

Abstract
Objective(s):
Carthamus tinctorius L. (CT) or safflower is widely used in traditional Chinese medicine. This study investigated the effects of CT extract (CTE) on ischemia–reperfusion (I/R) brain injury and elucidated the underlying mechanism.
Materials and Methods:
The I/R model was conducted by occlusion of both common carotid arteries and right middle cerebral artery for 90 min followed by 24 hr reperfusion in Sprague-Dawley rats. CTE (0.2-0.6 g/kg) was administered intraperitoneally before and during ischemia, and during reperfusion period. The cerebral infarction area, neurological deficit scores, free radicals (lucigenin chemiluminescence counts) and pro-inflammatory cytokines expression were measured.
Results:
Pretreatment and treatment with CTE significantly reduced the cerebral infarction area and neurological deficits. CTE (0.4 g/kg) also reduced blood levels of free radicals and expression of tumor necrosis factor-α and interleukin-1β in the cerebral infarction area.
Conclusion:
The reduction in I/R cerebral infarction caused by CTE is possibly associated with its antioxidation and anti-inflammatory properties.
Keywords: Carthamus tinctorius-extract, Free radicals, Ischemia-reperfusion Injury
Introduction
Acute ischemic stroke can lead to extensive brain damage and is also a major cause of morbidity and leading cause of mortality in developed countries (1). Recanalization by using tissue plasminogen activator through intravenous injection within 4.5 hr of symptoms onset and early endovascular treatment are the most effective approach to improve clinical outcomes in ischemic stroke patients (2-4). Reperfusion after cerebral ischemia is essential for brain tissue survival, however, it also resulted in additional brain damage, such as neurovascular injury, hemorrhagic transformation, fatal cerebral edema and neuronal death (5). Therefore, potential neuroprotective agents, including traditional Chinese herbal formulas, have been widely investigated as novel promising therapies in ischemic stroke (6, 7).
Of the mechanisms responsible for brain damage pathogenesis after cerebral infarction, oxidative stress and inflammation may be the most crucial pathophysiological mechanism involved in ischemic stroke (4, 8). The inappropriate activation of ionotropic N-methyl-d-aspartate (NMDA) receptors in the brain by excessive released glutamate induced “excitotoxicity” results in ischemic/reperfusion (I/R) brain injury (9). This excitotoxicity may further induce death in neurons through the overproduction of reactive oxygen species (ROS) released by inflammatory cells (10, 11). An inflammatory cascade initiated by ROS promotes glial activation and neutrophil infiltration from peripheral blood and leads to the release of proinflammatory cytokines such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α. These proinflammatory cytokines critically amplify inflammation in the damaged brain tissue (12, 13). Therefore, drugs with a free radical-scavenging function can have potential neuroprotective effects on I/R brain injury.
Carthamus tinctorius L. (CT), commonly known as Safflower, are widely used in traditional Chinese medicine (TCM) in treating cerebrovascular and cardiovascular disease. According to TCM theory, CT mainly promotes blood circulation and removes blood stasis. In China, safflower yellow injections are used clinically for coronary heart disease and unstable angina pectoris (14). In addition, in vivo and in vitro neuroprotective effects of CT extract (CTE) on cerebral ischemic injury were reported (15). Intravenous treatment using hydroxysafflor yellow A, the main component of safflower yellow pigments, significantly reduced neurological deficit scores in a rat infarction model (16). Kaempferol-3-O-rutinoside (K-3-R, Figure 1), a major flavonoid glycoside present in CT, has been used as a phytochemical marker and standard for qualitative and quantitative studies in China (17). K-3-R has neuroprotective effects on brain injury and neuroinflammation through the downregulation of the NF-κB signal pathway (18). However, the protective effect of CTE treatment against I/R brain injury remains unclear. Therefore, this study investigated the effects of CTE on an I/R model by using transient right middle cerebral artery occlusion, followed by reperfusion, in Sprague Dawley (SD) rats.
Figure 1.
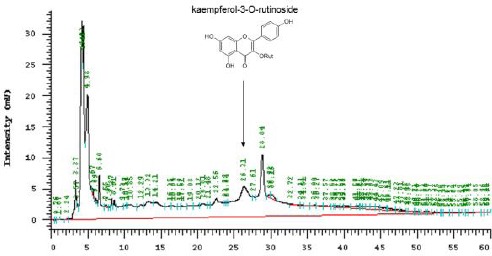
High-performance liquid chromatographic fingerprint analysis of subtle Carthamus tinctorius extract (CTE). Kaempferol-3-O-rutinoside was applied as the standard for this analysis. The arrow represents retention time
Materials and Methods
Plant material
CT, obtained from Sichuan, China, was prepared for extraction by Koda Pharmaceuticals (Taoyuan, Taiwan). In brief, 80 g of crude CT was boiled in 560 ml of water for 50 min, and the extract was then filtered. The residue was extracted again and filtered. The combined filtrate was then concentrated. The wet weight of the final product was 27.48 g (34.35%). The wet extract (25 g) was mixed with 131.7 ml of phosphate-buffered saline (PBS; pH 7.4). The concentration of the CTE solution was approximately 0.2 g/ml. K-3-R (Figure 1) was used as a phytochemical marker for the chromatographic fingerprint analysis. A high-performance liquid chromatographic (HP 1100, Agilent) method coupled with ultraviolet (UV, λ310 nm), was used for the qualitative determination of compounds in the extract: the samples were analyzed on the an Agilent Hypersil BDS C-18, 5 µm (250 × 4.6 mm) column, maintained at ambient room temperature. Next, gradient elution was performed using a mobile phase composed of methanol as solvent A and water as solvent B. The gradients were as follows: 0–15 min, 10% increasing to 20% solvent A; 15–30 min, 20% increasing to 30% solvent A; 30–40 min, 30% increasing to 50% solvent A; 40–60 min, 10% solvent A. The flow rate was 0.5 ml/min. The compounds were identified by comparing their retention times with those of known standards obtained from chromatograms (Figure 1).
Reagents
2,3,5-Triphenyltetrazolium chloride (TTC) and MK-801 (a highly potent and selective noncompetitive NMDA glutamate receptor antagonist) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Pathogen-free SD rats weighing 300– 400 g were obtained from the National Experimental Animal Center (Taipei, Taiwan). The animals were housed in the animal center of China Medical University (Taichung, Taiwan), and the room temperature was controlled at 25 °C±1 °C through air conditioning, with a 12 hr light–dark cycle and ad libitum food and water. All animal experiment procedures were conducted following the principles in the Guide for the Care and Use of Laboratory Animals.
Experimental I/R injury model
A rat I/R model of cerebral infarction was established by occluding both bilateral common carotid arteries (CCAs) and right middle cerebral artery (MCA) for 90 min, followed by reperfusion for 24 hr, in SD rats as reported previously (19, 20). The rats were anesthetized using an intraperitoneal (IP) injection of 400 mg/kg chloral hydrate. A PE-50 tube, connected to a heart rate–blood pressure monitoring apparatus (LE 5001 pressure meter; Panlab. S.L.L., Barcelona, Spain), was inserted in the right femoral artery. Both CCAs were then wrapped using a plastic line (0.1 mm in diameter). Next, the scalps of the rats were incised to expose the right MCA through the bone window. An 8-0 nylon line mediated via a surgical needle was placed under the right MCA at the immediate upper margin of the olfactory tract. A laser Doppler (DRT4; Moor Instruments Inc., Wilmington, USA) was used to monitor the perfusion of right MCA. When the blood flow of the bilateral CCAs was occluded, the perfusion decreased from 900 to 200 LD units. When the blood flow of the right MCA was occluded, the perfusion immediately decreased further from 200 to 50 LD units, indicating total occlusion over the right MCA territory. The ischemia period lasted for 90 min under bilateral CCA and right MCA occlusion; this was followed by reperfusion for 24 hr. Finally, the animals were sacrificed. During model establishment by our skilled researchers, the mortality rate was approximately 10%–20%.
Experiment design
Fifty-four SD rats were randomly divided into nine groups, with six rats in each of the following groups: (1) sham (S): rats underwent similar surgical procedures without occlusion; (2) control (C): occlusion of both CCAs and right MCA for 90 min, followed by reperfusion for 24 hr; (3) PBS: control rats pretreated with 0.2 ml/kg PBS IP; (4) MK: control rats pretreated with 1 mg/kg MK-801 IP 10 min before occlusion; (5) P-0.2: control rats pretreated with 0.2 g/kg CTE IP; (6) P-0.4: control rats pretreated with 0.4 g/kg CTE IP; (7) P-0.6: control rats pretreated with 0.6 g/kg CTE IP; (8) O-0.4: rats treated with 0.4 g/kg CTE IP 30 min after occlusion; and (9) R-0.4: rats treated with 0.4 g/kg CTE IP 30 min after reperfusion.
Neurological deficits evaluation
Neurological impairment after I/R injury was evaluated using the neurological examination grading system on a 4-point scale as described previously (19-21): 0, no significant deficits; 1, failure to completely extend the left forepaw; 2, decreased resistance to lateral push toward the paralytic limb; and 3, circling to the left. The neurological examination was performed by an examiner blinded to the experimental group classification after 24 hr reperfusion following ischemia.
Cerebral infarction area measurement
The rats were euthanized using 400 mg/kg chloral hydrate IP after evaluation of their neurological deficits, and their brains were rapidly removed and frozen at −20 °C for 15 min. Coronal brain sections of 2-mm thickness were stained with a 2% TTC solution at 37 °C for 15 min, followed by fixation with 4% formaldehyde for 24 hr. The cerebral infarction volumes of the first six sections from the frontal lobe were measured using a microscopic image analysis system (Image-Pro Lito Version 3.0, Media Cybernetics, USA). The ratio of infarction area to total brain area in each section was calculated and expressed as a percentage, similar to in our previous report (22).
Free radical measurement
Eighteen SD rats were randomly divided into three groups of six rats each: (1) C, (2) MK, and (3) P-0.4. Blood from the right femoral artery of each rat was analyzed three times for blood free radical levels. The sampling was performed 15 min before occlusion (Before), 30 min after occlusion (O-30), and 2 hr after reperfusion (R-2). Whole blood (0.3 ml) was sampled for a chemiluminescence (CL) assay. After the rats were sacrificed, their brains were immediately removed, fixed in 4% paraformaldehyde, embedded in paraffin, and finally cut into 20-μm-thick sections for immunohistochemistry.
CL assay for free radicals
Lucigenin CL counts were measured using the same method as in our previous studies (19, 22). Briefly, 0.2 ml of whole blood was mixed with 0.1 ml of PBS in a special chamber unit (Model CLD-110; Tohoku Electronic Industrial, Sendai, Japan). CL was then measured in an absolutely dark chamber of the CL Analyzing System (Tohoku Electronic Industrial). This system includes a photon detector (Model CLD-110), water circulator (Model CH-201), CL counter (Model CLD-110), and 32-bit IBM personnel computer. After 200 sec of continuous measurement, 1.0 ml of 0.01 mm lucigenin (Sigma-Aldrich) mixed in PBS was injected into the stainless steel cell to measure CL counts in the blood sample continuously. After 600 sec of continuous measurement, 0.2 ml of zymosan A (Sigma-Aldrich) was added in the aforementioned manner, and the CL counts were measured again for 1020 sec continuously. Total CL counts were calculated by integrating the area under the curve and subtracting it from the background level. The data is presented as CL counts/1000 white blood cells (WBCs), calculated by dividing the blood CL levels by the WBC count.
Immunohistochemical study
The primary antibodies against proinflammatory cytokines used in this study were mouse anti-TNF-α (1:200; BD Pharmingen) and anti-IL-1β (1:200; BD Pharmingen) antibodies purchased from Bender MedSystems (Vienna, Austria). Briefly, 5-µm-thick brain sections were washed with PBS and rinsed 3% H2O2 in methanol for 15 min to block endogenous peroxidase activity and then incubated with 10% normal animal serum (LsAB kit, Zymed, San Francisco, CA, USA) for 30 min to prevent nonspecific binding. Primary antibodies were applied to the brain sections for 30 min, which were then incubated with biotinylated secondary antibodies at 37 °C for 10 min. The tissue sections were subsequently incubated with a streptavidin-biotin peroxidase-labeled complex for 10 min, followed by 3,3’-diaminobenzidine tetrahydrochloride solution (Liquid DAB substrate kit, Zymed) for 2–10 min, and then counterstained with hematoxylin. The sections were observed under a light microscope by a researcher blinded to section grouping. TNF-α and IL-1β immunoreactive cell counts in the cerebral infarction zone of the third section (Figure 2) from the frontal lobe were calculated under a 40 x objective for semiquantitative analysis. The data are presented as cells/mm2.
Figure 2.
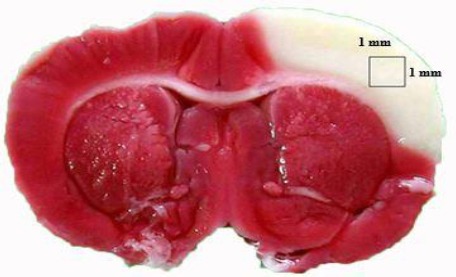
Third brain coronal section from the frontal lobe. The white area is the cerebral infarction lesion and the reddish-purple area indicates the noninfarction brain. The square within the cerebral infarction area indicates the region for immunostaining of tumor necrosis factor-α- and interleukin-1β-producing cells
Statistical analysis
All data are expressed as mean±SD. All statistical analyses were performed using the Prism 3.02 software (GraphicPad Software Inc., CA, USA), and one-way ANOVA was applied for multiple comparisons (post hoc Tukey test). P<0.05 was considered statistically significant.
Results
Effects of CTE on the neurological deficits in I/R rats
Figure 2 lists the neurological deficit scores of each group after 24 hr reperfusion following the 90-min cerebral infarction. All rats in the C and PBS groups developed severe neurological deficits, including left forelimb flexion, decreased resistance during lateral push toward the paretic side, and circling behavior as the rats moved around freely. Neurological deficit scores in the P-0.2, P-0.4, P-0.6, O-0.4, and R-0.4 groups were significantly lower than those in the C group (all P<0.05). MK-801 treatment, as the positive control, also yielded neurological deficit scores that were significantly lower than those of the controls (P< 0.05).
Effects of CTE on cerebral infarction in I/R rats
The infarction lesion remained white and the noninfarction area became reddish-purple after staining with 2% TTC (Figure 3). Figure 4 presents the brain infarction area in each group after I/R. All rats developed large infarction areas, reflecting severe I/R injury.
Figure 3.
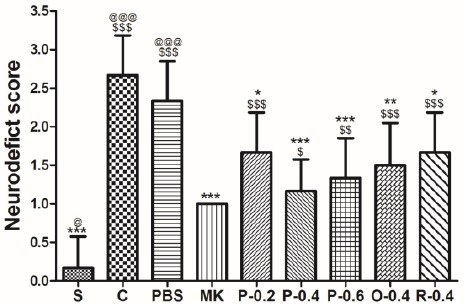
Effects of CT extract (CTE) on neurological deficit scores of ischemia–reperfusion (I/R)-injured rats. The scores were assessed 24 hr after reperfusion. Data are expressed as means±SD. S: sham group; C: control group; PBS: PBS group; MK: treatment with MK-801 group; P-0.2: pretreatment (P) with 0.2 g/kg CTE group; P-0.4: P with 0.4 g/kg CTE group; P-0.6: P with 0.6 g/kg CTE group; O-0.4: P with 0.4 g/kg CT E after occlusion group; R-0.4: P with 0.4 g/kg CTE after reperfusion group; $ P< 0.05, $$ P<0.01, $$$P<0.001 compared with the S group; * P<0.05, **P<0.01, ***P<0.001 compared with the C group; @ P<0.05, @@ P<0.01, @@@P<0.001 compared with the PBS group. n=6 in each group
Figure 4.
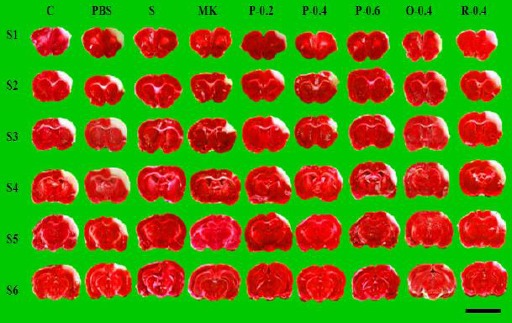
Effects of CTE on cerebral infarct in I/R rats. Representative brain coronal sections (S1–S6) from each group are shown. White indicates cerebral infarction, whereas reddish-purple represents noninfarcted brain tissue. Brain tissue was stained using 2,3,5-triphenyltetrazolium chloride. Scale bar=1 cm
The percentages of the cerebral infarction areas were 0.64%±0.85%, 16.08%±2.56%, 15.94%±2.36%, 5.77%± 2.02%, 9.66%±1.90%, 5.78%±2.64%, 6.82%±2.02%, 8.33%±2.59%, and 12.56%±3.53% in the S, C, PBS, MK, P-0.2, P-0.4, P-0.6, O-0.4, and R-0.4 groups, respectively. CTE pretreatment (P-0.2, P-0.4, and P-0.6) and treatment during the occlusive period (O-0.4) yielded significantly less cerebral infarction area compared with that of the C group (P<0.05). No differences were noted between the R-0.4 and C groups. Similarly, the effects did not differ significantly between the MK and P-0.2, P-0.4, P-0.6, and O-0.4 groups (all P>0.05; Figure 5).
Figure 5.
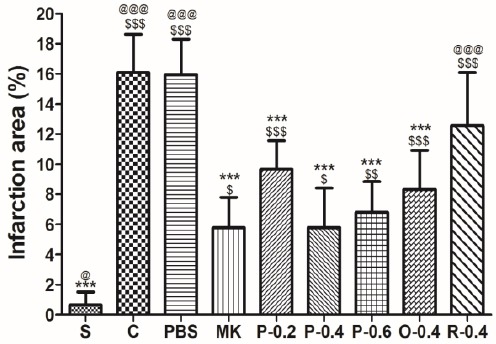
Effects of Effects of CT extract (CTE) on cerebral infarct in I/R rats. Data are expressed as means±SD.* P<0.05,**P<0.01, *** P<0.001 compared with the C. $ P<0.05, $$ P<0.01, $$$ P<0.001 compared with the S; @P<0.05, @@ P<0.01, @@@P<0.001 compared with the MK. n=6 in each group
Effect of CTE on free radical levels in I/R rats
The blood levels of free radicals were determined through the lucigenin CL counts (Figure 6). In the C group, the lucigenin CL counts were significantly higher during the reperfusion period (R-2; 29344±6263) than before the I/R injury (19767±3808; P<0.01). Both the MK and P-0.4 groups had significantly lower lucigenin CL counts in the R-2 period than did the C group (P<0.05).
Figure 6.
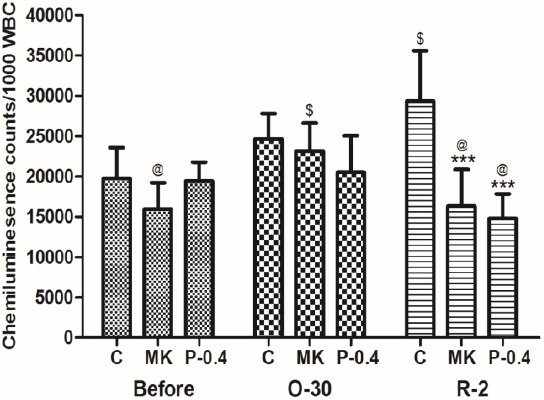
Effects of CTE on free radical levels (lucigenin chemiluminesence counts) in I/R rats. Data are expressed as means ±SD. C: control group; MK: treatment with MK-801 group; P-0.4: pretreatment with 0.4 g/kg CTE group; Before: 15 min before the occlusion of cerebral blood flow; O-30: 30 min after occlusion; R-2: 2 hr after reperfusion. *** P<0.001 compared with the C group, $P<0.05 compared with the Before group, @ P<0.05 compared with the O-30 group. n=6 in each group
Effects of CTE on cytokine expression in infarct lesions in I/R rats
Through immunohistochemistry for TNF-α and IL-1β, we assessed proinflammatory cytokine expression after I/R injury in rat brains (Figure 7). TNF-α-immunoreactive cells were significantly fewer in the MK (92.6±18.7/mm3) and P-0.4 (107.7±17.8/mm3) groups than in the C group (337.5±47.1/mm3; all P<0.001; Figure 8). Similarly, IL-1β-immunoreactive cells were significantly fewer in the MK (86.3±13.3/mm3) and P-0.4 (96.0±21.5/mm3) groups than in the C group (378.0± 53.7/mm3; all P<0.001).
Figure 7.
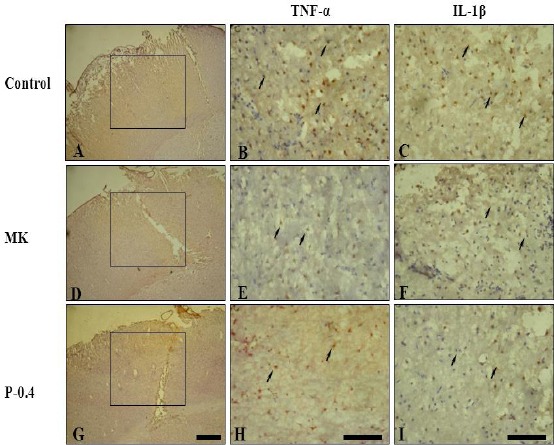
Effects of CTE on TNF-α- and IL-1β-producing cells in I/R rats. Squares in the cerebral infarction areas indicate immunostained regions. TNF-α- and IL-1β-immunoreactive cells (arrows) were significantly fewer in the MK and P-0.4 groups than in the C group. Scale bar = 30 µm
Figure 8.
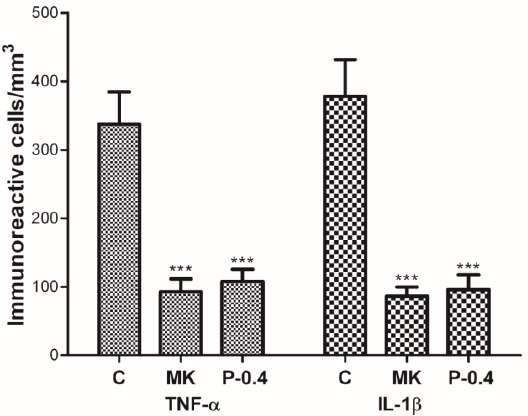
Effect of CTE on the number of TNF-α- and IL-1β-producing cells in I/R rats. The numbers of TNF-α- and IL-1β-immunoreactive cells were significantly lower in the MK and P-0.4 groups than in the C group. ***P<0.001 compared with the C group
Discussion
The present results indicated that CTE treatment 10 min before (P-0.2, P-0.4, and P-0.6) and 30 min after (O-0.4) the occlusion of blood flow reduced neurological deficit scores and cerebral infarction area in the I/R rat model. However, when CTE treatment was administered during the reperfusion period (R-0.4), the infarct lesion resolved slightly. Notably, CTE pretreatment reduced blood levels of free radicals and inflammatory cytokines in the infarct lesion, suggesting that the effects of CTE on I/R brain injury are associated with its antioxidation and anti-inflammatory properties.
Animal models mimicking the pathophysiology, including neurological deficits and pathological changes, after an episode of I/R injury have been developed (23-25). In this study, we used a rat model to investigate the effects of CTE on cerebral infarction by using the occlusion of bilateral CCAs and right MCA described in our previous reports (20, 22, 26). Consequently, the rats in the C and PBS groups developed cerebral infarct, as confirmed through TTC staining (26).
MK-801, a highly potent and selective noncompetitive NMDA glutamate receptor antagonist (the positive control here), inhibited free radical and cytokine production in this model, as reported previously (27, 28). Oxidative stress is a crucial factor in I/R injury in both animal models and humans (29, 30). Lucigenin CL has been widely used to monitor net mitochondrial superoxide generation in vivo (31, 32). Lucigenin CL counts mainly reflect myeloperoxidase-catalyzed reactions (33). In addition, lucigenin strongly reacts with superoxide anions generated by nicotinamide adenine dinucleotide phosphate hydrogen oxidase in neutrophils and monocytes (34-36). The results were similar to the previously reported steady increase in the ROS levels of MCAO rats (22). Notably, free radical levels were significantly lower in the P-0.4 and MK groups compared with those in the C group during reperfusion (R-2), suggesting that CTE and MK either suppress or scavenge superoxide anions. CTE contains numerous natural antioxidant flavonols including hydroxysafflor yellow A, safflor yellow B, and K-3-R (17, 18, 37, 38). These components might suppress or scavenge superoxide anion to reduce cerebral infarction and improve neurological deficits.
High levels of pro-inflammatory cytokines such as TNF-α and IL-1β play a central role in the initiation and propagation of the inflammatory cascade and gliosis in brain (39, 40). They modulate the volume of ischemic damage in experimental model of stroke (40). Microglia, intrathecal macrophages, and infiltrating monocyte-derived macrophages release TNF-α in response to numerous internal and external stimuli (41, 42). TNF-α mRNA and protein levels are elevated in the ischemic cortex as early as 1 hr and peak at 12 hr after MCAO in rats (43); furthermore, the administration of neutralizing anti-TNF-α antibodies or TNF-α-binding proteins can aid in preventing neurological damage in animal stroke models (44). In an ischemic cortex, IL-1β release is slightly delayed compared with that of TNF-α; however, it continues to increase from 12 to 24 hr after MCAO in rats (45, 46). The main sources of IL-1β after cerebral infarction are microglia and macrophages (39). IL-1β may regulate the expression of adhesion molecules and NO to affect the cerebral blood flow and blood–brain barrier integrity after brain insult (47). The administration of recombinant human IL-1Ra or inhibition of IL-1-converting enzyme also reduces the stroke volume in MACO rats (39). Our results showed that CTE pretreatment (P-0.4) reduced free radical levels as well as TNF-α- and IL-1β-producing cell numbers. However, the detailed molecular mechanisms involved in the effects of CTE require further clarification.
Conclusion
CTE has a neuroprotective role against cerebral I/R injury in rats. This effect may be partially attributable to the antioxidation and anti-inflammatory properties of CTE.
Conflict of Interest
The authors declare no financial or commercial conflict of interest.
Acknowledgment
The results reported in this paper were part of the master’s thesis of PKF. The authors acknowledge the grant (TCVGH-1034101B) and assistance from the Center for Translational Medicine, Taichung Veterans General Hospital, Taichung, Taiwan. This study was also supported by China Medical University grants under the Aim for the Top University Plan, Ministry of Education, and from the Clinical Trial and Research Center of Excellence (MOHW105-TDU-B-212-133019), Ministry of Health and Welfare, Taiwan, ROC.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullen MT, Pisapia JM, Tilwa S, Messe SR, Stein SC. Systematic review of outcome after ischemic stroke due to anterior circulation occlusion treated with intravenous, intra-arterial, or combined intravenous+intra-arterial thrombolysis. Stroke. 2012;43:2350–2355. doi: 10.1161/STROKEAHA.111.639211. [DOI] [PubMed] [Google Scholar]
- 3.van der Worp HB, van Gijn J. Clinical practice. Acute ischemic stroke. N Engl J Med. 2007;357:572–579. doi: 10.1056/NEJMcp072057. [DOI] [PubMed] [Google Scholar]
- 4.Musuka TD, Wilton SB, Traboulsi M, Hill MD. Diagnosis and management of acute ischemic stroke: speed is critical. CMAJ. 2015;187:887–893. doi: 10.1503/cmaj.140355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khatri R, McKinney AM, Swenson B, Janardhan V. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. 2012;79:S52–57. doi: 10.1212/WNL.0b013e3182697e70. [DOI] [PubMed] [Google Scholar]
- 6.Chen HS, Qi SH, Shen JG. One-compound-multi-target: combination prospect of natural compounds with thrombolytic therapy in acute ischemic stroke. Curr Neuropharmacol. 2016 doi: 10.2174/1570159X14666160620102055. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu M, Sun ZJ, Li LT, Ge HY, Song CQ, Wang AJ. The beneficial effects of the herbal medicine Di-huang-yin-zi (DHYZ) on patients with ischemic stroke: A Randomized, Placebo controlled clinical study. Complement Ther Med. 2015;23:591–597. doi: 10.1016/j.ctim.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Olmez I, Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem Int. 2012;60:208–212. doi: 10.1016/j.neuint.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong CH, Crack PJ. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr Med Chem. 2008;15:1–14. doi: 10.2174/092986708783330665. [DOI] [PubMed] [Google Scholar]
- 11.Hou YC, Liou KT, Chern CM, Wang YH, Liao JF, Chang S, et al. Preventive effect of silymarin in cerebral ischemia-reperfusion-induced brain injury in rats possibly through impairing NF-kappaB and STAT-1 activation. Phytomedicine. 2010;17:963–973. doi: 10.1016/j.phymed.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–342. doi: 10.1097/WCO.0b013e32813ba151. [DOI] [PubMed] [Google Scholar]
- 13.Yi JH, Park SW, Kapadia R, Vemuganti R. Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem Int. 2007;50:1014–1027. doi: 10.1016/j.neuint.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong D, Xia W, Zhang Z, Xiao L, Yuan D, Liu Y, et al. Safflower yellow injection combined with conventional therapy in treating unstable angina pectoris: a meta-analysis. J Tradit Chin Med. 2013;33:553–561. doi: 10.1016/s0254-6272(14)60021-2. [DOI] [PubMed] [Google Scholar]
- 15.Zhu HB, Zhang L, Wang ZH, Tian JW, Fu FH, Liu K, et al. Therapeutic effects of hydroxysafflor yellow A on focal cerebral ischemic injury in rats and its primary mechanisms. J Asian Natl Prod Res. 2005;7:607–613. doi: 10.1080/10286020310001625120. [DOI] [PubMed] [Google Scholar]
- 16.Wei X, Liu H, Sun X, Fu F, Zhang X, Wang J, et al. Hydroxysafflor yellow A protects rat brains against ischemia-reperfusion injury by antioxidant action. Neurosci Lett. 2005;386:58–62. doi: 10.1016/j.neulet.2005.05.069. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Chen P, Tang C, Li Y, Zhang H. Antinociceptive and anti-inflammatory activities of extract and two isolated flavonoids of Carthamus tinctorius L. J Ethnopharmacol. 2014;151:944–950. doi: 10.1016/j.jep.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Yu L, Chen C, Wang LF, Kuang X, Liu K, Zhang H, et al. Neuroprotective effect of kaempferol glycosides against brain injury and neuroinflammation by inhibiting the activation of NF-kappaB and STAT3 in transient focal stroke. PloS One. 2013;8:e55839. doi: 10.1371/journal.pone.0055839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng CY, Su SY, Tang NY, Ho TY, Chiang SY, Hsieh CL. Ferulic acid provides neuroprotection against oxidative stress-related apoptosis after cerebral ischemia/reperfusion injury by inhibiting ICAM-1 mRNA expression in rats. Brain Res. 2008;1209:136–150. doi: 10.1016/j.brainres.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 20.Tang NY, Liu CH, Hsieh CT, Hsieh CL. The anti-inflammatory effect of paeoniflorin on cerebral infarction induced by ischemia-reperfusion injury in Sprague-Dawley rats. Am J Chin Med. 2010;38:51–64. doi: 10.1142/S0192415X10007786. [DOI] [PubMed] [Google Scholar]
- 21.Cheng CY, Su SY, Tang NY, Ho TY, Lo WY, Hsieh CL. Ferulic acid inhibits nitric oxide-induced apoptosis by enhancing GABA(B1) receptor expression in transient focal cerebral ischemia in rats. Acta Pharmacol Sin. 2010;31:889–899. doi: 10.1038/aps.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh CL, Cheng CY, Tsai TH, Lin IH, Liu CH, Chiang SY, et al. Paeonol reduced cerebral infarction involving the superoxide anion and microglia activation in ischemia-reperfusion injured rats. J Ethnopharmacol. 2006;106:208–215. doi: 10.1016/j.jep.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 23.Aronowski J, Strong R, Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1048–1056. doi: 10.1097/00004647-199710000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Gursoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 2004;35:1449–1453. doi: 10.1161/01.STR.0000126044.83777.f4. [DOI] [PubMed] [Google Scholar]
- 25.Woitzik J, Schneider UC, Thome C, Schroeck H, Schilling L. Comparison of different intravascular thread occlusion models for experimental stroke in rats. J Neurosci Methods. 2006;151:224–231. doi: 10.1016/j.jneumeth.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Cheng CY, Ho TY, Lee EJ, Su SY, Tang NY, Hsieh CL. Ferulic acid reduces cerebral infarct through its antioxidative and anti-inflammatory effects following transient focal cerebral ischemia in rats. Am J Chin Med. 2008;36:1105–1119. doi: 10.1142/S0192415X08006570. [DOI] [PubMed] [Google Scholar]
- 27.Kuhlmann CR, Zehendner CM, Gerigk M, Closhen D, Bender B, Friedl P, et al. MK801 blocks hypoxic blood-brain-barrier disruption and leukocyte adhesion. Neurosci Lett. 2009;449:168–172. doi: 10.1016/j.neulet.2008.10.096. [DOI] [PubMed] [Google Scholar]
- 28.Neuhaus W, Burek M, Djuzenova CS, Thal SC, Koepsell H, Roewer N, et al. Addition of NMDA-receptor antagonist MK801 during oxygen/glucose deprivation moderately attenuates the upregulation of glucose uptake after subsequent reoxygenation in brain endothelial cells. Neurosci Lett. 2012;506:44–49. doi: 10.1016/j.neulet.2011.10.045. [DOI] [PubMed] [Google Scholar]
- 29.Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12:698–714. doi: 10.2174/1871527311312050015. [DOI] [PubMed] [Google Scholar]
- 30.Dominguez C, Delgado P, Vilches A, Martin-Gallan P, Ribo M, Santamarina E, et al. Oxidative stress after thrombolysis-induced reperfusion in human stroke. Stroke. 2010;41:653–660. doi: 10.1161/STROKEAHA.109.571935. [DOI] [PubMed] [Google Scholar]
- 31.Caraceni P, Rosenblum ER, Van Thiel DH, Borle AB. Reoxygenation injury in isolated rat hepatocytes: relation to oxygen free radicals and lipid peroxidation. Am J Physiol. 1994;266:G799–806. doi: 10.1152/ajpgi.1994.266.5.G799. [DOI] [PubMed] [Google Scholar]
- 32.Sun JS, Hang YS, Huang IH, Lu FJ. A simple chemiluminescence assay for detecting oxidative stress in ischemic limb injury. Free Radic Biol Med. 1996;20:107–112. doi: 10.1016/0891-5849(95)02011-x. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi R, Edashige K, Sato EF, Inoue M, Matsuno T, Utsumi K. Luminol chemiluminescence and active oxygen generation by activated neutrophils. Arch Biochem Biophys. 1991;285:325–330. doi: 10.1016/0003-9861(91)90367-r. [DOI] [PubMed] [Google Scholar]
- 34.McNally JA, Bell AL. Myeloperoxidase-based chemiluminescence of polymorphonuclear leukocytes and monocytes. J Biolumin Chemilum. 1996;11:99–106. doi: 10.1002/(SICI)1099-1271(199603)11:2<99::AID-BIO404>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Yang T, Peleli M, Zollbrecht C, Giulietti A, Terrando N, Lundberg JO, et al. Inorganic nitrite attenuates NADPH oxidase-derived superoxide generation in activated macrophages via a nitric oxide-dependent mechanism. Free Radic Biol Med. 2015;83:159–166. doi: 10.1016/j.freeradbiomed.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez-Perilli L, Alvarez MN, Prolo C, Radi R, Rubbo H, Trostchansky A. Nitroarachidonic acid prevents NADPH oxidase assembly and superoxide radical production in activated macrophages. Free Radic Biol Med. 2013;58:126–133. doi: 10.1016/j.freeradbiomed.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu YL, Liu YJ, Liu Y, Li XS, Liu SH, Pan YG, et al. Hydroxysafflor yellow A ameliorates lipopolysaccharide-induced acute lung injury in mice via modulating toll-like receptor 4 signaling pathways. Int Immunopharmacol. 2014;23:649–657. doi: 10.1016/j.intimp.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z, Yang J, Jia Y, Tian Y, Wen A. Pharmacokinetic properties of hydroxysafflor yellow A in healthy Chinese female volunteers. J Ethnopharmacol. 2009;124:635–638. doi: 10.1016/j.jep.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 39.Siniscalchi A, Gallelli L, Malferrari G, Pirritano D, Serra R, Santangelo E, et al. Cerebral stroke injury: the role of cytokines and brain inflammation. J Basic Clin Physiol Pharmacol. 2014;25:131–137. doi: 10.1515/jbcpp-2013-0121. [DOI] [PubMed] [Google Scholar]
- 40.Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32:1677–1698. doi: 10.1038/jcbfm.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cacci E, Claasen JH, Kokaia Z. Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res. 2005;80:789–797. doi: 10.1002/jnr.20531. [DOI] [PubMed] [Google Scholar]
- 42.Kogo J, Takeba Y, Kumai T, Kitaoka Y, Matsumoto N, Ueno S, et al. Involvement of TNF-alpha in glutamate-induced apoptosis in a differentiated neuronal cell line. Brain Res. 2006;1122:201–208. doi: 10.1016/j.brainres.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, et al. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 44.Lavine SD, Hofman FM, Zlokovic BV. Circulating antibody against tumor necrosis factor-alpha protects rat brain from reperfusion injury. J Cereb Blood Flow Metab. 1998;18:52–58. doi: 10.1097/00004647-199801000-00005. [DOI] [PubMed] [Google Scholar]
- 45.Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflamm. 2008;5:46. doi: 10.1186/1742-2094-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clausen BH, Lambertsen KL, Meldgaard M, Finsen B. A quantitative in situ hybridization and polymerase chain reaction study of microglial-macrophage expression of interleukin-1beta mRNA following permanent middle cerebral artery occlusion in mice. Neuroscience. 2005;132:879–892. doi: 10.1016/j.neuroscience.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 47.Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2:492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]