

Abstract
Objective(s)::
TLR-4 activates a number of inflammatory signaling pathways. Also, AMPK could be involved in anti-inflammatory signaling. The aim of this study was to identify whether stimulation of AMPK could inhibit LPS-induced Tlr-4 gene expression in mice hearts.
Materials and methods:
Heart AMPK activity and/or Tlr-4 expression was stimulated in different mice groups, using respectively IP injection of A-769662 (10 mg/kg) and LPS (2 mg/kg) or a combination of both agents. Moreover, compound-C (20 mg/kg), as an AMPK antagonist, was intraperitoneally co-administrated with both A-769662 and LPS in another group to investigate the role of AMPK activity on Tlr-4 regulation. After 8 hr, in addition to peripheral neutrophil cell count, myocardial p-AMPK, p-ACC as well as MyD88 protein contents and Tlr-4 expression was assessed by Western blotting and real-time qRT-PCR, respectively. TNF-α and IL-6 expression levels were also determined by ELISA.
Results:
LPS induced heart Tlr-4 expression (P<0.001) associating with an increase in the myocardial MyD88 protein content (P<0.001), elevation of heart TNF-α (P<0.01) and IL-6 (P<0.05) concentrations, and rise in the peripheral neutrophil cell count (P<0.001). Administration of A-769662 decreased LPS-induced Tlr-4 expression (P<0.01) and alleviated peripheral neutrophil cell count (P<0.01). The inhibitory effect of A-769662 on LPS-induced Tlr-4 expression was reversed by antagonizing AMPK with compound-C (P<0.001) which reduced p-AMPK (P<0.05) and p-ACC (P<0.01) myocardial protein contents in the LPS+A-769662 group.
Conclusion:
This study demonstrated that activation of AMPK, by A-769662 agent, could inhibit Tlr-4 expression and activity, suggesting a link between AMPK and Tlr-4 in heart tissue.
Keywords: ACC, A-769662, AMPK, Compound-C Lipopolysaccharide, TLR-4
Introduction
TLRs are transmembrane receptors that found on cell surfaces or on intracellular compartments (1). TLRs belong to the family of pattern recognition receptors diagnosing several pathogen-associated molecular patterns, such as cell-surface LPS (1, 2). They are also able to respond to stress and modulate inflammation and tissue damages following on non-infectious insults, like hypoxia and ischemia (1), in various tissues (3-6).
In human, 10 functional TLRs have been identified until now (2). Among this receptor family, TLR-2 and TLR-4 have major roles in myocardial Ischemia/-reperfusion (I/R) injury and some myocardial dysfunctions (7). hTLR-4 is generally expressed in macrophages and endothelial cells, as crucial contributor in the atherosclerotic plaque formation(8). We have previously shown a direct relationship between human monocyte TLR-4 expression and the respective cytokines, with a degree of coronary stenosis. In addition, it has been demonstrated that overexpression of hTLR-4 in the monocytes was associated with elevation of IL-1β and TNF-α levels in serum (9). We have also determined a positive correlation between hTLR-4 expression in monocytes and pro-inflammatory cytokine activities in patients with stable angina, who have undergone percutaneous coronary intervention (10). Furthermore, we currently showed that the heart is capable of producing TNF-α through Tlr-4 and MyD88 activation independent of classic immune system and suggest a local immune response in the heart (11).
Moreover, we have identified that activation of
AMPK prohibited Tlr-4 expression and activation in the heart suffering inflammatory conditions, including myocardial infarction (12, 13) or LPS-induced inflammation (14).
AMPK is a serine–threonine protein kinase regulating cellular metabolism and function (15). This enzyme, as a sensor of energy in cells, is activated as soon as shortage of nutrient supply and ATP generation or increased in cellular energy demand (16). Several studies have shown a correlation between AMPK and TLR-4 signaling. Santos et al reported (2013) that the effect of LPS on reduction of hypothalamic AMPK phosphorylation was TLR-4 dependent. Thus, this phosphorylation was increased by pharmacological hypothalamic AMPK activation (17). In this year, Park D et al also evaluated the effect of AMPK activation on neutrophil chemotaxis in vitro and in vivo models exposed to LPS. Their study indicated that the activation of AMPK decreased activity of TLR-4 downstream signals and, in contrast, enhanced neutrophil chemotaxis, both in vitro and in vivo (18). Although, correlation between TLR-4 and AMPK activity have previously been reported by Zhao et al in 2008. They demonstrated that AMPK function decreased TLR-4 induced neutrophil and pro-inflammatory cytokines activities (19). Whereas AMPK has potential anti-inflammatory effects, function of this enzyme was inhibited in TLR-4 activated cells due to translocation of HMGB1, a pro-inflammatory mediator, from nuclei to cytosol and inhibition of LKB1, as an upstream AMPK activator (20).
In the present study, we investigated the potential correlation of AMPK and Tlr-4 expressions in heart. For that, effect of the compound-C, as an AMPK antagonist, was evaluated on both myocardial AMPKα phosphorylation and Tlr-4 expression level in the administrated animals with A-769662+LPS agents. In this experiment, AMPK activity was initially stimulated by A-769662 agent and subsequently antagonized by compound-C in the LPS induced Tlr-4 expression group. In addition, we used metformin in some parts, as a well-known AMPK activator (12,13), to compare their effect with A-769662 agent, as a new AMPK activator.
Materials and Methods
Animals
Healthy male BALB/c mice (21) (25-30 g, 8-10 weeks of age) were used in this study. They were housed in standard polypropylene cages, six per cage, under a 12 hr light/dark at a controlled ambient temperature of 22±2 °C with 50±10% relative humidity. The present study was performed in accordance with the Guide for the Care and Use of Laboratory Animals of Tabriz University of Medical Sciences, Tabriz, Iran (National Institutes of Health Publication No. 85-23, revised 1985).
Chemicals
Escherichia coli (serotype k235) lipopolysaccharide was purchased from Sigma (Germany). A-769662 was from Tocris Bioscience (USA) and metformin was a gift from Osveh Pharmaceutical Inc. (Tehran, Iran). Compound-C was from Sigma (Germany). Rabbit monoclonal antibodies against phospho-AMPKα (Thr172), AMPKα and ACC, phospho-ACC (Ser79) and MyD88 were obtained from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibodies against GAPDH, peroxidase-conjugated goat anti-rabbit, and rabbit anti-mouse secondary antibodies were obtained from Abcam (Cambridge, MA). For ELISA, Rat TNF-α and IL-6 were bought from Bendermed (Austria). Protease inhibitor cocktail was from Roche (Mannheim, Germany).
Evaluation of required time and LPS concentration for MyD88 and Tlr-4 expression in mice
The mice model of LPS-induced inflammation was used in this study as previously reported by Ao et al (2007) (22) with modifications. Six groups of mice (n=3-4) were used for optimizing of endotoxin concentration and required time to achieve MyD88 and Tlr-4 expression: 1) two normal control groups were given vehicle intraperitoneally (normal saline) for 4 hr and 8 hr separately; 2) mice with endotoxemia were injected LPS (1 mg/kg and 2 mg/kg; IP) both for 4 hr and 8 hr. After LPS injection time set zero and at the end of the experiment, the heart was removed under anesthesia, rinsed in cold saline, snap-frozen in liquid nitrogen and stored at -70 °C for evaluating the level of MyD88 and Tlr-4 expression by Western blotting and real-time PCR, separately.
Dose response of compound-C inhibitory effects on AMPK
To evaluate the inhibitory dose of compound-C on AMPK, six groups of mice (n=3) were used: control who received vehicle (PBS and DMSO), IP; compound-C 10 and 20 mg/kg, separately (23), IP; compound-C (10 and 20 mg/kg, IP separately)+A-769662 (10 mg/kg, IP) and compound-C (20 mg/kg, IP)+metformin (100 mg/kg, IP). Eight hours later, the heart was removed under anesthesia, rinsed in cold saline and snap-frozen in liquid nitrogen and stored at -70 °C for evaluating the level of AMPK activation by Western blotting. The dose of A-769662 (24, 25) and metformin (23) used here, based on studies published in the literature, would be predicted to activate AMPK.
Experimental protocols to evaluate whether AMPK and TLR-4 are connected
The study was conducted on 11 groups of animals; each group consisted of six animals (Table 1). Eight hours following the IP injection of the drugs, the animals were anaesthetized by sodium pentobarbital. The heart, were taken out immediately by dissection, washed in ice-cold saline, snap-frozen in liquid nitrogen and were kept at −70°C for further analysis.
Table 1.
Animal groups and respective treatments
| Groups | Treatments | |
|---|---|---|
| 1 | Control (C) | Solvents (DMSO 80 µl+PBS1) |
| 2 | LPS (L) | 2 mg/kg |
| 3 | Compound-C (CC) | 20 mg/kg |
| 4 | Metformin (M) | 100 mg/kg |
| 5 | A-769662 (A) | 10 mg/kg |
| 6 | Metformin+compound-C (M+CC) | 100 mg/kg+20 mg/kg |
| 7 | A-769662+compound-C (A+CC) | 10 mg/kg+20 mg/kg |
| 8 | Metformin+LPS (M+L) | 100 mg/kg+2 mg/kg |
| 9 | A-769662+LPS (A+L) | 10 mg/kg+2 mg/kg |
| 10 | Metformin+LPS+compound-C (M+L+CC) | 100 mg/kg+2 mg/kg+20 mg/kg |
| 11 | A-769662+LPS+compound-C (A+L+CC) | 10 mg/kg+2 mg/kg+20 mg/kg |
phosphate buffer solution
Measurements of cardiac TNF-α and IL-6 concentration by ELISA
Cardiac level of TNF-α and IL-6 were quantified with the use of ELISA kit according to the manufacture’s instruction. Briefly, the samples were homogenized in an ice-cold solution containing 50 mM Tris–HCl, 150 mM NaCl, 5 mM sodium pyrophosphate (NaPPi), 50 mM NaF, 1 mM EDTA, 1 mM dithiothreitol (DTT), 0.1 %SDS (w/v), 1% TXT-100 (v/v), and protease inhibitor cocktail (Roche, Mannheim, Germany). The samples were then centrifuged twice at 10,000 (rpm) for 10 min at 4 °C. The resulting supernatants were used for assay. The concentration of the cytokines was expressed as pg/100 mg heart tissue.
Neutrophil count in blood
Prior to euthanization, venous blood sample was collected to determine the number of neutrophils in blood. Fresh blood sample was smeared on the clean lam and the percent of neutrophils were counted at 100×zooming in optical microscope after Gimsa coloring. The percentage of neutrophils was calculated as a percentage of total white blood cells.
Western blot analysis
Western blot analyses were performed as previously described by Soraya et al (2012) (12) with minor modifications. Following the experimental procedure, myocardial tissue was removed immediately and was deep-frozen in liquid nitrogen.
The tissue sample was homogenized in ice-cold solution, pH 7.4, containing 50 mM Tris–HCl, 150 mM NaCl, 5 mM sodium pyrophosphate (NaPPi), 50 mM NaF, 1 mM EDTA, 1 mM dithiothreitol (DTT), 0.1% SDS (w/v), 1% TXT-100 (v/v), and protease inhibitor cocktail. Homogenized sample was centrifuged at 10,000 rpm at 4 °C for 10 min. The supernatant was aliquoted and stored at -70 °C for further analysis. Bradford protein assay kit was used for evaluate the protein content of the supernatant. Same weight of the homogenate protein was loaded to SDS-polyacrylamide gel electrophoresis using Bio-Rad mini protean tetra system. The separated proteins were transferred to an Immobilon-P membrane and blocked in 5% non-fat milk. The membranes were probed using a range of primary antibodies raised against GAPDH (1:5000), MyD88, phospho-AMPKα (Thr172), AMPKα, ACC, and p-ACC (Ser79) (1:1000 dilution) at 4 ºC with gentle shaking, overnight. The membranes then were washed and incubated with peroxidase-conjugated goat anti-rabbit and rabbit anti-mouse secondary antibodies (1:5000 dilutions) at room temperature with gentle shaking. The antibodies were visualized using the BM Chemiluminescence kit. Densitometric analysis of the immunoblots was performed using image j software (Wayne Rasband, National Institute of Health, USA). The densitometric values for MyD88, p-AMPK and p-ACC were normalized to GAPDH, AMPK and ACC, respectively.
Detection of the Tlr-4 mRNA in cardiac tissue by quantitative real-time PCR
Myocardium total RNA was extracted using Trizol according to the manufacturer’s recommendations. Agarose electrophoresis was used to evaluate the integrity of the extracted RNA. Purity of RNA was determined by optical density measurement (A260/A280 Ratio) with nanodrop instrument. For cDNA synthesis one μg of the extracted RNA from each sample was used. Real time PCR was performed by dNTP mix (Biofluxbiotech), random hexamer primer, Ribonuclease Inhibitor, and M-MuLV Reverse Transcriptase (CinnaGen, Iran). All reactions were performed in a total volume of 25 μl reaction mixtures, triplicate using the iQ5 optical system. Negative control as well as no template control (NTC) was included in each experiment.
Reaction mixture contained: 1 μl cDNA, 1 μl primer (100 nM each primer), 12.5 μl 2X Power SYBR Green PCR Master Mix (Applied Biosystems, Foster city, USA), and 10.5 μl RNase/DNase free water. Target mRNA expression was normalized to GAPDH expression. The cycling conditions were as follow:
For both Tlr-4 and Gapdh: 1 cycle at 94 °C for 10 min, 40 cycles at 94 °C for 15 sec, 60 °C (annealing temperature) for 30 s and 72 °C for 30 s, and 1 cycle at 72 °C for 5 sec and 53 °C for 10 min.
The primers were designed for detection, as given below:
For Tlr-4,
forward: 5’-AAG TTA TTG TGG TGG TGT CTA G-3’;
reverse: 5’-GAG GTA GGT GTT TCT GCT AAG-3’.
For Gapdh,
forward: 5’-TTG TCA AGC TCA TTT CCT GGT ATG -3’;
reverse: 5’-GGA TAG GGC CTC TCT TGC TCA -3’.
The relative amount of Tlr-4 mRNA to the reference Gapdh gene was calculated using the 2-∆∆cT method (26) and expressed as an n-fold variation of Tlr-4 levels in the treated samples with respect to control samples (n=3-5).
Statistical analysis
In this experimental research, data were presented as mean±SEM. One-way ANOVA was used to make comparisons between the groups, by SPSS 12.0 software. If the ANOVA analysis indicated significant differences, LSD post hoc test was performed to compare the mean values between the treatment groups and the control. For the real time PCR, Pair Wise Fixed Reallocation Randomization Test using REST software was used to make comparisons between the groups. Any differences between the groups were considered significant at P<0.05.
Results
Required time and LPS concentration for MyD88 and Tlr-4 expression in mice
LPS acts as a ligand for TLRs, especially TLR-4. Binding LPS to TLR-4 activates either MyD88 or TRIF downstream pathways (27). Here, the concentration and the time spent for expression of Tlr-4 mRNA and so increasing MyD88 protein content by IP injection of LPS was evaluated. As demonstrated in Figure 1A increasing the level of MyD88 by LPS seems time and concentration dependent. As shown, there was no significant difference between the levels of MyD88/GAPDH content in the hearts obtained from mice injected LPS at 1 or 2 mg/kg after 4 hr and 1 mg/kg after 8 hr vs control. However, LPS at 2 mg/kg could elevate prominently the level of MyD88/GAPDH protein content after 8 hr (P<0.001 vs control 8h).
Figure 1.
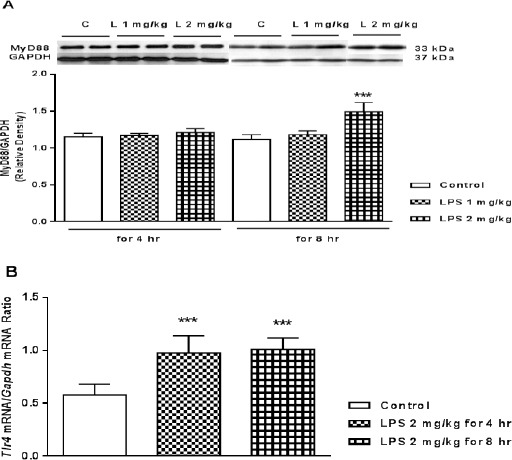
A) Representative immunoblots of heart myeloid differentiation factor 88 (MyD88) protein content (upper traces above the columns) in mice following 4 and 8 hr treatment with LPS (both 1 and 2 mg/kg, IP). Bars represent the ratio of MyD88 to GAPDH. ***P<0.001 vs. control 8 hr, using one way ANOVA with LSD post hoc test (L: LPS and C: control). B) Expression of Tlr-4 mRNA in mice myocardial tissues following 4 and 8 hr treatment, with LPS (2 mg/kg, IP). The expression of Tlr-4 mRNA was analyzed by real time PCR. Data are expressed as the mean±SEM of the three independent experiments with three repeat. The significant of results was tested by a Pair Wise Fixed Reallocation Randomization Test using REST software. Values are mean±SEM (n=4). ***P <0.001 from respective control value
In comparison with the control group, Tlr-4 mRNA expression significantly was elevated 4 and 8 hr after IP injection of LPS at 2 mg/kg (P<0.001, respectively) (Figure 1B).
Inhibitory concentration of compound-C on activated AMPK
A-769662 is a direct and metformin is indirect activator of AMPK. Also, compound-C is a potent, selective and reversible inhibitor of AMPK. So, it was subsequently investigated the required concentration of compound-C for inhibiting activated AMPK induced by A-769662 (10 mg/kg) and metformin (100 mg/kg).
As illustrated in Figure 2 both A-769662 (10 mg/kg, IP) and metformin (100 mg/kg, IP) increased the level of phosphorylated AMPKα at threonine residue 172 (P<0.05 and P<0.01, respectively) and adding compound-C with 20 mg/kg, IP could significantly decreased the level of AMPK activity (P<0.05). However, there was no significant difference between compound-C (20 mg/kg) and control group. Compound-C with 10 mg/kg, IP had no significant effect on the level of AMPK activity (data are not shown).
Figure 2.
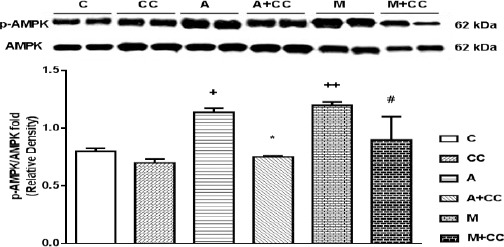
Representative immunoblots of p-AMPK and AMPK following 8 hr treatment, intraperitoneally, with CC (compound-C 20 mg/kg), A (A-769662 10 mg/kg), A+CC (A-769662 10 mg/kg+compound-C 20 mg/kg), M (metformin 100 mg/kg) and M+CC (metformin 100 mg/kg+compound-C 20 mg/kg) where C is control. Bars represent the ratio of p-AMPK to AMPK. Values are mean±SEM (n=3). +P<0.5 and ++P<0.01 from respective control value, *P<0.05 vs. A-769662, and #P<0.05 vs. metformin, using one way ANOVA with LSD post hoc test
The effect of compound-C on the level of cardiac AMPK and ACC activity in A-769662+LPS treated mice
A-769662 is a direct AMPK activator and LPS acts as a ligand for TLR-4. Furthermore, AMPK activation converts the ACC into an inactive form by phosphorylation on ser79 residue. For examining the possible connection between TLR-4 and AMPK activity, we determined the effect of compound-C on myocardial AMPKα and ACC phosphorylation level in the mice injected A-769662+LPS in which, activated AMPK (by A-769662) would be inhibited (by compound-C) in LPS induced Tlr-4 expression group. Western blot results in Figure 3A showed that both LPS and A-769662 induced a significant AMPK activation as the expressions of phospho (p)-AMPKα was increased after administration in the heart tissue (P<0.001) and there is no difference in the AMPK activity between LPS and A-769662 treated groups. Notably, p-AMPK/AMPK fold was elevated both in A-769662+LPS (P<0.001) and A-769662+LPS+compound-C (P<0.05). However, compound C decreased the heart p-AMPK/AMPK fold in A-769662+LPS+compound-C group in comparison with A-769662+LPS group (P<0.05). Besides, antagonizing AMPK with compound-C induced a significant reduction in ACC phosphorylation in comparison with LPS+A-769662 (P<0.01) which showed alleviating effects of AMPK activation (Figure 3B).
Figure 3.
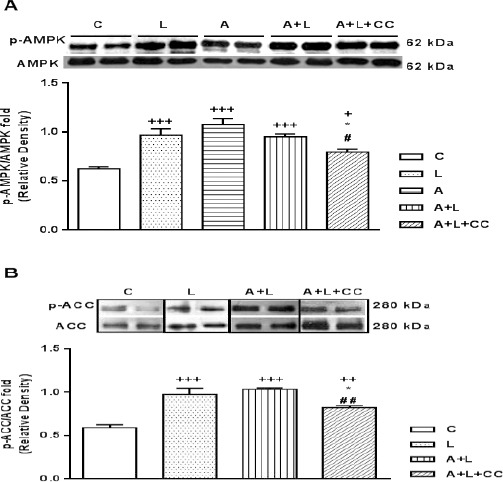
A) Representative immunoblots of phosphorylated AMPKα at residue threonine 172 (p-AMPKα) and total AMPKα. Bars represent the ratio of phosphorylated AMPKα to total AMPKα. B) Representative immunoblots of phosphorylated ACC at residue Serine 79 (p-ACC) and ACC. Bars represent the ratio of phosphorylated ACC to ACC. Following eight hr after IP injection of LPS (2 mg/kg), A (A-769662 10 mg/kg), A+L (A-769662 10 mg/kg+LPS 2 mg/kg) and A+L+CC (A-769662 mg/kg+2 mg/kg+compound-C 20 mg/kg) where C is control. Values are mean±SEM (n=5). +P<0.05, ++P<0.01 and +++P<0.001 from respective control value, *P<0.05 as compared with the LPS group, #P<0.05 and ##P<0.01 as compared with the A+L group, using one way ANOVA with LSD post hoc test
The effect of compound-C on the cardiac Level of Tlr-4 mRNA in A-769662+LPS treated mice
Real-time quantitative PCR results showed that the Tlr-4 mRNA level in the LPS and also A-769662+LPS+compound-C treated mice reached to more than 2 times its value in the control group (Figure 4; P<0.001, respectively). Where adding A-769662 along with LPS decreased the level of Tlr-4 mRNA in comparison to LPS treated alone (P<0.01) which reduced it to the level of Tlr-4 in control group. Besides, adding compound-C to A-769662+LPS group reversed the impact of A-769662 on LPS effects and increased the level of Tlr-4 mRNA expression in comparison with A-769662+LPS alone (P<0.001). Also, the level of myocardial Tlr-4 expression was elevated in LPS treated group in comparison with A-769662, compound-C (P<0.01, respectively) and A-769662 along with compound-C (P<0.05) and there is no significant differences between A-769662, compound-C, A-769662+compound-C and A-769662+LPS with control.
Figure 4.
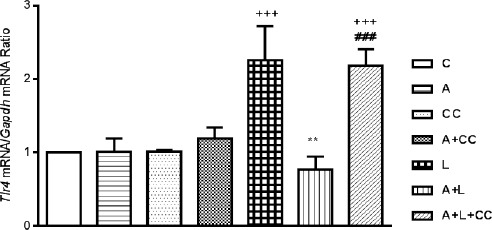
Expression of Tlr-4 mRNA in mice myocardial tissues following 8 hr treatment, intraperitoneally, with A (A-769662 10 mg/kg), CC (compound-C 20 mg/kg), A+CC (A-769662 10 mg/kg+compound-C.20 mg/kg), L (LPS 2 mg/kg), A+L (A-769662 10 mg/kg+LPS 2 mg/kg) and A+L+CC (A-769662 10 mg/kg+2 mg/kg+compound-C 20 mg/kg) where C is control. mRNA was isolated and converted to cDNA. The expression of Tlr-4 mRNA was analyzed by real time PCR. Data are expressed as the mean±SEM of the four independent experiments. The significant of results was tested by a Pair Wise Fixed Reallocation Randomization Test using REST software. +++P<0.001 from respective control value; **P<0.01 as compared with the LPS group, ###P<0.001 as compared with the A+L group
The effect of compound- C on the myocardial MyD88 protein expression in A-769662+LPS treated mice
The level of MyD88, TLR-4 adaptor protein for pro-inflammatory genes activation, was assessed to determine the effect of adminstratin of compound-C along with A-769662+LPS on the level of MyD88 in the heart tissue.
Compared with the normal control group, the MyD88 protein content in the heart of LPS and A-769662+LPS (P<0.001, respectively) and A-769662+LPS+compound-C (P<0.05) group, was elevated (Figure 5). Also, there is no difference between MyD88 protein content of LPS and A-769662+LPS group. The rise in MyD88 protein in A-769662+LPS group was significantly reduced by adding compound-C along with A-769662+LPS (P<0.001) where there is a significant differences among LPS and A-769662+LPS+compound-C (P<0.001).
Figure 5.
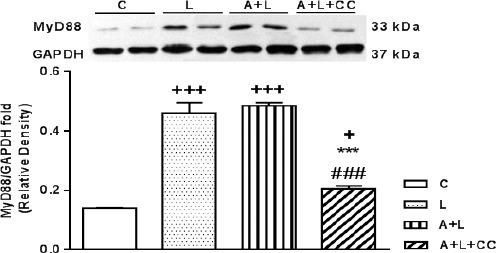
Representative immunoblots of MyD88 and GAPDH following 8 hr treatment, intraperitoneally, with L (LPS 2 mg/kg), A+L (A-769662 10 mg/kg+LPS 2 mg/kg) and A+L+CC (A-769662 10 mg/kg+2 mg/kg+compound-C 20 mg/kg) where C is control. Bars represent the ratio of MyD88 to GAPDH. Values are mean±SEM (n=5). +P<0.05, +++P<0.001 from respective control value, ***P<0.001 as compared with the LPS group and ###P<0.001 as compared with the A+L group, using one way ANOVA with LSD post hoc test
Data are expressed as the mean±SEM of the five independent experiments. The significant of results was tested by one way ANOVA with LSD post hoc test. +P<0.05, ++P<0.01 and +++P<0.001 from respective control value; **P<0.01 and ***P<0.001 as compared with the LPS group, ###P<0.001 as compared with the A+L/M+L group
The effect of compound-C on the cardiac TNF-α and IL-6 concentration in A-769662+LPS and metformin+LPS treated mice
The effect of compound-C along with A-769662 on the cardiac concentration of TNF-α in mice with LPS induced endotoxemia was assessed. As demonstrated in Figure 6, the cardiac level of TNF-α was profoundly increased by LPS from 527.9±38.5 pg/ml in the normal control to 731.8±36.7 pg/ml in the LPS group (P<0.001). The cardiac concentration of TNF-α in the mice that had received A-769662 immediately before LPS injection had no significant differences with LPS treated alone (819.6±57.7 pg/ml). Besides, inhibiting AMPK activity with compound-C increased the TNF-α concentration to 1117.3±20.7 pg/ml, higher than LPS or A-769662 along with LPS treated groups (P<0.001).
Figure 6.
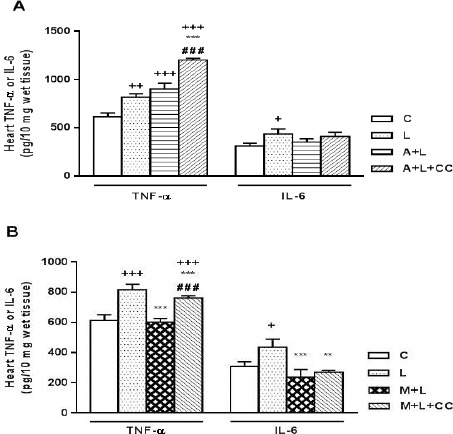
A) Concentration of TNF-α and IL-6 in mice myocardial tissues following 8 hr treatment, intraperitoneally, with L (LPS 2 mg/kg), A+L (A-769662 10 mg/kg+LPS 2 mg/kg) and A+L+CC (A-769662 10 mg/kg+2 mg/kg+compound-C 20 mg/kg) where C is control. Data are expressed as the mean±SEM of the four independent experiments. B) Concentration of TNF-α and IL-6 in mice myocardial tissues following 8 hr treatment, intraperitoneally, with L (LPS 2 mg/kg), M+L (metformin 100 mg/kg+LPS 2 mg/kg) and M+L+CC (metformin 100 mg/kg+2 mg/kg+compound-C 20 mg/kg) where C is control.
The effect of compound-C along with A-769662 on the cardiac concentration of IL-6 in mice with LPS induced endotoxemia was assessed. As shown in Figure 7, the cardiac level of IL-6 was profoundly increased by LPS from 285.84±31.4 pg/ml in the normal control to 412.3±53.5 pg/ml in the LPS group (P<0.05). The cardiac concentration of IL-6 in the mice that had received A-769662 immediately before LPS injection was alleviated to the value of normal control group (328.8±33.8 pg/ml), but there is no significant differences between LPS treated mice and LPS along with A-769662 group. Besides, inhibiting AMPK activity with compound-C reversed the effects of A-769662 on LPS induced IL-6 rise but not significantly in comparison with A-769662+LPS group (387.6±41.7 pg/ml).
Figure 7.
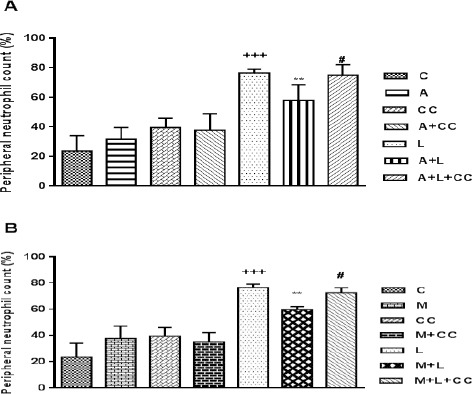
A) Peripheral neutrophil count following 8 hr treatment, intraperitoneally, with A (A-769662 10 mg/kg), CC (compound-C 20 mg/kg), A+CC (A-769662 10 mg/kg+compound-C.20 mg/kg), L (LPS 2 mg/kg), A+L (A-769662 10 mg/kg+LPS 2 mg/kg) and A+L+CC (A-769662 10 mg/kg+2 mg/kg+compound-C 20 mg/kg) where C is control. B) Peripheral neutrophil count following 8 hr treatment, intraperitoneally, with M (metformin 100 mg/kg), CC (compound-C 20 mg/kg), M+CC (metformin 100 mg/kg+compound-C. 20 mg/kg), L (LPS 2 mg/kg), M+L (metformin 100 mg/kg+LPS 2 mg/kg) and M+L+CC (metformin 100 mg/kg+2 mg/kg+compound-C 20 mg/kg) where C is control. Data are expressed as the mean±SEM of the five independent experiments. The significant of results was tested by one way ANOVA with LSD post hoc test. +++P<0.001 from respective control value, **P<0.01 as compared with the LPS group, #P<0.05 as compared with the A+L/M+L group
The effect of compound-C along with metformin (a well-known AMPK activator) on the cardiac concen-tration of TNF-α and IL-6 in mice with LPS induced endotoxemia was assessed, too. As demonstrated in Figure 6, the cardiac concentration of TNF-α in the mice that had received metformin immediately before LPS injection was significantly (P<0.001) alleviated to the value of normal control group (516.3±24 pg/ml). However, inhibiting AMPK activity with compound-C reversed the effects of metformin on LPS induced TNF-α rise and elevated cardiac TNF-α concentration to 676.5±14.5 pg/ml (P<0.001 vs metformin+LPS).
As illustrated in Figure 6, the cardiac concentration of IL-6 in the mice that had received metformin immediately before LPS injection was significantly (P<0.001) alleviated to the value of normal control group (215.5±50 pg/ml). Besides, inhibiting AMPK activity with compound-C reversed the effects of metformin on LPS induced IL-6 rise but not significantly (246.6±12.8 pg/ml).
The effect of compound-C on the peripheral neutrophil count in A-769662+LPS and metformin+LPS treated mice
As demonstrated in Figure 7, injection of LPS to animals caused a prominent elevation in the percentage of neutrophil from 33.5±10.6% of the total white blood cells in normal control to 76.5±2.6% (Figure 7). While A-769662 decreased percentage of peripheral neutrophil count from 76.5±2.6% in LPS group to 58±10.6% in A-769662+LPS group. Furthermore, using compound-C with A-769662+LPS reversed the reduction effect of A-769662 by significant reducing peripheral neutrophil count to 75±7.1% (P<0.05 vs. A-769662+LPS) (Figure 7). Besides, IP administration of metformin to LPS treated mice considerably (P<0.01) reduced the percentage of peripheral neutrophil to 59.8±2.1% and adding compound-C in metformin+LPS+compound-C group increased the percentage of peripheral neutrophil count to 72.7±3.7% (P<0.05 vs metformin+LPS group).
Discussion
In the present study, we evaluated the correlation of AMPK activity and Tlr-4 expression. As a critical finding, we determined that A-769662 suppressed LPS-induced expression of Tlr-4 in the heart tissue, by mediating AMPK activation. We also demonstrated that compound-C, as an inhibitor of AMPK, was able to harbor A-769662 effect on AMPK activity and Tlr-4 expression. In addition, administration of compound-C along with A-769662+LPS led to decreased level of p-AMPK and p-ACC compared to AMPK and ACC, respectively.
A-769662 is a non-nucleoside small thienopyridone molecule with high specificity for AMPK (28). At the molecular level, AMPK is a heterotrimer complex comprised of α, β, and γ subunits (29). A-769662 activates AMPK by directly binding to the β subunit of AMPK (28) independent of AMP: ATP ratio (24, 30). Several studies showed that AMPK activity prevented inflammatory reactions, while loss of this protein function could induce inflammation (19, 31, 32).
TLRs are transmembrane receptors found on the cell surface or intracellular compartments (1). TLRs activate a number of signaling pathways, among which MyD88, as myeloid differentiation primary response protein, is a critical adaptor protein. Activation of MyD88 promotes NFκB translocation into the nucleus and induces the gene expression of proinflammatory cytokines, particularly TNF-α and IL-6 (33). Thus far, potential relationship between AMPK activity and TLR-4 expression, particularly in vital tissues including heart, has not been appropriately elucidated and this is the first report demonstrating the effect of A-769662 agent on Tlr-4 gene expression, by activation of AMPK.
On the other hand, compound-C is a cell-permeable pyrazolo pyrimidine agent, functioning as a reversible and ATP competitive inhibitor of AMPK (34). This compound is incrementally utilized to inhibit AMPK activity in the cell-based assays, although it is not able to inhibit AMPK function in response to all stimuli. Thus, this pharmacological inhibitor blunted AICAR-induced, but not dinitrophenol-induced (35) or the LPS-induced (36), activity of AMPK. Consistent with the present findings, Kim et al (2014) showed the ability of compound-C to prohibit A-769662 induced AMPK activation (37). In addition, several investigations implicated on the inhibitory effects of compound-C on AMPK activity induced by metformin (38, 39).
The inflammatory responses in sepsis are primarily initiated by bacterial lipopolysaccharide as an endotoxin. During endotoxemia, LPS acts as a ligand to recognize the receptors which are expressed on immune cells and known as TLRs, particularly TLR-4 (27,40). It has been demonstrated that LPS function increased TLR-4 mRNA levels (41). Herein, using qRT-PCR, we showed that LPS had a strong effect on the Tlr-4 gene expression in the heart tissue. We determined that treatment with A-769662 profoundly suppressed the LPS induced Tlr-4 elevation and antagonizing AMPK with compound-C reversed the suppressive effects of A-769662 on LPS induced Tlr-4 expression. These results indicated the correlation of AMPK activity and Tlr-4 expression.
We have previously revealed that isoproterenol induced myocardial infarction was associated with a reduction of p-AMPK along with a significant increase in the Tlr-4 activity. Short term treatment with different doses of metformin increased the AMPK activity and, in contrast, remarkably reduced Tlr-4 activity (12). In addition, Zhao et al (2009) showed that deficiency of Tlr-4 had a cardio-protective effects on ischemic injury, in an regional ischemia induced in wild type c3H/HeN and Tlr-4 mutated C3H/HeJ mice, due to activation of AMPK and ERK signaling pathways (42). These findings are in agreement with Fang et al reports (2011) indicated that there was a correlation between TLR-4 and some other distinctive signaling pathways, like phosphatidyl inositol 3-kinase/protein kinase B and AMPK/ERK pathways, during myocardial I/R injury (1). Moreover, investigations of Dong et al (2012) on the heart triglyceride accumulation of wild type NOD and Tlr-4 deficient NOD mice were compatible to our findings. Thus, they found that triglyceride accumulation in the heart of Tlr-4 deficient NOD mice was lower than the other group. In addition, the level of MyD88 had been reduced, whereas the amount of p-AMPK had been increased in the early stages of diabetes in Tlr-4 deficient NOD mice (43). It has been determined that AMPK has potent anti-inflammatory effects, although this activity was inhibited in Tlr-4 activated cells due to nuclear to cytosolic translocation of the pro-inflammatory mediator high HMGB1 and inhibition of LKB1 (20).
As with the present study, Labuzek et al (2010) showed that LPS was able to stimulate AMPK (36). Furthermore, we have previously showed that metformin played a protective effect against LPS induced Tlr-4 expression as well as downstream proteins of this receptor, including MyD88. In these endotoxin treated mice, metformin significantly increased AMPK activity, while it decreased protein level of myocardial MyD88, as an adaptor protein for TLR-4 (44). Herein we showed that our previous results were associated with the decrease in cardiac TNF-α and IL-6 concentration and alleviation of peripheral neutrophil count. These results are in agreement with our previous studies where we showed the protective effects of metformin in subcutaneous isoproterenol injection induced myocardial infarction. Treatment with metformin decreased cardiac Tlr-4 expression and MYD88 protein content (12, 13). Those results were associated with reduction in TNF-α and IL-6 concentration levels both in the heart and serum (12, 13) and decrease in the peripheral neutrophil count and heart myeloperoxidase activity (45).
However, in this model we found some contro-versial effects of A-769662 on downstream of Tlr-4, in comparison with metformin. We determined that A-769662 significantly decreased the percentage of peripheral neurophil count in endotoxin injected mice and similar to metformin, antagonizing AMPK with compound-C reversed this protective effect. Nevertheless, treating LPS injected mice with A-769662 had no stimulatory effect on the cardiac MyD88 protein content or cardiac concentration of TNF-α. Although some promising, but not significant reversed effect on the level of cardiac IL-6 concentration was determined due to administration of compound-C.
To the best of our knowledge, effect of A-769662 on the TLR-4 expression or MyD88 protein content has not been appropriately investigated. However, there are some studies indicating the anti-inflammatory effects of A-769662 in some levels. In line with our findings, Guma et al (2015) demonstrated that AMPK activation, using A-769662, suppressed inflammatory arthritis in mice which were associated with decrease in IL-6 expression, but not TNF-α, in both serum and arthritic joints. This proposed that AMPK activation may have a considerable potential to use in IL-6 dependent inflammatory arthritis (46). However, we previously showed in rat model of LPS induced inflammation that concentration of TNF-α in the serum of the LPS+A-769662 group was reduced to the level of normal control group (14).
Conclusion
Altogether, these results demonstrated that A-769662 could block Tlr-4 expression, by activating AMPK. However, there are not sufficient studies about the effect of A-769662 on the MyD88 and inflammatory cytokines. This study proved our hypothesis that AMPK and Tlr-4 have some correlations. Ultimately, we propose that A-769662 might have some nonspecific effects other than inducing AMPK activity (Figure 8).
Figure 8.

Schematic diagram is showing the possible effects of A-769662-induced AMPK activation on TLR-4 expression and its downstream pathways. The effects of metformin, a well-known AMPK activator, are presenting for comparing.
AMPK: AMP-activated protein kinase; MyD88: myeloid differentia-tion primary-response protein 88; p-: phosphorylated-; TLR: toll like receptor.
blocking sign: 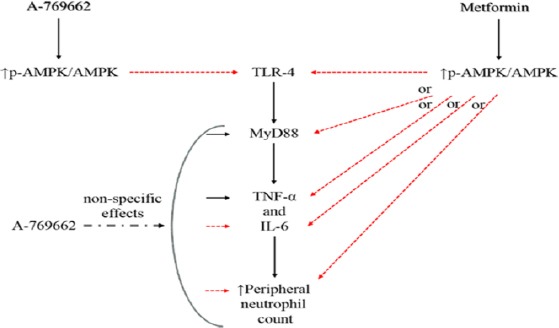
activating sign: 
Conflicts interests
There is no conflict of interest.
Acknowledgment
The present study was supported by a grant from the Research Vice Chancellors of Tabriz University of Medical Sciences; Tabriz, Iran. The results described in this paper were part of student thesis (M Rameshrad, NO 88).
References
- 1.Fang Y, Hu J. Toll-like receptor and its roles in myocardial ischemic/reperfusion injury. Medical science monitor : international medical journal of experimental and clinical research. 2011;17:RA100–109. doi: 10.12659/MSM.881709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timmers L, Pasterkamp G, de Hoog VC, Arslan F, Appelman Y, de Kleijn DP. The innate immune response in reperfused myocardium. Cardiovasc Res. 2012;94:276–283. doi: 10.1093/cvr/cvs018. [DOI] [PubMed] [Google Scholar]
- 3.Chao W. Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol-Heart C. 2009;296:H1–H12. doi: 10.1152/ajpheart.00995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 5.Prince JM, Levy RM, Yang R, Mollen KP, Fink MP, Vodovotz Y, et al. Toll-like receptor-4 signaling mediates hepatic injury and systemic inflammation in hemorrhagic shock. J Am Coll Surg. 2006;202:407–417. doi: 10.1016/j.jamcollsurg.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 6.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arumugam TV, Okun E, Tang S-C, Thundyil J, Taylor SM, Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32:4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 8.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–1161. [PubMed] [Google Scholar]
- 9.Bagheri B, Sohrabi B, Movassaghpur A, Mashayekhi S, Garjani A, Shokri M, et al. Association of monoctye expression of Toll-like receptor 4 and its related cytokines with coronary luminal stenosis. Adv Biosci Biotechnol. 2013;04:19–25. [Google Scholar]
- 10.Bagheri B, Sohrabi B, Movassaghpur A, Mashayekhi S, Garjani A, Shokri M, et al. Monocyte expression of Toll-like receptor-4 in patients with stable angina undergoing percutanoeus coronary intervention. Iran J Immunol. 2012;9:149–158. doi: 10.22034/iji.2012.16867. [DOI] [PubMed] [Google Scholar]
- 11.Rameshrad M, Maleki-Dizaji N, Vaez H, Soraya H, Nakhlband A, Garjani A. Lipopolysaccharide induced activation of toll like receptor 4 in isolated rat heart suggests a local immune response in myocardium. Iran J Immunol. 2015;12:104–116. [PubMed] [Google Scholar]
- 12.Soraya H, Farajnia S, Khani S, Rameshrad M, Khorrami A, Banani A, et al. Short-term treatment with metformin suppresses toll like receptors (TLRs) activity in isoproterenol-induced myocardial infarction in rat: are AMPK and TLRs connected? Int Immunopharmacol. 2012;14:785–791. doi: 10.1016/j.intimp.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Soraya H, Clanachan AS, Rameshrad M, Maleki-Dizaji N, Ghazi-Khansari M, Garjani A. Chronic treatment with metformin suppresses toll-like receptor 4 signaling and attenuates left ventricular dysfunction following myocardial infarction. Eur J Pharmacol. 2014;737:77–84. doi: 10.1016/j.ejphar.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Rameshrad M, Soraya H, Maleki-Dizaji N, Vaez H, Garjani A. A769662, a direct AMPK activator, attenuates lipopoly-saccharideinduced acute heart and lung inflammation in rats. Mol Med Rep. 2016;13:2843–2849. doi: 10.3892/mmr.2016.4821. [DOI] [PubMed] [Google Scholar]
- 15.Shirwany NA, Zou MH. AMPK in cardiovascular health and disease. Acta pharmacologica Sinica. 2010;31:1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young LH, Li J, Baron SJ, Russell RR. AMP-activated protein kinase: A key stress signaling pathway in the heart. Trends Cardiovas Med. 2005;15:110–118. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Santos GA, Moura RF, Vitorino DC, Roman EA, Torsoni AS, Velloso LA, et al. Hypothalamic AMPK activation blocks lipopolysaccharide inhibition of glucose production in mice liver. Mol Cell Endocrinol. 2013;381:88–96. doi: 10.1016/j.mce.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 18.Park DW, Jiang S, Tadie JM, Stigler WS, Gao Y, Abraham E, et al. Activation of AMPK enhances neutrophil chemotaxis and bacterial killing. Mol Med. 2013;1:00065. doi: 10.2119/molmed.2013.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, et al. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:27. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tadie JM, Bae HB, Deshane JS, Bell CP, Lazarowski ER, Chaplin DD, et al. Toll-like receptor 4 engagement inhibits adenosine 5’-monophosphate-activated protein kinase activation through a high mobility group box 1 protein-dependent mechanism. Mol Med. 2012;18:659–668. doi: 10.2119/molmed.2011.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1137–1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 22.Ao L, Song Y, Fullerton DA, Dinarello CA, Meng X. The interaction between myocardial depressant factors in endotoxemic cardiac dysfunction: role of TNF-alpha in TLR4-mediated ICAM-1 expression. Cytokine. 2007;38:124–129. doi: 10.1016/j.cyto.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdulrahman RM, Boon MR, Sips HC, Guigas B, Rensen PC, Smit JW, et al. Impact of Metformin and compound-C on NIS expression and iodine uptake in vitro and in vivo: a role for CRE in AMPK modulation of thyroid function. Thyroid. 2014;24:78–87. doi: 10.1089/thy.2013.0041. [DOI] [PubMed] [Google Scholar]
- 24.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell metabolism. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Takeda K. Evolution and integration of innate immune recognition systems: the Toll-like receptors. J Endotoxin Res. 2005;11:51–55. doi: 10.1179/096805105225006687. [DOI] [PubMed] [Google Scholar]
- 28.Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, et al. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:24–32. doi: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36:470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282:32549–32560. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan Q, et al. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol. 2010;80:1708–1717. doi: 10.1016/j.bcp.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 32.Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 33.Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 34.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fryer LG, Parbu-Patel A, Carling D. Protein kinase inhibitors block the stimulation of the AMP-activated protein kinase by 5-amino-4-imidazolecarboxamide riboside. FEBS letters. 2002;531:189–192. doi: 10.1016/s0014-5793(02)03501-9. [DOI] [PubMed] [Google Scholar]
- 36.Labuzek K, Liber S, Gabryel B, Buldak L, Okopien B. Ambivalent effects of compound-C (dorsomorphin) on inflammatory response in LPS-stimulated rat primary microglial cultures. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:41–57. doi: 10.1007/s00210-009-0472-2. [DOI] [PubMed] [Google Scholar]
- 37.Kim D, Kang D, Martin EA, Kim I, Carroll JL. Effects of modulators of AMP-activated protein kinase on TASK-1/3 and intracellular Ca(2+) concentration in rat carotid body glomus cells. Respir Physiol Neurobiol. 2014;195:19–26. doi: 10.1016/j.resp.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu W, Tang S, Shi J, Yin W, Cao S, Bu R, et al. Metformin attenuates palmitic acid-induced insulin resistance in L6 cells through the AMP-activated protein kinase/sterol regulatory element-binding protein-1c pathway. Int J Mol Med. 2015;35:1734–1740. doi: 10.3892/ijmm.2015.2187. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen TM, Seigneurin F, Froment P, Combarnous Y, Blesbois E. The 5’-AMP-activated protein kinase (AMPK) is involved in the augmentation of antioxidant defenses in cryopreserved chicken sperm. PloS one. 2015;10:e0134420. doi: 10.1371/journal.pone.0134420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 41.Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K, Webb GR, et al. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-γ: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood. 2002;99:3427–3431. doi: 10.1182/blood.v99.9.3427. [DOI] [PubMed] [Google Scholar]
- 42.Zhao P, Wang J, He L, Ma H, Zhang X, Zhu X, et al. Deficiency in TLR4 signal transduction ameliorates cardiac injury and cardiomyocyte contractile dysfunction during ischemia. J Cell Mol Med. 2009;13:1513–1525. doi: 10.1111/j.1582-4934.2009.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong B, Qi D, Yang L, Huang Y, Xiao X, Tai N, et al. TLR4 regulates cardiac lipid accumulation and diabetic heart disease in the nonobese diabetic mouse model of type 1 diabetes. Am J Physiol Heart Circ Physiol. 2012;303:H732–742. doi: 10.1152/ajpheart.00948.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaez H, Rameshrad M, Najafi M, Barar J, Barzegari A, Garjani A. Cardioprotective effect of metformin in lipopolysaccharide-induced sepsis via suppression of toll-like receptor 4 (TLR4) in heart. Eur J Pharmacol. 2015;772:115–123. doi: 10.1016/j.ejphar.2015.12.030. [DOI] [PubMed] [Google Scholar]
- 45.Soraya H, Rameshrad M, Mokarizadeh A, Garjani A. Metformin attenuates myocardial remodeling and neutrophil recruitment after myocardial infarction in rat. BioImpacts : BI. 2015;5:3–8. doi: 10.15171/bi.2015.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guma M, Wang Y, Viollet B, Liu-Bryan R. AMPK activation by A-769662 controls IL-6 expression in inflammatory arthritis. PloS one. 2015;10:e0140452. doi: 10.1371/journal.pone.0140452. [DOI] [PMC free article] [PubMed] [Google Scholar]