 20 and 21), a variety of Markov models are used that describe the substitution process between 61 of the 64 codons. Selection pressure is accommodated in the model by the ω parameter, which is the nonsynonymous/synonymous rate ratio (dN/dS). The ω parameter was estimated from the 75-PR data set by maximizing the likelihood of the data under a codon model with respect to the parameters of the ω distribution and any other free model parameters. All ML analyses were performed by using the codeml program of the paml package (12). The log likelihood was obtained for all models by performing multiple analyses by using a range of initial values for the ω parameter. Given ML estimates of the ω parameter consistent with positive Darwinian selection (ω > 1), and significant likelihood ratio tests (LRTs), it is desirable to infer which sites are under positive selection in each subset. We used an empirical Bayes approach (22) to predict which sites were most likely to have ω > 1 under model B. Results presented in this paper were obtained under model F3 × 4, which uses the nucleotide frequencies at the three positions of the codon to compute the expected codon frequencies (19). We obtained very similar results under the F61 model, which uses the 61 empirical codon frequencies as parameters (see Tables 4–6, which are published as supporting information on the PNAS web site).
Results and Discussion
To infer an evolutionary trajectory for spectral tuning, we assembled and analyzed 75 sequences representing samples from the central northern Pacific, eastern Pacific, and Southern oceans and the Mediterranean and Red seas. Phylogenetic analysis yielded the topology in Fig. 1. Genetic variation among the lineages in Fig. 1 is substantial. However, a recent study suggested amino acid variation at just a single site (position 105) functions as a “spectral tuning switch,” accounting for much of the difference between B-PR and G-PR light sensitivity (7). To determine when changes occurred at position 105, we inferred the ancestral PR sequences by using ML and mapped them to the phylogeny (Fig. 1). It was clear that changes at position 105 were discontinuous, being clustered in two different times during the history of the PR family (Fig. 1).
Fig. 1.

Phylogenetic tree inferred from nucleotide variation contained in the 75-PR data set. The branch lengths are scaled to the mean number of substitutions per codon site, as inferred under codon model M3. Branches along which amino acid substitutions had occurred at position 105 are highlighted in yellow and gray and are signified by FG. All other branches are signified by BG. The FG branch highlighted in gray corresponds to the evolution of B-PRs and is labeled B-FG. FG branches highlighted in yellow correspond to a period of diversification of green PRs and hence are labeled G-FG. Differences in selection pressure among BG and FG branches can be modeled by specifying different ω ratios for these sets of branches. Four hypotheses of variable selection pressure among BG and FG branches are specified as H0, H1, H2, and H3.
Next, we wanted to know whether selection pressures differed among branches where position 105 had changed (hereafter referred to as the foreground, or FG, branches) and branches where position 105 was invariant (hereafter referred to as the background, or BG, branches). To perform this study, we constructed a LRT for variation in selection pressure among branches (called a “branch model”), where selection pressure was measured by using the ratio (ω) of the nonsynonymous rate (dN) to the synonymous rate (dS) (reviewed in refs. 20 and 21). If amino acid changes are deleterious and subject to purifying selection, they will have a reduced fixation rate, and at such branches ω will be <1. Only when amino acid changes are selectively advantageous (i.e., positive Darwinian selection) will they be fixed at a rate greater than the neutral rate, and at such branches ω will be >1. The null model assumed one ω for all branches of the tree in Fig. 1. Three alternative models (H1, H2, and H3) were constructed that allowed for independent levels of selection pressure (ω) in two different sets of the FG branches (Fig. 1). LRTs of H1, H2, and H3 indicated that selection pressure along branches where substitutions had occurred at the spectral-switch site (position 105) differed significantly from selection pressure along branches where position 105 was invariant (Table 1). However, none of the estimated values of ω under H1, H2, and H3 were consistent with Darwinian selection.
Table 1. Parameter estimates and LRTs for models of variable selection pressure (ω) among lineages.
| Models | ωBG | ωG-FG | ωB-FG | 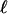 |
LRT |
|---|---|---|---|---|---|
| H0: ωBG = ωG-FG = ωB-FG | 0.07 | =ωBG | =ωBG | -6247.91 | NA |
| H1: ωBG = ωB-FG ≠ ωG-FG | 0.07 | 0.92 | =ωBG | -6244.26 | P = 0.007 |
| H2: ωBG = ωG-FG ≠ ωB-FG | 0.07 | =ωBG | 0.17 | -6245.56 | P = 0.030 |
| H3: ωBG ≠ ωG-FG = ωB-FG | 0.06 | 0.24 | 0.24 | -6242.97 | P = 0.007 |
The topology and branch specific ω ratios are presented in Fig. 1. The degrees of freedom for the LRTs are as follows: H0 vs. H1, df = 1; H0 vs. H2, df = 1; H0 vs. H3, df = 2. 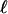 , log likelihood score; NA, not applicable.
, log likelihood score; NA, not applicable.
We were interested in assessing the possibility that a fraction of sites of the PR protein had evolved under Darwinian selection during the two periods of spectral tuning (Fig. 1), and the previously tested models were not capable of resolving such an episodic mode of adaptive evolution because they averaged ω over all codon sites in the data. Therefore, we used an LRT (23) to test for divergent selection pressure at sites along the G-FG and B-FG branches indicated in Fig. 1. The null hypothesis was specified as a model with two discrete categories of sites (called M3, k = 2, in ref. 24). The alternative model (called model B in ref. 23) assumed that selection pressure varied among sites, and at a subset of these sites, selection pressure changed in the FG branches. Four classes of sites were assumed. The first two classes (ω0 and ω1) were assumed to be homogenous over the entire phylogeny. The other two classes of sites were heterogeneous: Selection pressure changed from ω0 or ω1 in the BG branches to ω2 in the FG branches, i.e., one class had ω0 → ω2 and the other class had ω1 → ω2. The proportion of each class of sites in the gene and the ω parameters were estimated by ML (Table 2). We applied model B to the same three models (H1, H2, and H3) previously defined in Fig. 1. LRTs indicated significant support for a fraction of sites evolving under heterogeneous selection pressures between the BG and FG branches (Table 3).
Table 2. Parameter estimates and likelihood scores for the PR gene under different branch-site models.
| Codon model | Hypothesis | Estimates of ω parameters | Positive selection | 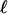 |
|---|---|---|---|---|
| M3 k = 2 | H0: BG = G-FG = B-FG | p0 = 0.81, ω0 = 0.02 | None | -6176.81 |
| (p1 = 0.18), ω1 = 0.33 | ||||
| Model B, ω2 = 1 | H1: BG = B-FG ≠ G-FG | p0 = 0.50, ω0 = 0.02 | G-FG: not allowed | -6167.16 |
| p1 = 0.12, ω1 = 0.31 | ||||
| (p2+3 = 0.38), (ω2 = 1) | BG+B-FG: none | |||
| H2: BG = G-FG ≠ B-FG | p0 = 0.78, ω0 = 0.03 | B-FG: not allowed | -6164.15 | |
| p1 = 0.13, ω1 = 0.38 | ||||
| (p2+3 = 0.08), (ω2 = 1) | BG+G-FG: none | |||
| H3: BG ≠ G-FG = B-FG | p0 = 0.75, ω0 = 0.03 | FG1 + FG2: not allowed | -6153.40 | |
| p1 = 0.13, ω1 = 0.36 | ||||
| (p2+3 = 0.12), (ω2 = 1) | BG: none | |||
| Model B | H1: BG = B-FG ≠ G-FG | p0 = 0.78, ω0 = 0.02 | G-FG: 5 sites | -6159.24 |
| p1 = 0.18, ω1 = 0.31 | ||||
| (p2+3 = 0.03), ω2 = 76 | BG+B-FG: none | |||
| H2: BG = G-FG ≠ B-FG | p0 = 0.82, ω0 = 0.03 | B-FG: 10 sites | -6156.29 | |
| p1 = 0.13, ω1 = 0.39 | ||||
| (p2+3 = 0.05), ω2 = 99 | BG+G-FG: none | |||
| H3: BG ≠ G-FG = B-FG | p0 = 0.80, ω0 = 0.03 | FG1+FG2: 16 sites | -6142.67 | |
| p1 = 0.13, ω1 = 0.37 | ||||
| (p2+3 = 0.06), ω2 = 14 | BG: none |
Tree topology, BG, G-FG, and B-FG are presented in Fig 1. 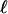 , log likelihood score. Boldface indicates an estimate value >1. Parentheses indicate a parameter value obtained by subtraction.
, log likelihood score. Boldface indicates an estimate value >1. Parentheses indicate a parameter value obtained by subtraction.
Table 3. LRTs for models of variable selection pressure (ω) among branches and sites.
| Null model ωFG | Alternative model ωFG | 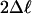 |
df | P |
|---|---|---|---|---|
| M3, k = 2 | Model B | |||
| H0: BG = G-FG = B-FG | H1: BG = B-FG ≠ G-FG | 35.14 | 2 | 2.3 × 10-8 |
| H0: BG = G-FG = B-FG | H2: BG = G-FG ≠ B-FG | 41.04 | 2 | 1.2 × 10-9 |
| H0: BG = G-FG = B-FG | H3: BG ≠ G-FG = B-FG | 68.28 | 2 | 1.5 × 10-15 |
| Model B1, ω2 = 1 | Model B2, ω2 ≥ 1 | |||
| H1: G-FG = 1 | H1: G-FG ≥ 1 | 14.90 | k | 3.5 × 10-5 |
| H2: B-FG = 1 | H2: B-FG ≥ 1 | 13.02 | k | 3.7 × 10-5 |
| H3: G-FG = B-FG = 1 | H3: G-FG = B-FG ≥ 1 | 19.87 | k | 1.9 × 10-6 |
Tree topology, BG, G-FG, and B-FG are identified in Fig. 1. k indicates that the LRT of model B1 against model B2 under the constraint of ω2 ≥ 1 was based on a 50:50 mixture of a chi-square distribution with one degree of freedom and a chi-square distribution with a point mass at zero.
In each case, parameter estimates under model B indicated that a small fraction of sites (3–5%) had evolved under very strong positive Darwinian selection in the FG branches (Table 2). The previous LRT tested for divergent selection pressure in the FG branch. To explicitly test the hypothesis that sites were evolving under positive selection in the FG branch, we implemented a modified LRT. The null was model B with ω2 = 1, and the alternative was model B with ω2 ≥ 1. Results were significant, indicating that selection pressure in the foreground branch was significantly greater than 1 (Table 3). Taken together, our results indicate an episodic mode of Darwinian evolution that is closely associated with the branches along which amino acids substitutions occurred at the spectral-switch site.
To determine the specific amino acid sites targeted for positive selection, we used an empirical Bayes approach (22). We compared the positive selection sites inferred under H1 and H2, because these two scenarios represent nonintersecting subsets of branches (Fig. 1). At a posterior probability >50%, 5 sites under H1 (positions 65, 68, 70, 101, and 105) and 10 sites under H2 (positions 25, 29, 40, 140, 145, 156, 157, 158, 169, and 188) were identified as potential targets of positive selection in the FG branches. None of the sites identified under H1 matched those identified under H2. These findings suggest that the sites targeted for positive selection shifted dramatically between the two episodes of Darwinian evolution.
Posterior probabilities for sites predicted to be evolving under positive Darwinian selection in the FG branches are included in Table 7, which is published as supporting information on the PNAS web site. Although the posterior probability provides a measure of reliability of this prediction, it is important that it is not misinterpreted, because systematic and random errors in parameter estimates affect the accuracy of inference; i.e., a posterior probability of 95% does not mean that there is only a 5% change that the site was not under positive selection (25, 26). However, simulation studies of site-models indicate that the type I error rate of Bayesian site prediction is most dependent on the number of lineages, and our sample of 75 sequences is well above the size required for reliable predictions (25). With regard to identifying positive selection sites (PS sites), the error rate can be excessively large when a fraction of sites is evolving under strict neutrality, e.g., ω = 1 (25–27). However, our LRT strongly rejected a model that included such sites, i.e., the rigid null with ω2 = 1, and the estimated value of ω2 was ≫1. Lastly, type I errors are expected to occur independent of any structural or functional features of the PR protein, yet the PS sites were nonrandomly distributed on the PR structural models. The G-PR sites were clustered very tightly within the center of the molecule, whereas the majority of B-PR sites were located on its external surfaces (Fig. 2).
Fig. 2.

Three-dimensional models of Béjà et al. (1) constructed by means of threading PR sequences on the 1.55-Å structure of bacteriorhodopsin (35). (A) Trace model of a G-PR. Amino acid sites inferred to be evolving under positive Darwinian selection are displayed in red as space-filled molecules. For reference, the lysine (position 232) that serves as the site for covalent attachment of retinal is shown in green. The amino acids serving as the primary proton donor (position 108) and acceptor (position 97) are shown in purple and blue, respectively. (B) Trace model of a B-PR. Amino acid sites inferred to be evolving under positive Darwinian selection are displayed in red as space-filled molecules. For clarity, positions 97 and 108 are not highlighted.
PR is an integral membrane protein composed of seven α-helices that form a pocket that encloses a single retinal chromophore (1, 4). The light-sensitive properties of PR proteins are derived from a covalent attachment of retinal to a lysine residue at position 232 by means of a protonated Schiff base linkage (7). The chromophore is activated by the absorption of a photon of light and functions to activate a biochemical cascade. Shifts in spectral sensitivity (absorption maxima) of PRs appear to result from altered interactions of the side chain of at least one amino acid of the retinal pocket and the retinal chromophore (7). We mapped the five amino acids with high posterior probabilities of being the targets of natural selection under H1 to the G-PR structural model; all five were highly localized, being <10 Å from the Schiff base (Fig. 2 A). Three are located at residues predicted to form the retinal binding pocket (positions 68, 101, and 105). Interestingly, all five formed a tight cluster between positions 97 and 108 (Fig. 2 A). Position 97 (Asp) functions as the primary proton acceptor, and position 108 (Glu) functions as the primary proton donor of the Schiff base proton (2, 3, 28). We hypothesize that the spatial distribution of positions 65, 68, 70, 101, and 105 in the G-PR structure is consistent with the action of natural selection to modulate the stability of the protonated Schiff base, thus altering the energy needed for photon absorption. We further speculate that spectral fine-tuning of the G-PRs resulted from the combined effect of those amino acids that form the retinal binding pocket (positions 68, 101, and 105), i.e., those that interact directly with the chromophore, and those nearby that could have an indirect effect by means of localized changes in the confirmation of the binding pocket (positions 65 and 70).
During the course of this study, Man-Aharonovich et al. (29) used site-directed mutagenesis to evaluate the effect of amino acid variation on the absorption spectrum of a Red Sea variant of G-PR (RS-29). This natural PR variant contains point mutations at positions 65, 68, 70, and 105. They found that the final spectrum appears to arise from the combined effect of a major blue shift (by means of a substitution at position 105) and two minor red shifts (by means of substitutions at positions 65 and 70). The biophysical analysis of Man-Aharonovich et al. (29) provides an independent verification of the functional importance of sites identified by our statistical approach. Our analysis demonstrates that amino acid variation at sites 65 and 70 had important fitness consequences in the natural oceanic environments of PR-bearing bacteria. Our findings, taken together with those of Man-Aharonovich et al. (29), demonstrate that amino acid substitutions that simply alter the intramolecular conformation of the binding pocket (i.e., at positions 65 and 70) can have important consequences for fine-tuning the phenotype and be subject to very strong selection pressures in the natural environment.
We also investigated the sites targeted by natural selection during the evolution of the B-PRs. Bayesian analysis identified 10 sites with high posterior probability of evolving under positive Darwinian selection during this period, hereafter referred to as blue-absorbing PS sites. In stark contrast to green-absorbing PS sites, the blue-absorbing PS sites were dispersed over the B-PR structure (Fig. 2B). With one exception, amino acids at the blue-absorbing PS sites appear unrelated to the retinal binding pocket (>10 Å from the Schiff base), and most are oriented toward the exterior of the protein. The exception is the amino acid at position 40, because it is closely associated with the Schiff base (232) (Fig. 2B). Biophysical analysis of a B-PR (28) suggests that in addition to differing in absorption maximum, the B-PR had a 10-fold slower photocycle. Interestingly, amino acid changes at position 105 do not account for the entire difference in photocycle; substituting a Q for an L at position 105 in G-PR did slow the photocycle by nearly 10-fold, but the reciprocal substitution in B-PR did not accelerate the photocycle, implying that differences at other sites can influence the photocycle speed (28). An intriguing hypothesis is that changes on these outward faces of the helix at B-PR sites are introducing conformational changes such as “kinks” that alter the distance between residues on the inward side, possibly influencing photocycle speed and spectral sensitivity. This hypothesis is not unprecedented; Chang et al. (30) hypothesized that a conserved proline is responsible for a kink in a helix of the vertebrate opsin that brings residues on the opposite side closer to each other and to the chromophore. Clearly, functional evolution of the B-PRs was accomplished by different means than was the diversification of G-PR spectral sensitivity (Fig. 2).
Interestingly, a significantly slower photocycle was measured in RS-29 as well as B-PR, leading to the hypothesis that they might have a sensory rather than a light-harvesting role (28, 29). PRs are likely to be complexed with other proteins in the natural environment. In the case of B-PR, the PS sites are distributed on the exterior of the protein structure, which is consistent with the possibility that Darwinian selection might have acted to convert its pumping activity into a sensory signal. The spatial pattern of sites targeted by positive selection within the G-PRs, however, is substantially different. One explanation of the G-PR pattern is that the combined effect of substitutions at sites with no detectable effect on absorption maximum, i.e., positions 68 and 101, were positively selected because they compensate for the negative effect on photocycle that changes at positions 65, 70, and 105 appear to have in RS-29. Further paleomolecular biochemistry on B- and G-PRs could test such questions directly.
Application of statistical methods has led to the discovery of many genes evolving under positive selection, with the majority relating to the immune response or sexual reproduction (20, 31). In such cases, these genes are typically called “fast” genes, because the evolutionary arms-race between host and parasite, or the evolutionary forces associated with sexual competition or conflict, leads to rapid evolution under diversifying selection. An alternative model exists for “directional” evolution for altered function in a novel environment; this model is strongly influenced by the extensive study of adaptive alteration of oxygen affinity in vertebrate hemoglobins (32, 33). Under the globin model, directional evolution proceeds by rare adaptive modifications of the protein at one or very few key positions, with all other substitutions being functionally neutral (32, 33). Indeed, direct examination of functional change in response to site-directed mutagenesis indicates that a large phenotypic change can arise in a variety of proteins, including PRs, from just one or a few amino acid changes (7, 30, 34). However, the results of site-directed mutagenesis do not constitute a proof of a hypothesis of natural selection. Observing a functional change in response to a substitution by site-directed mutagenesis does not mean that if an organism bore such a substitution it was subject to natural selection. Likewise, the lack of a detectable effect from a substitution is not proof that such a change did not have fitness consequences in the natural environment. All one can do is evaluate and refine a hypothesis about patterns and mechanisms of adaptive molecular evolution in light of all of the available evidence. In the case of PRs, the combination of phylogeny-based statistical analysis and site-directed mutagenesis suggested that substitutions with small or no measurable effect on absorption maxima might have had important fitness consequences in the oceanic environment. Although we suspect that much evolutionary change in proteins such as PRs is indeed neutral, we argue that the globin model for directional evolution, i.e., assuming that changes at other than the major-effect sites are selectively neutral (32, 33), should be treated cautiously.
Supplementary Material
Acknowledgments
We thank an anonymous referee for constructive comments on an earlier version of the manuscript. The work was supported in part by a start-up fund from Dalhousie University and by a grant from the Genome Atlantic Centre of Genome Canada (to J.P.B.). K.A.D. was supported by a Genome Atlantic postdoctoral fellowship. G.S. and O.B. were supported by Israel Science Foundation Grant 434/02 and Human Frontiers Science Program Grant P38/2002.
Author contributions: J.P.B. designed research; J.P.B., K.A.D., G.S., and O.B. performed research; J.P.B. and K.A.D. analyzed data; and J.P.B. and K.A.D. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BG, background; FG, foreground; LRT, likelihood ratio test; ML, maximum likelihood; PR, proteorhodopsin; G-PR, green-absorbing PR; B-PR, blue-absorbing PR; PS, positive selection.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AY250714, AY250716, AY250733, AY598757, AY598758, AY601905, and AY728898).
References
- 1.Béjà, O., Aravind, L., Koonin, E. V., Suzuki, M. T., Hadd, A., Nguyen, J. P., Jovanovich, S. B., Gates, C. M., Feldman, R. A., Spudich, J. L., et al. (2000) Science 289, 1902–1906. [DOI] [PubMed] [Google Scholar]
- 2.Dioumaev, A. K., Brown, L. S., Shih, J., Spudich, E. N., Spudich, J. L. & Lanyi, J. K. (2002) Biochemistry 41, 5348–5358. [DOI] [PubMed] [Google Scholar]
- 3.Friedrich, T., Geibel, S., Kalmback, R., Chizhov, I., Ataka, K., Heberle, J., Englehard, M. & Bamberg, E. (2002) J. Mol. Biol. 321, 821–838. [DOI] [PubMed] [Google Scholar]
- 4.Béjà, O., Spudich, E. N., Spudich, J. L., Leclerc, M. & DeLong, E. F. (2001) Nature 411, 786–789. [DOI] [PubMed] [Google Scholar]
- 5.Kolber, Z. S., VanDover, C. L., Niederman, R. A. & Falkowski, P. G. (2000) Nature 407, 177–179. [DOI] [PubMed] [Google Scholar]
- 6.Sabehi, G., Ramon, M., Bielawski, J. P., Rosenberg, M., Delong, E. F. & Béjà, O. (2003) Environ. Microbiol. 5, 842–849. [DOI] [PubMed] [Google Scholar]
- 7.Man, D., Wang, W., Sabehi, G., Aravind, L., Post, A. F., Massana, R., Spudich, E. N., Spudich, J. L. & Béjà, O. (2003) EMBO J. 22, 1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D., Paulsen, I., Nelson, K. E., Nelson, W., et al. (2004) Science 304, 58–60. [DOI] [PubMed] [Google Scholar]
- 9.Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. (1997) Nucleic Acids Res. 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whelan, S. & Goldman, N. (2001) Mol. Biol. Evol. 18, 691–699. [DOI] [PubMed] [Google Scholar]
- 11.Yang, Z. (1994) J. Mol. Evol. 39, 306–314. [DOI] [PubMed] [Google Scholar]
- 12.Yang, Z. (1997) Comput. Appl. Biosci. 13, 555–556. [DOI] [PubMed] [Google Scholar]
- 13.Xia, X. & Xie, Z. (2001) J. Hered. 92, 371–373. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa, M., Kishino, H. & Yano, T. (1985) J. Mol. Evol. 22, 160–174. [DOI] [PubMed] [Google Scholar]
- 15.Swofford, D. L. (2000) PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods) (Sinauer, Sunderland, MA), Version 4.0.
- 16.Yang, Z., Kumar, S. & Nei, M. (1995) Genetics 141, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGuire, G., Wright, F. & Prentice, M. J. (1997) Mol. Biol. Evol. 14, 1125–1131. [DOI] [PubMed] [Google Scholar]
- 18.McGuire, G. & Wright, F. (2000) Bioinformatics 16, 130–134. [DOI] [PubMed] [Google Scholar]
- 19.Goldman, N. & Yang, Z. (1994) Mol. Biol. Evol. 11, 725–736. [DOI] [PubMed] [Google Scholar]
- 20.Yang, Z. & Bielawski, J. P. (2000) Trends Ecol. Evol. 15, 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bielawski, J. P. & Yang, Z. (2003) J. Struct. Funct. Genomics 3, 201–212. [PubMed] [Google Scholar]
- 22.Nielsen, R. & Yang, Z. (1998) Genetics 148, 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang, Z. & Nielsen, R. (2002) Mol. Biol. Evol. 19, 908–917. [DOI] [PubMed] [Google Scholar]
- 24.Yang, Z., Nielsen, R., Goldman, N. & Pedersen, A.-M. K. (2000) Genetics 155, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anisimova, M., Bielawski, J. P. & Yang, Z. (2002) Mol. Biol. Evol. 19, 950–958. [DOI] [PubMed] [Google Scholar]
- 26.Zhang, J. (2004) Mol. Biol. Evol. 21, 1332–1339. [DOI] [PubMed] [Google Scholar]
- 27.Haydon, D. T., Bastos, A. D., Knowles, N. J. & Samuel, A. R. (2001) Genetics 157, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang, W.-W., Sineshchekov, O. A., Spudich, E. N. & Spudich, J. L. (2003) J. Biol. Chem. 278, 33985–33991. [DOI] [PubMed] [Google Scholar]
- 29.Man-Aharonovich, D., Sabehi, G., Sineshchekov, O. A., Spudich, E. N., Spudich, J. L. & Béjà, O. (2004) Photochem. Photobiol. 3, 459–462. [DOI] [PubMed] [Google Scholar]
- 30.Chang, B., Crandall, K. A., Carulli, J. P. & Hartl, D. L. (1995) Mol. Biol. Evol. 4, 31–43. [DOI] [PubMed] [Google Scholar]
- 31.Swanson, W. J. &Vacquier, V. D. (2002) Nat. Rev. Genet. 3, 137–144. [DOI] [PubMed] [Google Scholar]
- 32.Perutz, M. F. (1983) Mol. Biol. Evol. 1, 1–28. [DOI] [PubMed] [Google Scholar]
- 33.Poyart, C., Wajcman, H. & Kister, J. (1992) Respir. Physiol. 90, 3–17. [DOI] [PubMed] [Google Scholar]
- 34.Jessen, T.-H., Weber, R. E., Fermi, G., Tame, J. & Braunitzer, G. (1991) Proc. Natl. Acad. Sci. USA 88, 6519–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luecke, H., Schobert, B., Richter, H. T., Cartailler, J. P. & Lanyi, J. K. (1999) J. Mol. Biol. 291, 899–911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}