

Abstract
Paracellular ion flux across epithelia occurs through selective and regulated pores in tight junctions; this process is poorly understood. Mutations in the kinase WNK4 cause pseudohypoaldosteronism type II (PHAII), a disease featuring hypertension and hyperkalemia. Whereas WNK4 is known to regulate several transcellular transporters and channels involved in NaCl and K+ homeostasis, its localization to tight junctions suggests it might also regulate paracellular flux. We performed electrophysiology on mammalian kidney epithelia with inducible expression of various WNK4 constructs. Induction of wild-type WNK4 reduced transepithelial resistance by increasing absolute chloride permeability. PHAII-mutant WNK4 produced markedly larger effects, whereas kinase-mutant WNK4 had no effect. The electrochemical and pharmacologic properties of these effects indicate they are attributable to the paracellular pathway. The effects of WNK4 persist when induction is delayed until after tight-junction formation, demonstrating a dynamic effect. WNK4 did not alter the flux of uncharged solutes, or the expression or localization of selected tight-junction proteins. Transmission and freeze-fracture electron microscopy showed no effect of WNK4 on tight-junction structure. These findings implicate WNK signaling in the coordination of transcellular and paracellular flux to achieve NaCl and K+ homeostasis, explain PHAII pathophysiology, and suggest that modifiers of WNK signaling may be potent antihypertensive agents.
Keywords: protein serine-threonine kinases, ion transport
The flux of solutes across epithelia is highly regulated, with permeating species traversing by either the transcellular route through cells or the paracellular route between cells. Transcellular transport is mediated by channels, exchangers, cotransporters, and pumps on apical and basolateral membranes. The paracellular barrier to flux occurs at tight junctions, apical points of contact between epithelial cells (1, 2).
Tight junctions of different epithelia are selectively permeable for different ions and solutes (3). Recent work has demonstrated the key role of the claudin family of tight-junction proteins in determining paracellular permeability (4-6). Mutations in claudin members confer specific defects in electrolyte homeostasis (7, 8). In vitro studies show that claudins act as channels or pores in the intercellular space, exhibiting size and charge selectivity, and dependence on ion concentration and pH (9, 10). Physiologic evidence has indicated that paracellular pathways are not static, but can be dynamically regulated (1, 3, 11-13). However, the factors and mechanisms that regulate paracellular permeability are not well defined (13).
The study of rare inherited diseases has the potential to identify integral components of complex regulatory networks. Pseudohypoaldosteronism type II (PHAII) is a rare genetic disease featuring hypertension and hyperkalemia (14). Positional cloning identified mutations in genes encoding the serinethreonine kinases WNK1 and WNK4 as a cause of this disease (15). Recent work has demonstrated that these proteins regulate diverse transcellular ion transport pathways; in the kidney, they inhibit the thiazide-sensitive NaCl cotransporter and the renal outer medullary K+ channel (16-18). Importantly, the missense mutations in WNK4 that cause PHAII relieve inhibition of the NaCl cotransporter (16, 17), but increase inhibition of the renal outer medullary K+ channel (18). These findings establish that WNKs act as a molecular switch that allows the kidney to alternate between NaCl-reabsorbing and K+-secreting states (18, 19). In extrarenal epithelia, WNK4 regulates other transporters and exchangers, establishing pleiotropic roles for these proteins (20).
In many epithelia, including kidney, WNKs localize tight junctions (15, 20, 21). This localization, the role of WNK4 in regulating the balance between NaCl reabsorption and K+ secretion (15-20), and the Cl- dependence of PHAII phenotypes (22, 23), suggest WNK4 might also play regulate paracellular Cl- flux, because this pathway plays a major role in NaCl homeostasis in the distal nephron. We report an investigation of WNK4 in the regulation of paracellular permeability in mammalian kidney epithelia.
Methods
Generation and Culture of Madin-Darby Canine Kidney (MDCK) II tet-off Wild-Type and Mutant WNK4 Cell Lines. cDNAs encoding full-length mouse WT-WNK4, WNK4-Q562E, WNK-E559K, and WNK-D318A (16), each tagged at the carboxyl terminus with hemagglutinin (HA), were subcloned into the pTRE2hyg expression vector (Clontech) and transfected into MDCK II tet-off cells. Transfected clones were selected for hygromycin B resistance. Resistant clones were maintained in high-glucose DMEM with hygromycin and 50 ng/ml doxycycline. Cell lines were screened for induction of WNK4 by incubation without doxycycline, followed by cell lysis and Western blotting with a rabbit anti-HA antibody (16).
Immunofluorescence Microscopy. For immunofluorescence and electrophysiological experiments, cells were plated onto porous filters (clear polyester or opaque polycarbonate Transwell filters; Corning Life Sciences, Corning, NY) either with doxycycline (50 ng/ml) or without doxycycline. Experiments were performed on day 4 after plating, corresponding with peak induction of WNK4 (data not shown). WNK4 was detected by immunofluorescence microscopy after staining with mouse (Roche Diagnostics, Basel) or rabbit (Santa Cruz Biotechnology) anti-HA antibody. Other antibodies included mouse anti-ZO-1 (Zymed Laboratories, South San Francisco, CA), mouse anti-occludin (Zymed Laboratories), rabbit antibodies to claudins 1, 2, and 3 (Zymed Laboratories), and mouse anti-claudin-4 (Zymed Laboratories). Affinity-purified donkey anti-mouse or rabbit secondary antibodies were conjugated to CY2 or CY3 (Jackson ImmunoResearch). Western blotting using the same antibodies was performed after the same induction protocol (16).
Electrophysiology. Current-voltage (I-V) relationships were acquired by voltage clamping monolayers between -50 and +50 mV and transepithelial resistance (TER) was calculated from Ohm's law. The resistance of blank filters was 21.7 ± 0.1 Ω cm2 and was subtracted from the values obtained for monolayers. TER was measured with buffer A, composed of 145 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM Hepes (pH 7.4) in both apical and basolateral chambers.
Dilution potentials were measured with apical buffer (buffer A), and a basal buffer B that differed from buffer A by the substitution of 72.5 for 145 mM NaCl; isoosmolarity was maintained by addition of 145 mM mannitol. All transepithelial voltages are reported as V1-V2, where the V1 electrode is in the apical compartment and V2 is in the basal compartment. The junction potential at the Ag/AgCl voltage electrodes exposed to a change in Cl- activity was measured before and after dilution potential determination. Junction potentials were measured by first recording the offset potential with the Ag/AgCl electrode of interest bathed in 145 mM NaCl and connected through an agar bridge (3% agar in 3 M KCl) to the reference electrode bathed in 3 M KCl. The solution bathing the Ag/AgCl electrode was then substituted with 72.5 mM NaCl buffer and the junction potentials were recorded. The observed junction potentials corresponded well with those calculated from the Nernst equation for a perfect Cl- selective conductance (24).
The ion permeability ratio (β) for the monolayer was calculated from the dilution potential by using the Goldman-Hodgkin-Katz equation (24), where β is the ratio of the permeability of the monolayer to Cl- over the permeability to Na+ ions (β = PCl/PNa).
The absolute permeabilities of Na+ (PNa) and Cl- (PCl) were calculated by using a simplified Kimizuka-Koketsu equation (25),
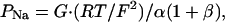 |
1 |
where G is conductance per unit of surface area and α is the NaCl concentration. It follows that
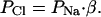 |
2 |
TER and dilution potential measurements were performed in triplicate on each of three different clonal cell lines in at least three independent experiments; results from independent clones of each construct showed no significant differences and were treated as independent measures. For each construct, the mean TER of uninduced monolayers is adjusted to a value of 100%, and TER after induction is expressed as the percentage of this value. Dilution potentials are shown as raw data. All data are expressed as mean ± SEM, and statistical comparisons between groups are made by a paired t test (induced vs. uninduced), or by an unpaired t test for comparison of effects of different constructs. Two-tailed P values are reported.
Inhibitors of Transcellular Transport Studies. Baseline TER and dilution potentials were measured in buffer A, after which the same buffer containing the indicated drug [amiloride (0.1 mM), bumetanide (0.01 mM), 4-acetamido-4′-isothiocyanato-stilbene-2,2′-disulfonic acid (0.1 mM), or hydrochlorothiazide (0.1 mM) each from Sigma] was substituted in the apical and basal chambers. Measurements were repeated, and the apical buffer A was replaced with buffer B, containing the indicated drug. TER and dilution potentials were again determined.
Dextran Flux Studies. Paracellular dextran flux was measured (26) by using monolayers prepared as above. Fluorescein-labeled 3-kDa dextran (Molecular Probes) was added to apical chambers to 3.5 μM. Aliquots were taken from the basal chamber at intervals, and fluorescence emission at 518 nm was measured after excitation at 498 nm.
Transmission and Freeze-Fracture Electron Microscopy (EM). WNK4-Q562E and WNK4-E559K monolayers were grown on plastic or porous filters with or without doxycycline for 4 days, and transmission (27) or freeze-fracture (28) EM was performed. Replica sets from two separate uninduced and induced MDCK II cell clones were examined for each construct.
Results
Regulated Expression and Localization of WNK4 in MDCK II Cells. The MDCK epithelial II tet-off cell line (MDCK II tet-off) stably expresses the tetracycline transactivator. The tetracycline transactivator induces transcription at cytomegalovirus promoters in the absence of doxycycline; this induction is repressed by doxycycline (29). We cloned HA-tagged wild-type (WT-WNK4), two different HA-tagged PHAII-mutant WNK4s (WNK4-Q562E and WNK4-E559K; ref. 16), and a WNK4 mutant with a single-point mutation at an essential residue of the kinase catalytic domain (WNK4-D318A; ref. 16) into the pTRE2hyg expression vector. Constructs were transfected into MDCK II tet-off cells, and stable transfectant clones were selected (see Methods). Clones showing low transgene expression in the presence of doxycycline, and robust transgene induction in the absence of doxycycline, were selected for further study (Fig. 1a).
Fig. 1.

Regulated expression of WNK4 in MDCK II tet-off cell lines. Clones showing doxycycline-regulated expression of WT and mutant WNK4 were produced as described in Methods. (a) Immunoblot analysis of HA-tagged WT-WNK4, WNK4-E559K, and WNK4-Q562E. For each construct, independent clones (1, 2, and 3) were cultured in the presence (+) or absence (-) of doxycycline. Samples were fractionated by SDS/PAGE, followed by immunoblotting for WNK4 with anti-HA antibody. HA-tagged WT and mutant WNK4 migrate as ≈150-kDa species. (b and c) Induction and localization of WT-WNK4 in MDCK II cells. dox, Doxycycline. Cells were cultured, stained for WNK4 with anti-HA antibody, and visualized by immunofluorescence microscopy (see Methods). WNK4 expression is low in the presence of doxycycline (b) and is induced in its absence (c). WNK4 localizes to points of cell-cell contact. WNK4-D318A, WNK4-Q562E, and WNK4-E559K each showed a similar staining pattern (data not shown). (d) Localization of ZO-1 and WNK4. Cells were induced and stained for ZO-1 (green) and WNK4 (red). comp, composite. Both proteins localize to cell-cell junctions. (e) As in d, WNK4 signal only. (f) As in d, ZO-1 signal only.
Immunofluorescence microscopy with anti-HA antibodies was performed to determine the cellular localization of wild-type and mutant WNK4 in these cell lines (Fig. 1 b and c). Staining was predominant at cell-cell borders in clones expressing either wild-type (Fig. 1c) or mutant WNK4 (data not shown). Immunofluorescence light microscopy after costaining cells with antibodies specific for ZO-1, a marker of the tight junction, and HA-WNK4, demonstrated significant overlap of WNK4 and ZO-1 at tight junctions (Fig. 1 d-f). This localization is consistent with the immunolocalization of endogenous WNK4 in kidney (15, 20) and other tissues (20).
WNK4 Decreases TER. In monolayers with low TER, like MDCK II, the paracellular pathway is the major determinant of transepithelial conductance (30, 31). The TER of MDCK II monolayers was determined before and after induction of wild-type and mutant WNK4. For each construct, three independent clones were studied, and measurements were performed in triplicate with and without induction in at least three independent experiments (see Methods). Cells transfected with the empty pTRE2hyg vector had TERs of ≈100 Ω cm2 in both the presence and absence of doxycycline, similar to previously reported values (32-35). In contrast, induction of WT-WNK4 led to a 26% decrease in TER (P < 0.0001; Fig. 2a, and Table 1, which is published as supporting information on the PNAS web site). Current-voltage curves were linear in the presence and absence of doxycycline (data not shown), consistent with conductance being attributable to the paracellular pathway. Moreover, inhibition of the major mediators of transcellular Na+ and Cl- flux (amiloride for the epithelial Na+ channel, bumetanide for the Na+-K+-2 Cl- cotransporter, 4-acetamido-4′-isothiocyanatostilbene-2,2′-disulfonic acid for anion exchangers, and hydrochlorothiazide for the NaCl cotransporter; see Methods) had no significant effect on TER in induced or uninduced cells (data not shown). These findings indicate that wild-type WNK4 induction increases paracellular ion conductance.
WNK4 has both kinase domain-dependent and -independent functions (16-18, 20). We tested the effect of mutation of an amino acid required for WNK kinase activity (16, 36) on TER. Expression of WNK4-D318A had no significant effect on the TER of MDCK II cells (Fig. 2a). These data demonstrate the specificity of the effect of wild-type WNK4 and suggest that the ability of WNK4 to alter TER depends on its kinase activity.
Fig. 2.

Expression of WT and PHAII-mutant WNK4 decreases TER. (a) TER was measured on indicated monolayers as described in Methods. Mean ± SEM from three different clones for each construct is shown. *, P < 0.0001 vs. matched uninduced monolayer; **, P < 0.0001 vs. matched uninduced monolayer and vs. uninduced WT-WNK4 monolayer. The mean TER in the uninduced state (+dox) is expressed as 100% and the induced state (-dox) is indicated as a percentage of this value. (b) Effect of PHAII-mutant WNK4-Q562E on flux of 3-kDa dextran. Monolayers were prepared in the presence (red circles) or absence (blue triangles) of doxycycline, fluorescent dextran was applied to the apical surface, and its appearance in the basolateral media was measured over time.
We next tested the effects of WNK4 mutations that cause PHAII on TER. Induction of either WNK4-Q562E or WNK4-E559K decreased TER by ≈57% (Fig. 2a); this TER was significantly lower than that seen in uninduced monolayers (P < 0.0001 for both WNK4-Q562E and WNK4-E559K), and was also significantly lower than the TER seen in monolayers expressing WT-WNK4 (P < 0.0001 for both WNK4-Q562E and WNK4-E559K). Thus, PHAII-mutant WNK4 causes a greater reduction in TER than wild-type WNK4, consistent with a gain-of-function effect of these mutations.
WNK4 could increase paracellular conductance by nonspecifically altering the integrity of epithelial tight junctions. To test this possibility, we measured the unidirectional diffusion of fluorescein-labeled 3-kDa dextran, a small uncharged solute (26), across MDCK II monolayers in the presence or absence of WNK4-Q562E. The flux of dextran was not significantly altered by this mutant WNK4 (Fig. 2b), indicating that the effect of WNK4 on paracellular flux is not the result of a general disruption of tight-junction integrity.
WNK4 Selectively Increases Paracellular Cl- Conductance. The increase in paracellular conductance induced by WNK4 could result from a selective or nonselective increase in permeability for cations and anions. To address this hypothesis, we measured the dilution potential (ΔVt) resulting from application of a transepithelial NaCl gradient in the presence or absence of WNK4. Determinants of the dilution potential include the absolute permeabilities for Na+ (PNa) and Cl- (PCl) and the ratio of their permeabilities (PCl/PNa). PCl/PNa can be calculated from the observed dilution potential (ref. 24; see Methods). Absolute PNa and PCl can be calculated from the measured TER and PCl/PNa (ref. 25; see Methods).
Similar to previous reports (32-35), the parental MDCK II monolayers showed higher permeability for Na+ than Cl- (Na+ permeability was 2.6-fold higher than Cl- permeability); doxycycline had no significant effect on the dilution potential in the parental cell line (Fig. 3a). However, induction of WT-WNK4 resulted in a highly significant change in the dilution potential (Fig. 3a), with an increase in PCl/PNa (P < 0.0001; Fig. 3b). This increase was attributable to an ≈2-fold increase in absolute PCl (P < 0.0001), while the absolute value of PNa displayed minor changes (14% increase, P < 0.0001; Fig. 3 c and d). Kinase-mutant WNK4 affected none of these parameters (Fig. 3).
Fig. 3.

WT and PHAII-mutant WNK4 increase paracellular Cl- permeability. Indicated monolayers were cultured in the presence or absence of doxycycline for 4 days; dilution potentials and TER were measured as described in Methods. For each construct, the mean ± SEM of measurements is shown. *, P < 0.0001 vs. matched uninduced monolayer; **, P < 0.0001 vs. matched uninduced monolayer and vs. induced WT-WNK4 monolayer; #, P < 0.002 vs. matched uninduced monolayer. (a) Dilution potentials. Expression of WT-WNK4 has a significant effect on the dilution potential, and the effect is larger with PHAII-mutant WNK4. (b) Relative permeability of Cl- and Na+ (PCl/PNa). PCl/PNa of the indicated monolayers was determined. Wild-type and PHAII-mutant WNK4 markedly increases the ratio. (c) Absolute permeability for Na+ (PNa) in cm/s. (d) Absolute permeability for Cl- (PCl) in cm/s. WT-WNK4 markedly increases PCl but not PNa. PHAII-mutant WNK4 has a larger effect. (e) WNK4 alters the paracellular permeability of preformed tight junctions. Twelve replicates of a clone containing PHAII-mutant WNK4 (WNK4-Q562E) were plated and allowed to form monolayers in the presence of doxycycline for 4 days. TER and dilution potentials (DP) were measured. Half the monolayers were then incubated for 2 more days in the presence of doxycycline, whereas the other half were incubated in the absence of doxycycline, thereby inducing WNK4-Q562E. Measurements were repeated. Data from individual wells are shown for TER measurements and mean ± SEM of dilution potentials is shown for each group. Induction of WNK4-Q562E markedly reduced the TER and altered the dilution potential of monolayers whose tight junctions have already assembled.
Dilution potentials were also measured before and after induction of PHAII-mutant WNK4. Induction of WNK4-Q562E or WNK4-E559K resulted in a marked increase in PCl/PNa (Fig. 3b). For both WNK4 mutants, this increased ratio resulted from a marked increase in PCl (≈4.5-fold, P < 0.0001; Fig. 3d). Again, the increase in PNa was minor, although statistically significant (Fig. 3c). The PCl seen with PHAII-mutant WNK4 is significantly greater than both the PCl in uninduced cells (P < 0.0001) and the PCl seen after induction of WT-WNK4 (P < 0.0001). Importantly, dilution potentials were indistinguishable when the ionic gradient was directed apically or basally, and antagonists of transcellular NaCl flux pathways did not alter the effect of PHAII-mutant WNK4 on dilution potentials (data not shown). These findings indicate that the observed effects are attributable to the paracellular pathway.
WNK4 Alters the Function of Preformed Tight Junctions. In the above experiments, WNK4 was expressed during tight-junction assembly/formation; this leaves open whether WNK4 can alter the function of tight junctions that have already been formed. To address this hypothesis, we first repressed WNK4-Q562E expression until after tight-junction formation was complete by incubating cells in doxycycline from the time cells were plated to day 4. We then measured the TER and dilution potential and subsequently either continued repression with doxycycline or induced WNK4-Q562E, and repeated measurements after 2 additional days. Monolayers maintained in the presence of doxycycline showed no significant changes in TER or dilution potential (Fig. 3e). In contrast, induction of WNK4-Q562E resulted in a marked decrease in TER (>45% decrease, P < 0.0001; Fig. 3e), with changes in dilution potential (-7.0 ± 0.7 mV to 2.1 ± 0.5 mV, P < 0.0001; Fig. 3e). This change was due to a 4-fold increase in PCl, quantitatively similar to that seen when WNK4-Q562E was induced at the time of cell plating (Fig. 3e). Similar results were seen with WNK4-E559K (data not shown). These data demonstrate that PHAII-mutant WNK4 is able to markedly alter the properties of preformed tight junctions.
Effect of WNK4 on Tight-Junction Proteins and Structure. WNK4 could regulate paracellular permeability by altering the expression, localization, or function of proteins that determine paracellular ion permeability and selectivity. We compared the expression and localization of claudins 1-4, occludin, and ZO-1 before and after induction of WT-WNK4 or WNK4-Q562E (see Methods). Induction of either protein had no significant effect on the localization or expression levels of these tight-junction proteins (Fig. 5, which is published as supporting information on the PNAS web site). We cannot rule out effects on other components of this complex structure.
Overexpression of various claudins has been shown to alter paracellular conductance in MDCK cells; these changes have often been accompanied by gross alterations in tight-junction structure (32, 34, 35). To assess the effect of WNK4 on tight-junction structure, we performed transmission (Fig. 4 a-d) and freeze-fracture (Fig. 4 e and f) EM of monolayers in the presence and absence of WNK4-E559K. The depth of tight junctions by transmission EM and the number and complexity of tight-junction strands by freeze-fracture was unchanged by induction of PHAII-mutant WNK4. Indistinguishable results were obtained with WT-WNK4. These data demonstrate that WNK4 does not impart its effect on paracellular permeability by eliciting gross structural changes on the tight-junction complex.
Fig. 4.

Effect of WNK4 on structure of tight junctions. (a-d) Transmission EM of WNK4-Q562E cells. Monolayers were grown in the presence and absence of doxycyline, and were subjected to transmission EM. The depth of tight junctions is unaffected by WNK4-Q562E induction. Similar results were seen for WT-WNK4 (data not shown). ZA, zonula adherens; TJ, tight junctions (zonula occludens). (e and f) Freeze-fracture EM of WNK4-Q562E cells. Monolayers were treated as above and subjected to freeze-fracture EM as described in Methods. The number and complexity of tight-junction strands (indicated by arrows) are unchanged by induction of WNK4-Q562E. Similar results were seen for WT-WNK4 (data not shown). WNK4 does not impart its effect on paracellular permeability by eliciting gross structural changes on the tight-junction complex.
Discussion
The claudins play a key role in determining selective paracellular permeability. The factors that allow dynamic regulation of these flux pathways have been unknown. The present findings indicate that WNK4 can function as a specific regulator of the paracellular pathway, selectively increasing paracellular Cl- permeability. This evidence is derived from significant effects of WNK4 on both TER and dilution potential. The magnitude of the observed effects are large, with up to 2-fold increases in monolayer conductance and 4-fold increases in PCl. The magnitude of this effect is sufficiently large that monolayers are switched from a state that favors Na+ flux, to a state that is more permeable to Cl-.
In addition to showing that WNK4 can alter paracellular function when it is expressed during tight-junction formation, we have shown that WNK4 can also alter the function of preformed tight junctions, with effects that are quantitatively similar to those found when WNK4 is expressed from the time of cell plating. This finding reveals that WNK4 is capable of dynamic regulation of paracellular ion flux, consistent with the notion that signaling pathways acting through WNK4 can modify paracellular Cl- flux in response to physiologic demand.
Because they constitute paracellular channels, the claudins are logical potential targets for WNK4 action at the tight junction. Yamauchi et al. (37) have shown increased phosphorylation of claudins in the presence of PHAII-mutant WNK4, and we have shown that the effect of WNK4 on paracellular ion flux is abolished by missense mutations that impair WNK kinase function. These findings together implicate WNK4-mediated phosphorylation of claudins in the mechanism of altered paracellular flux. How such phosphorylation imparts its effect is presently unknown. Both we and Yamauchi et al. (37) have seen no change in the abundance or localization of claudins 1-4, ZO-1, or occludin after expression of WNK4. In addition, our structural studies by both transmission and freeze-fracture EM show no difference in tight-junction architecture, indicating that altered paracellular flux is not achieved by gross changes in tight-junction structure. These observations leave open the question of mechanism. One possibility is alteration of claudin function (e.g., altered conductance) by posttranslational modification, as occurs with many transcellular ion channels (38). Alternatively, WNK signaling could alter the claudin composition of the tight junction. Further investigation will be required to distinguish these possibilities.
In addition to its role in the kidney, WNK4 and other WNKs are expressed in other tissues, predominantly those involved in epithelial Cl- flux (20, 21). In many of these epithelia, WNKs localize to intercellular junctions (20, 21), suggesting that WNKs may play a general role in the regulation of paracellular Cl- flux.
These findings have implications for normal and disease physiology. We have previously proposed that WNK4 is a molecular switch that integrates physiologic signals to vary the balance between net renal NaCl reabsorption and K+ secretion (18). We have suggested that these effects explain the mechanism by which the kidney responds differently to aldosterone signaling in the setting of hypovolemia vs. hyperkalemia (18). The present findings are consistent with and extend this proposal. Parcellular Cl- flux plays an important role in NaCl and K+ homeostasis in the connecting tubule and collecting duct, where 70% of Cl- reabsorption occurs via the paracellular path (39). In these nephron segments, Na+ reabsorption via the epithelial Na+ channel is accompanied by either paracellular Cl- reabsorption or transcellular K+ secretion. In the presence of PHAII-mutant WNK4, Na+ reabsorption via the epithelial Na+ channel would be accompanied by increased paracellular Cl- reabsorption and reduced K+ secretion due to the effect of WNK4 to both reduce the lumen-negative potential and to directly inhibit the renal outer medullary K+ channel. Thus, the multiple renal effects of PHAII-mutant WNK4 can all be viewed as serving a coordinated effort to expand plasma volume by increasing NaCl reabsorption, while maintaining serum K+.
Although thiazide diuretics have efficacy equal to any class of antihypertensive agents (40), the magnitude of their effect is limited by activation of the renin-angiotensin system, which increases distal NaCl reabsorption through increased epithelial Na+ channel activity and paracellular Cl- flux. The demonstration that WNK signaling regulates NaCl reabsorption at both the thiazide target NaCl cotransporter and the more distal paracellular Cl- pathway suggests that modification of WNK signaling has the potential to produce larger effects than can be achieved with thiazides or other agents that attack single-flux mediators. The WNKs may consequently present opportunities for the development of uniquely efficacious antihypertensive agents.
Note. While preparing this manuscript, Yamauchi et al. (37), using 22Na+ and 36Cl- flux, came to similar general conclusions (37). They observed that a PHAII-mutant WNK4 increased transepithelial 36Cl- flux >2-fold (37). In contrast to Yamauchi et al. (37), we found a much larger effect of PHAII-mutant WNK4 on PCl [PCls of 25 and 35 × 10-6 cm/s for two different PHAII-mutants, vs. 5.5 × 10-6 cm/s for the mutant tested by Yamauchi et al. (37)]. In addition, we show that wild-type WNK4 also has a highly significant effect on PCl. These differences may be attributable to greater sensitivity of electrophysiologic measurement of TER and dilution potential and to differences in the starting cell lines. The parental MDCK II line of Yamauchi et al. (37) showed a much higher selectivity for Na+ than ours (PNa/PCl ≈12.9 vs. 2.8, respectively).
Supplementary Material
Acknowledgments
We thank Keith Choate, Jim Anderson, Christina Van Itallie, Yin Lu, and Peter Aronson for helpful discussions. This work was supported by a Specialized Center of Research in Hypertension grant from the National Heart, Lung, and Blood Institute. R.P.L. is an Investigator of the Howard Hughes Medical Institute.
Author contributions: K.T.K, F.H.W., A.N.V.H., S.C.H., E.L.B., and R.P.L. designed research; K.T.K., G.G.M., A.N.V.H., and T.A. performed research; K.T.K., G.G.M., F.H.W., A.N.V.H., D.B., T.A., M.K., and R.P.L. contributed new reagents/analytic tools; K.T.K., G.G.M., F.H.W., A.N.V.H., D.B., M.K., G.G., S.C.H., E.L.B., and R.P.L. analyzed data; and K.T.K., G.G., E.L.B., and R.P.L. wrote the paper.
Abbreviations: PHAII, pseudohypoaldosteronism type II; HA, hemagglutinin; TER, transepithelial resistance; MDCK, Madin-Darby canine kidney; EM, electron microscopy.
References
- 1.Anderson, J. M. & Van Itallie, C. M. (1995) Am. J. Physiol. 32, G467-G475. [DOI] [PubMed] [Google Scholar]
- 2.Tsukita, S., Furuse, M. & Itoh, M. (2001) Nat. Rev. Mol. Cell Biol. 4, 285-293. [DOI] [PubMed] [Google Scholar]
- 3.Madara, J. L. (1998) Annu. Rev. Physiol. 60, 143-159. [DOI] [PubMed] [Google Scholar]
- 4.Tsukita, S. & Furuse, M. (2002) Curr. Opin. Cell Biol. 14, 531-536. [DOI] [PubMed] [Google Scholar]
- 5.Mitic, L. L. & Anderson, J. M. (1998) Annu. Rev. Physiol. 60, 121-142. [DOI] [PubMed] [Google Scholar]
- 6.Peter, Y. & Goodenough, D. (2004) Curr. Biol. 14, R293-R294. [DOI] [PubMed] [Google Scholar]
- 7.Simon, D. B., Lu, Y., Choate, K. A., Velazquez, H., Al-Sabban, E., Praga, M., Casari, G., Bettinelli, G., Colussi, J., Rodriguez-Soriano, J., et al. (1999) Science 285, 103-106. [DOI] [PubMed] [Google Scholar]
- 8.Wilcox, E. R., Burton, Q. L., Naz, S., Riazuddin, S., Smith, T. N., Ploplis, B., Belyantseva, I., Ben-Yosef, T., Liburd, N. A., Morell, R. J., et al. (2001) Cell 104, 165-172. [DOI] [PubMed] [Google Scholar]
- 9.Colegio, O. R., Van Itallie, C. M., McCrea, H. J., Rahner, C. & Anderson, J. M. (2002) Am. J. Physiol. 283, C14-C147. [DOI] [PubMed] [Google Scholar]
- 10.Tang, V. W. & Goodenough, D. A. (2003) Biophys. J. 84, 1660-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopkins, A. M., Li, D., Marsny, R. J., Walsh, S. V. & Nusrat, A. (2000) Adv. Drug Delivery Rev. 41, 329-340. [DOI] [PubMed] [Google Scholar]
- 12.Clarke, H., Marano, C. W., Peralta-Soler, A. & Mullin, J. M. (2000) Adv. Drug Delivery Rev. 43, 283-301. [DOI] [PubMed] [Google Scholar]
- 13.Schneeberger, E. & Lynch, R. (2004) Am. J. Physiol. 286, C1213-C1228. [DOI] [PubMed] [Google Scholar]
- 14.Gordon, R. D., Klemm, S. A., Tunny, T. J. & Stowasser, M. (1995) in Hypertension: Pathophysiology, Diagnosis, and Management, eds. Laragh, J. H. & Brenner, B. M. (Raven, New York), pp. 2111-2123.
- 15.Wilson, F. H., Disse-Nicodeme, S., Choate, K. A., Ishikawa, K., Nelson-Williams, C., Desitter, I., Gunel, M., Milford, D. V., Lipkin, G. W., Achard, J. M., et al. (2001) Science 293, 1107-1112. [DOI] [PubMed] [Google Scholar]
- 16.Wilson, F. H., Kahle, K. T., Sabath, E., Lalioti, M. D., Rapson, A. K., Hoover, R. S., Hebert, S. C., Gamba, G. & Lifton, R. P. (2003) Proc. Natl. Acad. Sci. USA 100, 680-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang, C. L., Angell, J., Mitchell, R. & Ellison, D. H. (2003) J. Clin. Invest. 111, 1039-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahle, K. T., Wilson, F. H., Leng, Q., Lalioti, M. D., O'Connell, A. D., Dong, K., Rapson, A. K., MacGregor, G. G., Giebisch, G., Hebert, S. C., et al. (2003) Nat. Genet. 35, 372-376. [DOI] [PubMed] [Google Scholar]
- 19.Bindels, R. J. (2003) Nat. Genet. 35, 302-303. [DOI] [PubMed] [Google Scholar]
- 20.Kahle, K. T., Gimenez, I., Hassan, H., Wilson, F. H., Wong, R. D., Forbush, B., Aronson, P. S. & Lifton, R. P. (2004) Proc. Natl. Acad. Sci. USA 101, 2064-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choate, K. A., Kahle, K. T., Wilson, F. H., Nelson-Williams, C. & Lifton, R. P. (2003) Proc. Natl. Acad. Sci. USA 100, 663-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schambelan, M., Sebastian, A. & Rector, F. C., Jr. (1981) Kidney Int. 19, 716-727. [DOI] [PubMed] [Google Scholar]
- 23.Take, C., Ikeda, K., Kurasawa, T. & Kurokawa, K. (1991) N. Engl. J. Med. 324, 472-476. [DOI] [PubMed] [Google Scholar]
- 24.Hille, B. (2001) in Ion Channels of Excitable Membranes (Sinauer, Sunderland, MA), p. 184.
- 25.Kimizuka, H. & Koketsu, K. (1964) J. Theor. Biol. 6, 290-305. [DOI] [PubMed] [Google Scholar]
- 26.Meyer, T. N., Schwesinger, C. & Denker, B. M. (2002) J. Biol. Chem. 277, 24855-24858. [DOI] [PubMed] [Google Scholar]
- 27.Zahler, R., Sun, W., Ardito, T., Zhang, Z. T., Kocsis, J. D. & Kashgarian, M. (1996) Circ. Res. 78, 870-879. [DOI] [PubMed] [Google Scholar]
- 28.van Hoek, A. N., Yang, B., Kirmiz, S. & Brown, D. (1998) J. Membr. Biol. 165, 243-254. [DOI] [PubMed] [Google Scholar]
- 29.Gossen, M. & Bujard, H. (1992) Proc. Natl. Acad. Sci. USA 89, 5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powell, D. W. (1981) Am. J. Physiol. 241, G275-G288. [DOI] [PubMed] [Google Scholar]
- 31.Diamond, J. (1977) Physiologist 20, 10-18. [PubMed] [Google Scholar]
- 32.Van Itallie, C. M., Fanning, A. S. & Anderson, J. M. (2003) Am. J. Physiol. 285, F1078-F1084. [DOI] [PubMed] [Google Scholar]
- 33.Singh, A. B. & Harris, R. C. (2004) J. Biol. Chem. 279, 3543-3552. [DOI] [PubMed] [Google Scholar]
- 34.Yu, A. S., Enck, A. H., Lencer, W. I. & Schneeberger, E. E. (2003) J. Biol. Chem. 278, 17350-17359. [DOI] [PubMed] [Google Scholar]
- 35.Van Itallie, C. M, Rahner, C. & Anderson, J. M. (2001) J. Clin. Invest. 107, 1319-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu, B., English, J. M., Wilsbacher, J. L., Stippec, S., Goldsmith, E. J. & Cobb, M. H. (2001) J. Biol. Chem. 22, 16795-16801. [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi, K., Rai, T., Kobayashi, K., Sohara, E., Suzuki, T., Itoh, T., Suda, S., Hayama, A., Sasaki, S. & Uchida, S. (2004) Proc. Natl. Acad. Sci. USA 101, 4690-4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marban, E., Yamagishi, T. & Tomaselli, G. F. (1998) J. Physiol. (London) 508, 647-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuster V. L. & Stokes J. B. (1987) Am. J. Physiol. 253, F203-F212. [DOI] [PubMed] [Google Scholar]
- 40.ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group (2002) J. Am. Med. Assoc. 288, 2981-2997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}