

Abstract
Glutamate-evoked Na+ increase in astrocytes has been identified as a signal coupling synaptic activity to glucose consumption. Astrocytes participate in multicellular signaling by transmitting intercellular Ca2+ waves. Here we show that intercellular Na+ waves are also evoked by activation of single cultured cortical mouse astrocytes in parallel with Ca2+ waves; however, there are spatial and temporal differences. Indeed, maneuvers that inhibit Ca2+ waves also inhibit Na+ waves; however, inhibition of the Na+/glutamate cotransporters or enzymatic degradation of extracellular glutamate selectively inhibit the Na+ wave. Thus, glutamate released by a Ca2+ wave-dependent mechanism is taken up by the Na+/glutamate cotransporters, resulting in a regenerative propagation of cytosolic Na+ increases. The Na+ wave gives rise to a spatially correlated increase in glucose uptake, which is prevented by glutamate transporter inhibition. Therefore, astrocytes appear to function as a network for concerted neurometabolic coupling through the generation of intercellular Na+ and metabolic waves.
Glutamate, released in the synaptic cleft during neuronal activity, is rapidly removed by surrounding astrocytes (1). One of the roles of glutamate clearance by astrocytes is to trigger a cascade of molecular mechanisms that provides metabolic substrates to neurons (2). Glutamate is cotransported with three Na+ ions by excitatory amino acid transporters expressed in astrocytes (3, 4), inducing an intracellular 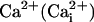 elevation. This
elevation. This 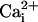 elevation results in an activation of the Na+/K+-ATPase, causing an increased energy demand in astrocytes (5, 6), which in turn enhances cellular glucose utilization and glycolysis (7). Lactate, the end-product of glycolysis, is released by astrocytes and could serve as metabolic substrate for neurons in conjunction with glucose (2).
elevation results in an activation of the Na+/K+-ATPase, causing an increased energy demand in astrocytes (5, 6), which in turn enhances cellular glucose utilization and glycolysis (7). Lactate, the end-product of glycolysis, is released by astrocytes and could serve as metabolic substrate for neurons in conjunction with glucose (2).
Astrocytes can also release glutamate in response to neuro-active agents such as prostaglandins (8), ATP (9), bradykinin (10), or glutamate itself (11). Several release mechanisms have been identified such as release through swelling-activated anion channels (12), P2X7 channels (13), or hemichannels (14). Growing evidence suggests that astrocytic glutamate release is mediated by intracellular (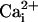 -dependent vesicular exocytosis (8, 10, 15, 16).
-dependent vesicular exocytosis (8, 10, 15, 16).
Astrocytes can communicate with each other by the propagation of  elevation (17). These so-called Ca2+ waves have been extensively described in primary cell culture and brain slices (18). The release of ATP by astrocytes appears to be the main signaling mechanism of the
elevation (17). These so-called Ca2+ waves have been extensively described in primary cell culture and brain slices (18). The release of ATP by astrocytes appears to be the main signaling mechanism of the 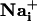 wave (19, 20). After diffusion into the extracellular space, ATP binds to purinoceptors of neighboring astrocytes inducing inositol 1,4,5-triphosphate (IP3)-mediated mobilization of Ca2+ from internal stores. Glutamate is released in association with Ca2+ waves (21), but does not appear to have a primary role in the propagation of the wave itself (22). Gap junctions also play a role in the transmission mechanism by the diffusion of IP3 from cytosol to cytosol (17). The function of astrocytic Ca2+ waves is unclear and could represent a form of multicellular, bidirectional communication with neurons (23-25) or microarterioles (26).
wave (19, 20). After diffusion into the extracellular space, ATP binds to purinoceptors of neighboring astrocytes inducing inositol 1,4,5-triphosphate (IP3)-mediated mobilization of Ca2+ from internal stores. Glutamate is released in association with Ca2+ waves (21), but does not appear to have a primary role in the propagation of the wave itself (22). Gap junctions also play a role in the transmission mechanism by the diffusion of IP3 from cytosol to cytosol (17). The function of astrocytic Ca2+ waves is unclear and could represent a form of multicellular, bidirectional communication with neurons (23-25) or microarterioles (26).
In this study, we unveiled a role of 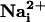 and glutamate as mediators of a concerted multicellular metabolic response, through the generation of intercellular Na+ waves and of spatially correlated enhanced glucose utilization.
and glutamate as mediators of a concerted multicellular metabolic response, through the generation of intercellular Na+ waves and of spatially correlated enhanced glucose utilization.
Materials and Methods
Cell Culture. Cortical astrocytes in primary culture were obtained from 1- to 3-day-old OF1 mice as described (5). Cells were grown for 2-5 weeks on glass coverslips in DME medium supplemented with 10% FCS.
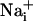 and
and 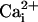 Imaging.
Imaging. 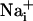 was measured by using the Na+-sensitive fluorescent probe sodium-binding benzofuran isophthalate (SBFI) (Teflabs, Austin, TX), and
was measured by using the Na+-sensitive fluorescent probe sodium-binding benzofuran isophthalate (SBFI) (Teflabs, Austin, TX), and 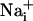 was measured by using the Ca2+-sensitive fluorescent probe Fluo-4 (Teflabs). Fluorescence was sequentially excited at 340 and 380 nm for SBFI, and at 490 nm for Fluo-4. Emitted fluorescence was detected through a 520-nm (40-nm bandwidth) filter (Omega Optical). Unless specified, experiments were carried out with astrocytes loaded at 37°C with 15 μM SBFI-acetoxymethyl ester (AM) and 6 μM Fluo-4-AM in a Hepes-buffered solution (see below). Cells were then mounted in an open perfusion chamber (Warner Instrument, Hamden, CT) at room temperature or at 37°C as indicated.
was measured by using the Ca2+-sensitive fluorescent probe Fluo-4 (Teflabs). Fluorescence was sequentially excited at 340 and 380 nm for SBFI, and at 490 nm for Fluo-4. Emitted fluorescence was detected through a 520-nm (40-nm bandwidth) filter (Omega Optical). Unless specified, experiments were carried out with astrocytes loaded at 37°C with 15 μM SBFI-acetoxymethyl ester (AM) and 6 μM Fluo-4-AM in a Hepes-buffered solution (see below). Cells were then mounted in an open perfusion chamber (Warner Instrument, Hamden, CT) at room temperature or at 37°C as indicated.
Experiments were performed on the stage of an inverted epifluorescence microscope (Nikon) and observed through a 20× 0.8 numerical aperture (NA) glycerol-immersion or a 40× 1.3 NA oil-immersion objective lens (Nikon). Fluorescence excitation wavelengths were selected by using a fast filter wheel (Sutter Instruments) fed to the microscope by a liquid light guide. Fluorescence was detected by using a Gen III+ intensified charge-coupled device (CCD) Camera (VideoScope International, Washington, DC). Acquisition and digitization of images as well as time series was computer controlled by using metafluor software (Universal Imaging). Images were recorded without video frame averaging, allowing Na+ plus Ca2+ monitoring (i.e., acquisition of three images) at 0.7 Hz.
When indicated, at the end of the experiment, a 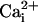 calibration was performed as described (5) by permeabilizing cells for monovalent cations using 6 μg/ml gramicidin and 10 μM monensin with simultaneous inhibition of the Na+/K+ATPase using 1 mM ouabain. Cells were then sequentially perfused with standard solutions containing 0, 5, 10, 20, 50, and 100 mM Na+. A calibration curve was computed for each selected cell and used to convert fluorescence ratio values into Na+ concentrations.
calibration was performed as described (5) by permeabilizing cells for monovalent cations using 6 μg/ml gramicidin and 10 μM monensin with simultaneous inhibition of the Na+/K+ATPase using 1 mM ouabain. Cells were then sequentially perfused with standard solutions containing 0, 5, 10, 20, 50, and 100 mM Na+. A calibration curve was computed for each selected cell and used to convert fluorescence ratio values into Na+ concentrations.
Experimental solutions (pH 7.4) contained 135 mM NaCl, 5.4 mM KCl, 20 mM Hepes, 1.3 mM CaCl2, 0.8 mM MgSO4, 0.78 mM NaH2PO4, and 5 mM glucose. The solution for dye loading contained 20 mM glucose and 0.1% Pluronic F127.
Cell Stimulation. Electrical stimulation was applied by one 10-V square twin pulse (10 ms) using a fine tip diameter concentric bipolar electrode (World Precision Instruments, Sarasota, FL, or FHC, Bowdoinham, ME) placed 20 μm above the cell surface. For mechanical stimulations, a patch-clamp micropipette was gently brought into contact with the astrocyte membrane. To ensure that  or
or 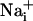 responses were not initiated by cell membrane damage, the contact between the membrane and the pipette tip was controlled by monitoring the pipette resistance and seal formation with an Axopatch 200A amplifier (Axon Instrument, Union City, CA). Electrical stimulation enabled us to reproducibly trigger several waves from the same cells, showing that it had no harmful effect on the stimulated cell. However, we considered any loss of fluorescence signal from a stimulated cell as a sign of cell membrane injury and discarded the experiment. Cell stimulation was always performed without superfusing the cells. Astrocytes were first electrically stimulated in the control medium described above. After recovery of
responses were not initiated by cell membrane damage, the contact between the membrane and the pipette tip was controlled by monitoring the pipette resistance and seal formation with an Axopatch 200A amplifier (Axon Instrument, Union City, CA). Electrical stimulation enabled us to reproducibly trigger several waves from the same cells, showing that it had no harmful effect on the stimulated cell. However, we considered any loss of fluorescence signal from a stimulated cell as a sign of cell membrane injury and discarded the experiment. Cell stimulation was always performed without superfusing the cells. Astrocytes were first electrically stimulated in the control medium described above. After recovery of 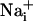 and
and 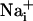 to basal level, test compounds or enzymes were introduced by superfusion. Unless specified, after a 3-min incubation with the test compound, a second stimulation of the same cells was performed.
to basal level, test compounds or enzymes were introduced by superfusion. Unless specified, after a 3-min incubation with the test compound, a second stimulation of the same cells was performed.
Wave Speed Measurement. Image sequences were analyzed by using metamorph software (Universal Imaging) to measure the propagation speed of the Na+ and Ca2+ waves. SBFI fluorescence excitation ratios (F340 nm/F380 nm) were computed for each pixel to produce ratio-images that were proportional with  . Fluo-4 and SBFI image stacks were built separately for the first 30 time points after the stimulation. Then, a 20 × 20 pixel median filter was applied on every third pixel of all images of this stack, and an initial reference image was subtracted. Both stacks were finally thresholded and binarized. The pixel number in binary masks corresponding to areas of increased
. Fluo-4 and SBFI image stacks were built separately for the first 30 time points after the stimulation. Then, a 20 × 20 pixel median filter was applied on every third pixel of all images of this stack, and an initial reference image was subtracted. Both stacks were finally thresholded and binarized. The pixel number in binary masks corresponding to areas of increased 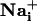 or
or 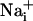 was measured over time and approximated to a circular area for each time point. The speed of wave propagation (μm/sec) was calculated from the rate of radius change.
was measured over time and approximated to a circular area for each time point. The speed of wave propagation (μm/sec) was calculated from the rate of radius change.
Imaging of Glucose Uptake. After acquisition of a background image, the fluorescent glucose analogue 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBD-glucose) (500 μM) was added to the experimental solution. Electrical stimulation was then immediately performed as described above and, after a 2-min delay, 2-NBD-glucose was rapidly washed out and images of the cell monolayer were captured. Fluorescence of 2-NBD-glucose and of background was excited at 490 nm and observed through a 520-nm (40-nm bandwidth) filter.
Data and Statistics. Data are presented as means ± SEM. A paired Student t test was performed to assess the statistical significance of results with P < 0.05 considered as significant. The number n represents independent experiments performed on different coverslips.
Materials. Threo-β-benzyloxyaspartate (TBOA) and suramin hexasodium salt were from Tocris-Anawa Trading (Zurich), 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate-BAPTA)-AM, 2-NBD-glucose, and Pluronic F-127 were from Molecular Probes, and all other compounds and enzymes were from Sigma.
Results
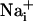 Wave.
Wave. 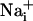 was monitored microspectrofluorimetrically by using the Na+-sensitive fluorescent probe SBFI loaded in astrocytes. Both mechanical and electrical stimulation of a single cultured astrocyte induced an intracellular Na+ elevation in the stimulated cell that spread to the surrounding cells (Fig. 1A). In a given direction, the
was monitored microspectrofluorimetrically by using the Na+-sensitive fluorescent probe SBFI loaded in astrocytes. Both mechanical and electrical stimulation of a single cultured astrocyte induced an intracellular Na+ elevation in the stimulated cell that spread to the surrounding cells (Fig. 1A). In a given direction, the 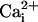 increases propagate as a wave (Fig. 1B). The extent of wave spreading appeared to depend on the strength of stimulation. The Na+ wave front had a radial propagation immediately after stimulation (Fig. 1C). Interestingly, farther away from the stimulated cells, the wave did not spread in all directions with the same speed (Fig. 1D).
increases propagate as a wave (Fig. 1B). The extent of wave spreading appeared to depend on the strength of stimulation. The Na+ wave front had a radial propagation immediately after stimulation (Fig. 1C). Interestingly, farther away from the stimulated cells, the wave did not spread in all directions with the same speed (Fig. 1D).
Fig. 1.

Astrocytes generate intercellular 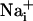 waves. (A) Intensity-modulated-display images of SBFI excitation ratio recorded in cultured astrocytes showing the
waves. (A) Intensity-modulated-display images of SBFI excitation ratio recorded in cultured astrocytes showing the 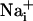 elevation over time after single-cell mechanical stimulation. The top image shows the pipette tip position over the cell. The false colors represent SBFI ratio values corresponding to
elevation over time after single-cell mechanical stimulation. The top image shows the pipette tip position over the cell. The false colors represent SBFI ratio values corresponding to 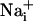 ranging from dark green for resting values to red for elevated values. (Scale bar, 200 μm.) (B) Single cell SBFI ratio values plotted against time. The corresponding cells (white regions, Right) were randomly selected along a line (dotted line) starting from the stimulated cell 1. The red arrows represent the onset of response in each cell defined as the timepoint preceding a signal increase >2 SD over baseline. The graph shows that the
ranging from dark green for resting values to red for elevated values. (Scale bar, 200 μm.) (B) Single cell SBFI ratio values plotted against time. The corresponding cells (white regions, Right) were randomly selected along a line (dotted line) starting from the stimulated cell 1. The red arrows represent the onset of response in each cell defined as the timepoint preceding a signal increase >2 SD over baseline. The graph shows that the 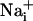 increase occurred sequentially in a cell-by-cell manner along the line. (C)Na+ increase for regions selected at a radial distance of 180 μm (dotted circle, Right). The arrows shows that at an equal given distance,
increase occurred sequentially in a cell-by-cell manner along the line. (C)Na+ increase for regions selected at a radial distance of 180 μm (dotted circle, Right). The arrows shows that at an equal given distance, 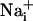 increase is synchronous. (D)
increase is synchronous. (D) 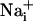 increase for cells selected 210 μm from the stimulated spot (dotted circle, Right). The red arrows indicate that, at this distance,
increase for cells selected 210 μm from the stimulated spot (dotted circle, Right). The red arrows indicate that, at this distance, 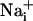 increases are no longer synchronous (n = 6 experiments).
increases are no longer synchronous (n = 6 experiments).
Na+ and Ca2+ Waves. The Na+ waves described above are reminiscent of the Ca2+ waves in astrocytes reported earlier by several groups (17). To investigate whether these two types of waves could coexist, a method was developed to simultaneously monitor  and
and 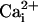 by using the fluorescent probes SBFI and Fluo-4, respectively. Both a Ca2+ and a Na+ wave were simultaneously observed after mechanical stimulation (data not shown) or electrical stimulation (Fig. 2 and see Movie 1, which is published as supporting information on the PNAS web site). The kinetics of
by using the fluorescent probes SBFI and Fluo-4, respectively. Both a Ca2+ and a Na+ wave were simultaneously observed after mechanical stimulation (data not shown) or electrical stimulation (Fig. 2 and see Movie 1, which is published as supporting information on the PNAS web site). The kinetics of 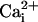 and
and 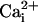 increase were found to be markedly different in the cells reached by the wave (Fig. 2B). Moreover, during the first 4-8 sec, the initial Na+ wave speed was ≈40% slower than the Ca2+ wave speed (Fig. 2C).
increase were found to be markedly different in the cells reached by the wave (Fig. 2B). Moreover, during the first 4-8 sec, the initial Na+ wave speed was ≈40% slower than the Ca2+ wave speed (Fig. 2C).
Fig. 2.

Coexistence and characteristics of Na+ and Ca2+ waves. (A) Simultaneous Na+ and Ca2+ imaging showed that a Ca2+ wave is evoked by electrical stimulation and parallels the observed Na+ wave. (Upper) Intensity-modulated display image series of SBFI ratio (Na+ wave). (Lower) Fluorescence image series of Fluo-4 recorded simultaneously (Ca2+ wave). An initial reference image was subtracted from this image series to represent only the Ca2+ elevation over time after stimulation. (Scale bar, 200 μm.) (B) Fluo-4 fluorescence and SBFI ratio were plotted against time for four selected cells (circles in A Upper Right). (D) The speed of the Na+ wave was 40% slower than that of the Ca2+ wave. Data are means ± SEM (*, P < 0.05; n = 9 experiments).
Mechanism of Wave Transmission. Two consecutive electrical stimulations on the same astrocyte yielded reproducible Na+ and Ca2+ wave speeds (Fig. 3), confirming that cell integrity is maintained throughout the experiment. Therefore, in subsequent protocols, we could safely use the wave evoked by the first stimulation as the experimental control value. Numerous studies have supported a role for gap junctional coupling in intercellular Ca2+ signaling in astrocytes. However, octanol and carbenoxolone, two compounds known to block gap junctions in astrocytes (17, 27), had a modest (if any) inhibitory effect on Ca2+ wave propagation, but a 40-45% reduction of the Na+ wave (Fig. 3). ATP has been identified as a primary extracellular messenger involved in the transmission of Ca2+ waves (19, 20, 28). We found that both Ca2+ and Na+ waves were inhibited by >70% by the P2Y1-2 purinergic receptor antagonist suramin (100 μM) (Fig. 3), confirming the involvement of ATP. Chelating 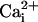 with BAPTA has been reported to inhibit Ca2+ waves (21, 23). To evaluate whether the Na+ wave depended directly on
with BAPTA has been reported to inhibit Ca2+ waves (21, 23). To evaluate whether the Na+ wave depended directly on 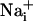 elevation, after a first control stimulation, cells were incubated for 30 min with the membrane-permeant BAPTA-AM (50 μM) in the presence of Ca2+ in the bath. This maneuver, which buffers
elevation, after a first control stimulation, cells were incubated for 30 min with the membrane-permeant BAPTA-AM (50 μM) in the presence of Ca2+ in the bath. This maneuver, which buffers 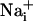 increases, led to a massive inhibition of both Ca2+ and Na+ waves (Fig. 3) to 86% and 89% of the control values, respectively.
increases, led to a massive inhibition of both Ca2+ and Na+ waves (Fig. 3) to 86% and 89% of the control values, respectively.
Fig. 3.

Mechanism of Ca2+ and Na+ wave propagation. Results from Na+ (Upper) or Ca2+ (Lower) waves triggered by a first electrical stimulation as a control (white bars) and a second consecutive stimulation (black bars) are shown. Data are presented as percentage of the control wave speed. The waves triggered by two consecutive stimulations were not significantly different (2nd stim) (n = 9 experiments). Blocking gap junctions with 500 μM octanol (n = 8 experiments) or 20 μM carbenoxolone (CBX, n = 6 experiments) had a modest, if any, effect on the Ca2+ wave speed, but a more pronounced effect on the Na+ wave. Both Ca2+ and Na+ waves were inhibited up to 70% by the purinergic receptor antagonist suramin (100 μM, n = 6 experiments). Chelating intracellular Ca2+ by treatment with 50 μM BAPTA-AM massively inhibited both waves (n = 6 experiments). Application of the glutamate transporter inhibitor TBOA (500 μM) resulted in a strong inhibition of the Na+ wave. By contrast, the Ca2+ wave was not influenced by the presence of TBOA (n = 7 experiments). Na+ waves were ≈50% reduced by application of either glutamate oxidase (GLOD; 1 unit/ml) in the extracellular medium (n = 6 experiments) or glutamate decarboxylase (GAD; 40 units/ml) (n = 6 experiments). Ca2+ waves were not influenced by either GLOD or GAD. Data are means ± SEM (*, P < 0.05; **, P < 0.01; ***, P < 0.001). n.s., Not significant.
It has been described that astrocytes release glutamate in association with Ca2+ waves (21); however, the released glutamate does not appear to play a primary role in the propagation of the Ca2+ waves themselves (22). Glutamate is also avidly taken up by astrocytes with three Na+ ions, resulting in an increase of the astrocytic 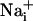 concentration (5). Fig. 3 shows that application of the glutamate transporter inhibitor TBOA (500 μM) resulted in a strong (≈70%) inhibition of the Na+ wave, which was fully reversible (data not shown), without significantly affecting the Ca2+ wave. To confirm the involvement of glutamate in the generation of the Na+ wave, l-glutamate oxidase was added to the extracellular medium to oxidize the released glutamate to 2-oxoglutarate. The Na+ wave speed was inhibited by 50% in the presence of extracellular glutamate oxidase without slowing down the Ca2+ wave (Fig. 3). Another enzyme, glutamate decarboxylase, applied under the same conditions, had a similar effect as glutamate oxidase. These results demonstrate that glutamate release and its subsequent Na+-dependent uptake mediate the regenerative propagation of the Na+ wave.
concentration (5). Fig. 3 shows that application of the glutamate transporter inhibitor TBOA (500 μM) resulted in a strong (≈70%) inhibition of the Na+ wave, which was fully reversible (data not shown), without significantly affecting the Ca2+ wave. To confirm the involvement of glutamate in the generation of the Na+ wave, l-glutamate oxidase was added to the extracellular medium to oxidize the released glutamate to 2-oxoglutarate. The Na+ wave speed was inhibited by 50% in the presence of extracellular glutamate oxidase without slowing down the Ca2+ wave (Fig. 3). Another enzyme, glutamate decarboxylase, applied under the same conditions, had a similar effect as glutamate oxidase. These results demonstrate that glutamate release and its subsequent Na+-dependent uptake mediate the regenerative propagation of the Na+ wave.
Metabolic Consequences of the Na+ Wave. Because glutamate-evoked 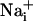 increase has been shown to increase the energy demands in astrocytes and mediate the metabolic response of astrocytes (5) in response to synaptic activity (3), we investigated whether Na+ waves would affect glucose metabolism. To this aim, electrical stimulations of astrocytes were performed under the same conditions as those described above in the presence of the fluorescent glucose analogue 2-NBD-glucose, which was used as a tracer to evaluate the spatial distribution of glucose uptake. This fluorescent glucose analogue has been shown to undergo rapid transport in astrocytes, which was enhanced by the presence of glutamate (29). Fig. 4 shows that stimulations that evoke Na+ and Ca2+ waves increased cellular 2-NBD-glucose in a circular region around the stimulated cell. Remarkably, the pattern of spatial spreading of 2-NBD-glucose closely follows that of
increase has been shown to increase the energy demands in astrocytes and mediate the metabolic response of astrocytes (5) in response to synaptic activity (3), we investigated whether Na+ waves would affect glucose metabolism. To this aim, electrical stimulations of astrocytes were performed under the same conditions as those described above in the presence of the fluorescent glucose analogue 2-NBD-glucose, which was used as a tracer to evaluate the spatial distribution of glucose uptake. This fluorescent glucose analogue has been shown to undergo rapid transport in astrocytes, which was enhanced by the presence of glutamate (29). Fig. 4 shows that stimulations that evoke Na+ and Ca2+ waves increased cellular 2-NBD-glucose in a circular region around the stimulated cell. Remarkably, the pattern of spatial spreading of 2-NBD-glucose closely follows that of 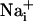 spreading, measured in separate experiments and represented on the same graph. It should be noted that, under these conditions, the stimulated cell responded with a
spreading, measured in separate experiments and represented on the same graph. It should be noted that, under these conditions, the stimulated cell responded with a 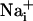 increase of 64.2 ± 11.2 mM from a resting
increase of 64.2 ± 11.2 mM from a resting 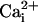 value of 6.6 ± 0.7 mM, whereas the amplitude of
value of 6.6 ± 0.7 mM, whereas the amplitude of 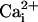 response in cells reached by the wave decreased monotonically along the propagation axes reaching 4.6 ± 1.1 mM at a distance of 213 μm away from the wave origin (Fig. 4 A, C, and D). To verify that the increased 2-NBD-glucose uptake was indeed caused by the glutamate-mediated Na+ wave, we repeated the experiment in the presence of the glutamate transporter inhibitor TBOA (500 μM). Under these conditions, where the Na+ waves are selectively inhibited, only basal 2-NBD-glucose uptake could be observed (Fig. 4 B and E). These experiments demonstrate that the Na+ waves bring about a wave of increased glucose uptake with tight spatial correlation.
response in cells reached by the wave decreased monotonically along the propagation axes reaching 4.6 ± 1.1 mM at a distance of 213 μm away from the wave origin (Fig. 4 A, C, and D). To verify that the increased 2-NBD-glucose uptake was indeed caused by the glutamate-mediated Na+ wave, we repeated the experiment in the presence of the glutamate transporter inhibitor TBOA (500 μM). Under these conditions, where the Na+ waves are selectively inhibited, only basal 2-NBD-glucose uptake could be observed (Fig. 4 B and E). These experiments demonstrate that the Na+ waves bring about a wave of increased glucose uptake with tight spatial correlation.
Fig. 4.

Na+ waves enhance cellular glucose uptake. (A) 2-NBD-glucose uptake (dotted line, filled diamonds) expressed as percentage of the 2-NBD-glucose fluorescence intensity measured in cells >200 μm away from the electrode tip (equivalent to basal 2-NBD-glucose uptake). 2-NBD-glucose intensity was plotted against distance from the electrically stimulated spot in regions depicted in the image (C). The amplitude of 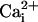 (in mM), obtained in separate experiments (solid line, open diamonds), shows the high degree of correspondence of the spatial spreading of Na+ wave and the enhanced glucose uptake. Resting
(in mM), obtained in separate experiments (solid line, open diamonds), shows the high degree of correspondence of the spatial spreading of Na+ wave and the enhanced glucose uptake. Resting 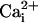 values were 6.6 ± 0.7 mM (n = 36 cells, four experiments). (B) 2-NBD-glucose uptake measured in the absence (dotted line, filled diamonds) or the presence of 500 μM TBOA (solid line, open diamonds) as described in A. Data are means ± SEM (n = 4 experiments each). The linear slopes of fluorescent signal in the range of 48-165 μM were -1.9 ± 0.7 μM-1 for control and -0.08 ± 0.06 μM-1 for TBOA (P < 0.05). (C) Transmitted light image of cells showing the electrode tip position and an overlay of the regions selected for analysis. (D and E) Images of 2-NBD-glucose fluorescence after electrical stimulation. The two images shown were acquired on the same cells, first in the presence of TBOA (E) and then in control solution after washout of the drug (D). (Scale bar, 200 μm.)
values were 6.6 ± 0.7 mM (n = 36 cells, four experiments). (B) 2-NBD-glucose uptake measured in the absence (dotted line, filled diamonds) or the presence of 500 μM TBOA (solid line, open diamonds) as described in A. Data are means ± SEM (n = 4 experiments each). The linear slopes of fluorescent signal in the range of 48-165 μM were -1.9 ± 0.7 μM-1 for control and -0.08 ± 0.06 μM-1 for TBOA (P < 0.05). (C) Transmitted light image of cells showing the electrode tip position and an overlay of the regions selected for analysis. (D and E) Images of 2-NBD-glucose fluorescence after electrical stimulation. The two images shown were acquired on the same cells, first in the presence of TBOA (E) and then in control solution after washout of the drug (D). (Scale bar, 200 μm.)
Discussion
Glutamate-evoked Na+ uptake in astrocytes and the resulting activation of the Na+/K+-ATPase have been identified as a signal coupling excitatory neuronal activity to increased glucose utilization (5, 7). In addition, astrocytes have the ability of transmitting 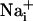 elevations from cell to cell (17). These so-called Ca2+ waves have revealed that astrocytes are active participants in multicellular signaling in the central nervous system. In this study, we show that astrocytes can also generate intercellular Na+ waves through a mechanism involving glutamate as a key mediator in addition to ATP,
elevations from cell to cell (17). These so-called Ca2+ waves have revealed that astrocytes are active participants in multicellular signaling in the central nervous system. In this study, we show that astrocytes can also generate intercellular Na+ waves through a mechanism involving glutamate as a key mediator in addition to ATP, 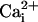 , and gap junctions. The Na+ wave described here mediates the spreading of the cellular metabolic responses within groups of astrocytes. This observation has important implications for brain imaging techniques based on deoxyglucose uptake as an index of glucose utilization.
, and gap junctions. The Na+ wave described here mediates the spreading of the cellular metabolic responses within groups of astrocytes. This observation has important implications for brain imaging techniques based on deoxyglucose uptake as an index of glucose utilization.
An intercellular Na+ wave could be evoked by both electrical and mechanical stimulation in cultured mouse astrocytes. Pharmacological data indicate that the release of both ATP and glutamate are primarily responsible for the transmission mechanism as well as, to a lesser extent, gap junctions, inasmuch as 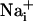 can equilibrate among astrocytes through gap junctions (30). The Na+ waves were found to coexist with Ca2+ waves. The propagation speed of the Ca2+ waves observed here are comparable to those previously reported for astrocytes, with values ranging from 5 to 30 μm/s (17). The kinetics of the initial phase of the Ca2+ wave (23.9 ± 2.7 μm/s) and Na+ wave (14.4 ± 2.7 μm/s) were markedly different, as was the kinetics of single cell ion concentration changes (Fig. 4), excluding a crosstalk between the Na+ and Ca2+ signals.
can equilibrate among astrocytes through gap junctions (30). The Na+ waves were found to coexist with Ca2+ waves. The propagation speed of the Ca2+ waves observed here are comparable to those previously reported for astrocytes, with values ranging from 5 to 30 μm/s (17). The kinetics of the initial phase of the Ca2+ wave (23.9 ± 2.7 μm/s) and Na+ wave (14.4 ± 2.7 μm/s) were markedly different, as was the kinetics of single cell ion concentration changes (Fig. 4), excluding a crosstalk between the Na+ and Ca2+ signals.
Although these lines of evidence demonstrate that Na+ and Ca2+ waves are distinct cellular phenomena, we evaluated the nature of their relationship. Whereas the kinetics of Na+ and Ca2+ responses are markedly different, inhibiting purinoceptors or chelating 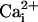 have similar inhibitory effects on both waves. Therefore, one could argue that Ca2+ and Na+ share the same transmission mechanism, and that the
have similar inhibitory effects on both waves. Therefore, one could argue that Ca2+ and Na+ share the same transmission mechanism, and that the 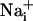 elevation is mainly due to the Na+/Ca2+ exchanger. This hypothesis is unlikely because a
elevation is mainly due to the Na+/Ca2+ exchanger. This hypothesis is unlikely because a 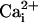 elevation is not accompanied by a significant
elevation is not accompanied by a significant 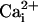 elevation as shown by the application of the α-adrenergic agonist phenylephrine, and because ATP does not induce a direct intracellular Na+ increase in astrocytes (data not shown). Furthermore, the fact that application of the Na+/glutamate transporter inhibitor TBOA resulted in a strong inhibition of the Na+ wave without significantly affecting the Ca2+ wave excludes the Na+/Ca2+ exchanger as the main mediator of Na+ waves. However, our data show that Na+ waves require at some level an ATP-evoked intracellular Ca2+ elevation.
elevation as shown by the application of the α-adrenergic agonist phenylephrine, and because ATP does not induce a direct intracellular Na+ increase in astrocytes (data not shown). Furthermore, the fact that application of the Na+/glutamate transporter inhibitor TBOA resulted in a strong inhibition of the Na+ wave without significantly affecting the Ca2+ wave excludes the Na+/Ca2+ exchanger as the main mediator of Na+ waves. However, our data show that Na+ waves require at some level an ATP-evoked intracellular Ca2+ elevation.
Because Ca2+ elevations have been shown to trigger the release of glutamate in astrocytes (8, 15, 16), we tested whether glutamate could be the element that links our observations. Indeed, the fact that inhibiting Na+/glutamate transporter selectively blocked the Na+ wave indicated that extracellular glutamate was involved in its mechanism of transmission. This conclusion was supported by the fact that extracellular glutamate degradation, using specific enzymes, also selectively inhibited the Na+ wave without affecting the Ca2+ wave. The observation that each maneuver that inhibited the Ca2+ wave also inhibited the Na+ wave in a equivalent manner can be explained by the fact that glutamate release is triggered by 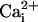 elevations (8, 15); it also implies that the Na+ wave is “driven” by the Ca2+ wave.
elevations (8, 15); it also implies that the Na+ wave is “driven” by the Ca2+ wave.
The data presented here support the following transmission mechanism for the two waves (Fig. 5): mechanical or electrical stimuli induce Ca2+ release from the intracellular stores in the stimulated cell as described previously. 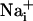 elevation is followed by the release of ATP, by a possibly Ca2+-independent mechanism (28), and by the release of glutamate in the extracellular space. Extracellular ATP then binds to purinoceptors on neighboring astrocytes, which induce a
elevation is followed by the release of ATP, by a possibly Ca2+-independent mechanism (28), and by the release of glutamate in the extracellular space. Extracellular ATP then binds to purinoceptors on neighboring astrocytes, which induce a 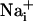 response and participate in the propagation of the Ca2+ signals in addition to inositol 1,4,5-triphosphate (IP3) (17). In parallel, the released glutamate is taken up by Na+/glutamate cotransporters, resulting in
response and participate in the propagation of the Ca2+ signals in addition to inositol 1,4,5-triphosphate (IP3) (17). In parallel, the released glutamate is taken up by Na+/glutamate cotransporters, resulting in 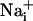 increases. Na+ has also the ability to diffuse through gap junctions (30) and participate in the Na+ wave extension. In essence, it appears that glutamate release/reuptake sustains the regenerative propagation mechanism of the Na+ signal, whereas gap junctions mediate the passive spreading of the Na+ signal.
increases. Na+ has also the ability to diffuse through gap junctions (30) and participate in the Na+ wave extension. In essence, it appears that glutamate release/reuptake sustains the regenerative propagation mechanism of the Na+ signal, whereas gap junctions mediate the passive spreading of the Na+ signal.
Fig. 5.

Model for Na+ and metabolic wave transmission mechanism. 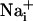 elevation is followed by the release of ATP and the release of glutamate in the extracellular space. Extracellular ATP then binds to purinoceptors on neighboring astrocytes, which induce a
elevation is followed by the release of ATP and the release of glutamate in the extracellular space. Extracellular ATP then binds to purinoceptors on neighboring astrocytes, which induce a  response and participate in the propagation of the Ca2+ signals in addition to inositol 1,4,5-triphosphate (IP3) (17). In parallel, the released glutamate is taken up by Na+/glutamate cotransporters, resulting in
response and participate in the propagation of the Ca2+ signals in addition to inositol 1,4,5-triphosphate (IP3) (17). In parallel, the released glutamate is taken up by Na+/glutamate cotransporters, resulting in 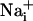 increases. Na+ has also the ability to diffuse through gap junctions and participate in the Na+ wave extension. The
increases. Na+ has also the ability to diffuse through gap junctions and participate in the Na+ wave extension. The  elevation is sufficient to enhance Na+/K+-ATPase activity and, therefore, glucose consumption in astrocytes implicated by the wave.
elevation is sufficient to enhance Na+/K+-ATPase activity and, therefore, glucose consumption in astrocytes implicated by the wave.
The 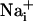 concentration in the cells adjacent to the stimulated one reaches 41.1 ± 6.5 mM and decreases monotonically along the propagation axes of the wave to a value of 10.8 ± 4.1 mM at a distance of 165 μm from the stimulated cell. Our laboratory has shown that the amplitude of Na+ rise is proportional to the extracellular concentration of glutamate, reaching maximal values of 30-40 mM (5). In addition, an extracellular glutamate concentration of 10 μM, inducing an
concentration in the cells adjacent to the stimulated one reaches 41.1 ± 6.5 mM and decreases monotonically along the propagation axes of the wave to a value of 10.8 ± 4.1 mM at a distance of 165 μm from the stimulated cell. Our laboratory has shown that the amplitude of Na+ rise is proportional to the extracellular concentration of glutamate, reaching maximal values of 30-40 mM (5). In addition, an extracellular glutamate concentration of 10 μM, inducing an 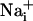 elevation of ≈8 mM, already markedly increases the energy demands of the astrocytes by activating the Na+/K+ ATPase (5, 6). Therefore, the
elevation of ≈8 mM, already markedly increases the energy demands of the astrocytes by activating the Na+/K+ ATPase (5, 6). Therefore, the 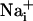 elevation should be sufficient to enhance Na+/K+-ATPase activity up to 165 μm away from the stimulated cell, and consequently glucose consumption in the astrocytes implicated by the wave. Indeed, we showed that there was a quasi-perfect match between the spread of the Na+ wave and the pattern of increased 2-NBD-glucose uptake, which was increased compared to basal uptake >100 μm from the initiation site. Therefore, we can propose that the Na+ wave could serve to mediate the spreading of the cellular metabolic response within groups of astrocytes and confirm the role of
elevation should be sufficient to enhance Na+/K+-ATPase activity up to 165 μm away from the stimulated cell, and consequently glucose consumption in the astrocytes implicated by the wave. Indeed, we showed that there was a quasi-perfect match between the spread of the Na+ wave and the pattern of increased 2-NBD-glucose uptake, which was increased compared to basal uptake >100 μm from the initiation site. Therefore, we can propose that the Na+ wave could serve to mediate the spreading of the cellular metabolic response within groups of astrocytes and confirm the role of 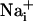 as a second messenger in the neurometabolic coupling (5-7).
as a second messenger in the neurometabolic coupling (5-7).
Glutamate released during synaptic transmission is taken up by astrocytes but can also initiate the emergence of a Ca2+ wave as described (31). We found that local photoactivation of caged glutamate was able to evoke a propagating Ca2+ response in cultured astrocytes (data not shown). However, in the experiments presented here, mechanical and electrical stimulations were preferred to exclude the exogenously applied glutamate's stimulation of other, neighboring cells by diffusion, therefore potentially masking glutamate actively released by cells, as discussed by others (31, 32). Although Ca2+ signals can mediate the astrocytic control of brain microcirculation, allowing astrocytes to participate in neurovascular coupling (26), the physiological role of Ca2+ waves is unclear. We propose here that Ca2+ waves, through the concomitant glutamate release and generation of Na+ waves, send a metabolic signal in register to synaptic activity and provide energy substrates, e.g., under the form of lactate, to neurons at a significant distance from the activated synapse. One could also envisage that inhibitory synapses more distant from the glutamate release sites could be fed through the astrocytic network, considering the fact that the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (also taken up by Na+-dependent transporters into astrocytes) does not couple inhibitory neuronal activity with glucose utilization (6). In addition, the existence of Na+ waves, demonstrated in this study, may have implications for brain imaging techniques based on glucose consumption such as positron emission tomography (PET). Even though the current resolution of 2-18FDG PET imaging is limited by the scanner resolution to 3-5 mm (33), our data may contribute to the interpretation of such signals and could indicate that the metabolic response to local synaptic activity probably extends to a larger spatial domain than initially thought.
Supplementary Material
Acknowledgments
We thank Corinne Moratal for helpful technical assistance and Igor Allaman and Andrea Volterra for stimulating discussions. This work was supported by Novartis Research Foundation and Swiss National Science Foundation Grant 3100-067116 (to J.-Y.C.).
Author contributions: Y.B., P.J.M., and J.-Y.C. designed research; Y.B. and J.-Y.C. performed research; Y.B. and J.-Y.C. analyzed data; and Y.B., P.J.M., and J.-Y.C. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SBFI, sodium-binding benzofuran isophthalate; 2-NBD-glucose, 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose; TBOA, threo-β-benzyloxyaspartate; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate; 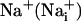 , intracellular Na+;
, intracellular Na+; 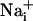 , intracellular Ca2+; AM, acetoxymethyl ester.
, intracellular Ca2+; AM, acetoxymethyl ester.
References
- 1.Robinson, M. B. & Dowd, L. A. (1997) Adv. Pharmacol. 37, 69-115. [DOI] [PubMed] [Google Scholar]
- 2.Magistretti, P. J., Pellerin, L., Rothman, D. L. & Shulman, R. G. (1999) Science 283, 496-497. [DOI] [PubMed] [Google Scholar]
- 3.Voutsinos-Porche, B., Bonvento, G., Tanaka, K., Steiner, P., Welker, E., Chatton, J. Y., Magistretti, P. J. & Pellerin, L. (2003) Neuron 37, 275-286. [DOI] [PubMed] [Google Scholar]
- 4.Nedergaard, M., Takano, T. & Hansen, A. J. (2002) Nat. Rev. Neurosci. 3, 748-755. [DOI] [PubMed] [Google Scholar]
- 5.Chatton, J.-Y., Marquet, P. & Magistretti, P. J. (2000) Eur. J. Neurosci. 12, 3843-3853. [DOI] [PubMed] [Google Scholar]
- 6.Chatton, J.-Y., Pellerin, L. & Magistretti, P. J. (2003) Proc. Natl. Acad. Sci. USA 100, 12456-12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pellerin, L. & Magistretti, P. J. (1994) Proc. Natl. Acad. Sci. USA 91, 10625-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bezzi, P., Carmignoto, G., Pasti, L., Vesce, S., Rossi, D., Rizzini, B. L., Pozzan, T. & Volterra, A. (1998) Nature 391, 281-285. [DOI] [PubMed] [Google Scholar]
- 9.Jeremic, A., Jeftinija, K., Stevanovic, J., Glavaski, A. & Jeftinija, S. (2001) J. Neurochem. 77, 664-675. [DOI] [PubMed] [Google Scholar]
- 10.Parpura, V., Basarsky, T. A., Liu, F., Jeftinija, K., Jeftinija, S. & Haydon, P. G. (1994) Nature 369, 744-747. [DOI] [PubMed] [Google Scholar]
- 11.Bezzi, P. & Volterra, A. (2001) Curr. Opin. Neurobiol. 11, 387-394. [DOI] [PubMed] [Google Scholar]
- 12.Kimelberg, H. K., Rutledge, E., Goderie, S. & Charniga, C. (1995) J. Cereb. Blood Flow Metab. 15, 409-416. [DOI] [PubMed] [Google Scholar]
- 13.Duan, S., Anderson, C. M., Keung, E. C., Chen, Y. & Swanson, R. A. (2003) J. Neurosci. 23, 1320-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye, Z. C., Wyeth, M. S., Baltan-Tekkok, S. & Ransom, B. R. (2003) J. Neurosci. 23, 3588-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montana, V., Ni, Y., Sunjara, V., Hua, X. & Parpura, V. (2004) J. Neurosci. 24, 2633-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bezzi, P., Gundersen, V., Galbete, J. L., Seifert, G., Steinhauser, C., Pilati, E. & Volterra, A. (2004) Nat. Neurosci. 7, 613-620. [DOI] [PubMed] [Google Scholar]
- 17.Charles, A. C. & Giaume, C. (2002) in The Tripartite Synapse, eds. Volterra, A., Magistretti, P. J. & Haydon, P. G. (Oxford Univ. Press, New York), pp. 110-126.
- 18.Schipke, C. G., Boucsein, C., Ohlemeyer, C., Kirchhoff, F. & Kettenmann, H. (2002) FASEB J. 16, 255-257. [DOI] [PubMed] [Google Scholar]
- 19.Guthrie, P. B., Knappenberger, J., Segal, M., Bennett, M. V., Charles, A. C. & Kater, S. B. (1999) J. Neurosci. 19, 520-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson, C. M., Bergher, J. P. & Swanson, R. A. (2004) J. Neurochem. 88, 246-256. [DOI] [PubMed] [Google Scholar]
- 21.Innocenti, B., Vladimir, P. & Haydon, P. G. (2000) J. Neurosci. 5, 1800-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venance, L., Stella, N., Glowinski, J. & Giaume, C. (1997) J. Neurosci. 17, 1981-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araque, A., Sanzgiri, R. P., Parpura, V. & Haydon, P. G. (1998) J. Neurosci. 18, 6822-6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiacco, T. A. & McCarthy, K. D. (2004) J. Neurosci. 24, 722-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, Q. S., Xu, Q., Arcuino, G., Kang, J. & Nedergaard, M. (2004) Proc. Natl. Acad. Sci. USA 101, 3172-3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zonta, M., Augulo, M. C., Gobbo, S., Rosengarten, B., Hossmann, K.-A., Pozzan, T. & Carmignoto, G. (2003) Nat. Neurosci. 6, 43-50. [DOI] [PubMed] [Google Scholar]
- 27.Rouach, N., Segal, M., Koulakoff, A., Giaume, C. & Avignone, E. (2003) J. Physiol. 553, 729-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang, Z., Haydon, P. G. & Yeung, E. S. (2000) Anal. Chem. 72, 2001-2007. [DOI] [PubMed] [Google Scholar]
- 29.Loaiza, A., Porras, O. H. & Barros, L. F. (2003) J. Neurosci. 23, 7337-7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose, C. R. & Ransom, B. R. (1997) Glia 20, 299-307. [DOI] [PubMed] [Google Scholar]
- 31.Araque, A., Carmignoto, G. & Haydon, P. G. (2001) Annu. Rev. Physiol. 63, 795-813. [DOI] [PubMed] [Google Scholar]
- 32.Charles, A. C., Merrill, J. E., Dirksen, E. R. & Sanderson, M. J. (1991) Neuron 6, 683-992. [DOI] [PubMed] [Google Scholar]
- 33.Kessler, R. M. (2003) Neurobiol. Aging 24, S21-S35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}