

Abstract
There is little genomic information regarding gene expression differences at the whole blood transcriptome level of different pig breeds at the neonatal stage. To solve this, we characterized differentially expressed genes (DEGs) in the whole blood of Dapulian (DPL) and Landrace piglets using RNA-seq (RNA-sequencing) technology. In this study, 83 DEGs were identified between the two breeds. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses identified immune response and metabolism as the most commonly enriched terms and pathways in the DEGs. Genes related to immunity and lipid metabolism were more highly expressed in the DPL piglets, while genes related to body growth were more highly expressed in the Landrace piglets. Additionally, the DPL piglets had twofold more single nucleotide polymorphisms (SNPs) and alternative splicing (AS) than the Landrace piglets. These results expand our knowledge of the genes transcribed in the piglet whole blood of two breeds and provide a basis for future research of the molecular mechanisms underlying the piglet differences.
1. Introduction
The domestic pig is a major livestock animal in China, where the long-term breeding and geographical separation have generated over 80 indigenous pig breeds, many of which have particular features [1]. Among them, Dapulian (DPL) pig is an indigenous breed, which is mainly distributed in Jining, Shandong Province, China. It has many desirable idioplasmatic traits such as high prolificacy, strong disease resistance, good meat flavor, and good adaptability to crude feed [1, 2]. However, its growth rate is slow and fat content is high. Landrace pigs have a better growth rate and higher lean content than DPL pigs but are weaker in disease resistance compared to DPL [1, 2]. Pork producers have great economic interest to improve breeding stocks with better growth performance and immune capacity, thus to meet the increasing consumer demand for higher quality pork and lower production cost. However, the genetic basis of such differences is still not well understood. Therefore, it will be very valuable to unravel the genetic mechanisms underlying the differences between these two breeds.
Growth performance and immune capacity play essential roles in the economics of pig production. However, understanding the gene expression variation during body growth and immune capacity development at young ages is in its primitive stage. Blood cells constitute the first line of the immune defense system and pervade the whole body [3]. Moreover, peripheral blood cells share more than 80% of the transcriptome with nine tissues including brain, colon, heart, kidney, liver, lung, prostate, spleen, and stomach [4]. Thus, immune status and metabolic changes in other tissues may manifest through whole blood gene expression. Therefore, blood is now widely used as a surrogate tissue to identify the extent and kinetics for various economically important traits [4].
The advent of next-generation sequencing (NGS) technology and progresses in bioinformatics have provided a useful platform to easily explore DEGs [5]. Moreover, NGS technology can simultaneously assess mRNA transcription patterns for all the genes in various species. RNA-seq using NGS technology has become widely used for high-throughput researches of gene expression. RNA-seq has been used to identify genes associated with economically important quantitative traits, such as body growth, immune, disease resistance, and meat quality [6–9]. Although RNA-seq technology has been applied in many species, including pigs [10, 11], limited information is available regarding its application in the transcriptome of newborn piglets. SNPs can be found inside candidate genes for artificial or natural selection and might provide a cost-effective way to obtain more information than other molecular markers. AS is an important mechanism to increase the genomic and proteomic diversity, which occurs widely in the process of gene transcription in eukaryotes. Up to date, many new AS events have been discovered in humans [12], Arabidopsis [13], and Drosophila melanogaster [14] and other organisms. There are six common types of AS events: Skipped Exon (SE), Intron Retention (IR), Alternative 3′ Splice Sites (A3SS), Alternative 5′ Splice Sites (A5SS), Alternative First Exon (AFE), and Alternative Last Exon (ALE) [15].
To the best of our knowledge, few studies have been performed to investigate blood transcriptome for divergence in gene expression in newborn piglets [16, 17]. The growth performance and immune capacity are the main factors that affect pig herd productivity [18]. Newborn piglets of DPL and Landrace were used to discover DEGs, SNP, and ASs between two breeds, which may help to explain the growth/production and immune differences of the two breeds. It will advance our understanding of the mechanism of piglet breed differences and may be helpful to improve stock breeding with better growth performance and immune capacity.
2. Materials and Methods
2.1. Experimental Design and Ethical Statement
Three female piglets from each purebred DPL and Landrace were used in this study. After birth, the piglets were weaned at once and sent to a common nursery with the ambient temperature set at 25 ± 1°C. For one day, cow milk was supplied for these piglets to meet the nutrient needs. 1 mL whole blood samples were collected from the external jugular vein of one-day-old piglets and then immediately mixed with 3 mL RNALock Reagent (Tiangen Biotech Co., Beijing, China). The mixture was kept at room temperature for 2 h and then stored at −80°C until use. All animal experiments in this study were approved by the Animal Care and Use Committee of Shandong Agricultural University (Approval number: 2004006).
2.2. Total RNA Extraction and RNA Quality Analysis
Total RNA was extracted using a RNAsimple Total RNA kit (Tiangen Biotech Co., Beijing, China) according to the manufacturer's instructions. RNA concentration and purity were assessed using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). RNA integrity was assessed using a RNA Nano 6000 Assay Kit on an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
2.3. cDNA Library Construction and Next-Generation Sequencing (RNA-seq)
Poly(A) mRNAs were further purified from the extracted total RNA using magnetic oligo (dT) beads (Invitrogen, Carlsbad, CA, USA). Sequencing libraries were constructed using the NEBNext® Ultra RNA Library Prep Kit from Illumina (New England Biolabs, Ipswich, MA, USA) with multiplexing primers, following the manufacturer's instructions. The average insert size for the paired-end libraries was 200 bp (150~250 bp). AMPure XP Beads (Beckman Coulter Inc., Brea, CA, USA) were used for cDNA purification. The purified double-stranded cDNAs were subjected to end repair and adapter ligation. Agencourt AMPure XP beads (Beckman Coulter Inc.) were then used for the selection of suitable fragments. Finally, the cDNA libraries were generated through PCR enrichment. A 125 bp paired-end run was performed on the Illumina HiSeq2500 platform at the Biomarker Technologies Corporation (Beijing, China). The RNA-seq data was deposited into NCBI database, and its accession is SAMN04423129.
2.4. Bioinformatics Analysis
2.4.1. Reads Trimming and Reference Genome Alignment
After discarding low quality reads (reads with adaptors, unknown nucleotides larger than 5%, or Q20 < 20%) by perl script, clean reads that were filtered from the raw reads were mapped to the reference genome of Sus scrofa (Sscrofa 10.2; ftp://ftp.ensembl.org/pub/release-75/fasta/sus_scrofa/) using the TopHat2 software. Gene expression levels were measured using the FPKM (fragments per kilobase of exon per million fragments mapped) values by the Cufflinks software [22]. Genes (FPKM > 0.1) that were measured commonly in all the six samples were assigned as cogenes.
2.4.2. Gene Annotation and DEGs Analysis
The FPKM value ratio was used to estimate gene expression level between those samples. The false discovery rate (FDR) control method was used to identify the p value in multiple tests. Fold changes (log 2 ratio) were estimated according to the normalized gene expression level in each sample. The DEGs were analyzed using the R package DESeq [23] between the two breeds. The input data for gene differential expression are the readcount data obtained from the gene expression level analysis. The DESeq software internally performs an FPKM conversion on the count value. The genes with fold changes (log 2 ratio) ≥ 2 and FDR significance score < 0.01 were considered as DEGs. The p value of the original hypothesis test was corrected by the accepted Benjamini-Hochberg correction method. Finally, FDR was used as the key index for DEGs screening.
The identified DEGs were used for subsequent GO and KEGG pathway analysis. To annotate the genes with GO terms, the Nr BLAST results were imported into the blast2 GO program. GO annotations for the genes were obtained by blast2GO. This analysis mapped all of the annotated genes to GO terms in the database and counted the number of genes associated with each term. Perl script was then used to plot GO functional classification for the unigenes with a GO term hit to view the distribution of gene functions. The obtained annotation was enriched and refined using the TopGo (R package) with the “elim” method and Kolmogorov–Smirnov test. The genes were classified into three categories including biological process, cellular component, and molecular function. Moreover, the enriched pathways of DEGs were analyzed using the KEGG database (http://www.genome.ad.jp/kegg/) using the right sided Fisher's exact test.
2.4.3. Identification of SNPs and AS
RNA-seq technology is an effective way to quickly and reliably discover SNPs and AS. Comparisons were made between the two pig breed transcriptomes and the reference genome of Sus scrofa. Single base mismatches between sequencing samples and the reference genomes were identified using the SAMtools software [24] to find potential SNPs in the gene region. The criteria for SNPs identification included nucleotide with reads mapping quality score ≥ 50; consensus quality score ≥ 20; and sequence depth ≥ 5. For the identification of AS, the Cufflinks method was used to predict AS events [22]. To detect AS events, the blasted results using TopHat were assembled using Cufflinks and compared with initial annotated results using Cuffcompare. The variable splicing types and corresponding expression levels were obtained using the ASprofile software [25].
2.5. Quantitative Real-Time PCR (qRT-PCR)
To validate the RNA-seq data, qRT-PCR analysis was conducted using the MX3000p qPCR system (Stratagene, La Jolla, CA, USA) with SYBR green as the fluorescent dye, according to the manufacturer's instructions (Takara Biotechnology, Dalian, China). Six functionally important genes were selected from the identified DEGs for qRT-PCR validation. The primers for qRT-PCR were designed either using the online Prime3 program (http://primer3.ut.ee/) or according to the published references [19–21, 26]. All the primers used in this study were listed in Table 1. All qRT-PCR reactions were conducted in triplicate with corresponding negative controls. Using the B2M and PPIA gene for normalization [27], the expression fold differences of the selected DEGs were calculated using the 2−ΔΔCT method [28].
Table 1.
Primer sequences for qRT-PCR.
| Gene | Sequence 5′-3′ | Size (bp) | Tm | Reference or gene accession number |
|---|---|---|---|---|
| G0S2 | F: gtcgccttacgtttggacttgc | 156 | 58 | [19] |
| R: caggtaactccgctcaggtgc | ||||
| ATP4B | F: cccctgcaggtggaatactt | 160 | 58 | NM_001001258.1 |
| R: ggatcttgcacacgatgacc | ||||
| GSTO1 | F: attacctcatctggccctgg | 150 | 58 | NM_214050.1 |
| R: ctcgaaggtctctcggttca | ||||
| CST3 | F: cgagtacaacaaagcgagca | 193 | 60 | NM_001044602.1 |
| R: cagcgttttcttctgcaggt | ||||
| FECH | F: gatcgcgtttaccagtgacc | 180 | 60 | NM_001170523.1 |
| R:cgctcattggactggatgtg | ||||
| MX2 | F: aggcaaccaagagggaaatc | 131 | 60 | NM_001097416.1 |
| R: cacactgatatgcccgatga | ||||
| B2M | F: ttcacaccgctccagtag | 166 | 60 | [20] |
| R: ccagatacatagcagttcagg | ||||
| PPIA | F: cacaaacggttcccagtttt | 171 | 60 | [21] |
| R: tgtccacagtcagcaatggt |
3. Results
3.1. Overview of the Sequencing Data
Across all 6 samples, the total read length was 30.2 gigabases (Gb). The fold change of reading per animal compared to the pig genome (2.8 Gb) is 2.04-, 1.93-, 1.82-, 1.63-, 1.80-, and 1.57-fold, respectively. The main statistical data of the six samples were described in Table 2. Sequencing read length of all the samples was 125 bp paired-end reads, with Q30 > 88%. More than two-thirds of the reads could be mapped to the reference genome, with mapped ratio from 67.7% to 79.41%. In addition, more than two-thirds of the mapped reads had unique genomic locations (except DPL 3). The variation between individual blood samples was evaluated through pairwise correlation. Except one sample (DPL 3), a strong correlation among other five samples was found (Spearman correlation coefficient across all genes ranging from 0.850 to 0.985); therefore, the DPL 3 sample was excluded from further analysis.
Table 2.
Summary of the RNA-sequencing and mapping.
| Sample | Landrace 1 | Landrace 2 | Landrace 3 | DPL 1 | DPL 2 | DPL 3 |
|---|---|---|---|---|---|---|
| Total reads | 45768956 | 43112026 | 40759848 | 36558806 | 40206856 | 35205066 |
| Total base pairs | 5766888456 | 5432115276 | 5135740848 | 4606409556 | 5066063856 | 4435838316 |
| Mapped reads | 35504697 (77.57%) | 34236396 (79.41%) | 31893141 (78.25%) | 26752.117 (73.18%) | 31099885 (77.35%) | 23833962 (67.70%) |
| Unique mapped reads | 34286547 (74.91%) | 33172939 (76.95%) | 30790776 (75.54%) | 25714679 (70.34%) | 29884275 (74.33%) | 22,473,200 (63.84%) |
| % ≥ Q30 | 89.68 | 89.65 | 89.12 | 88.97 | 89.67 | 89.00 |
| GC content% | 58.35 | 59.19 | 58.53 | 57.77 | 57.22 | 56.25 |
3.2. Identification and Analysis of DEGs
By comparing the transcriptome data of the two breeds, a total of 83 DEGs were identified, including 57 genes with significantly higher expression and 26 genes with significantly lower expression in the Landrace piglets compared to the DPL piglets. The major DEGs are listed in Supplementary Table S1 in Supplementary Material available online at http://dx.doi.org/10.1155/2016/7907980 including their log fold difference and FDR values. A heatmap that depicted the majority of DEGs was presented in Figure 1.
Figure 1.

Heatmap of the DEGs between the DPL and Landrace piglets. Columns represent individual samples. Rows indicate genes with significant expression differences between the two breeds (DPL, left columns 1 and 2; Landrace, right columns 1, 2, and 3).
3.3. GO and KEGG Pathway Enrichment Analysis of DEGs
The GO and KEGG enrichment analysis were performed to gain insights into the biological implications of the identified DEGs. GO analysis was performed using the blast2GO software [29]. The GO analysis showed that the DEGs clustered in molecular functions, biological processes, and cellular components (Figure 2). The GO annotation indicated that the DEGs were involved in many biological processes, such as organ morphogenesis, skeletal system development, immune response, protein binding, and integral component of membrane.
Figure 2.

GO analysis of DEGs between the DPL and Landrace piglets. The DEGs are classified into three categories: cellular component, molecular function, and biological process. The percentage of genes in each category and the number of genes are shown above.
The DEGs were also mapped into the KEGG pathway database to predict the significantly enriched pathways and the detailed information was shown in Supplementary S2. The major enriched pathways included genetic information processing, metabolism, human disease, cellular processes, environmental information processing, and organismal systems.
3.4. DEGs Related to Immunity, Growth, and Metabolism
Among the DEGs, we found many genes related to immunity, which may be possible reasons for the different immune capability between the two breeds. For instance, myxovirus resistance 2 (MX2), a novel innate immunity restriction factor with important roles in defensing virus infection, had higher expression in the DPL piglets than the Landrace piglets (1.56-fold). CEBPE, a member of the basic-leucine zipper transcription factor family, mediating innate immune response and systemic lipid metabolism, had higher expression in the DPL piglets compared to the Landrace piglets (2.27-fold). Other immune-related genes including GNLY, GZMA, CSF1R, ADAM8, and CD59 were all differentially expressed between the two breeds.
We also detected some DEGs relevant with lipid metabolism and growth (such as GOS2 and RGS2) had higher expression in the DPL piglets compared to the Landrace piglets (6.0-fold and 1.58-fold, resp.). However, body growth relevant genes PHOSPHO1, KIT, and CST3 had higher expression in the Landrace piglets compared to the DPL piglets (2.32-fold, 3.92-fold, and 1.66-fold, resp.).
3.5. Statistical Analysis of SNPs
The average SNPs in the DPL piglets were 37344, twofold more than the Landrace piglets. The genic SNPs were higher in DPL piglets; however, the intergenic SNPs were higher in the Landrace piglets. The frequencies of transition, transversion, and heterozygosity were similar in the two breeds. The details of different SNPs in the two breeds were listed in Table 3 and Supplementary S3.
Table 3.
The distribution of the SNPs in the two breeds.
| Animal | SNP number | Genic SNP | Intergenic SNP | Transition | Transversion | Heterozygosity |
|---|---|---|---|---|---|---|
| L1 | 16,943 | 13,104 | 3,839 | 73.77% | 26.23% | 54.84% |
| L2 | 10,135 | 8,148 | 1,987 | 73.34% | 26.66% | 58.59% |
| L3 | 13,754 | 11,011 | 2,743 | 73.68% | 26.32% | 57.93% |
| D1 | 38,153 | 27,022 | 11,131 | 74.53% | 25.47% | 44.02% |
| D2 | 36,535 | 27,461 | 9,074 | 75.19% | 24.81% | 58.32% |
3.6. Identification of AS
AS is a mechanism that brings remarkable diversity to proteins and allows a gene to generate different mRNA transcripts that are translated into distinct proteins. Skipped Exon, Intron Retention, Alt3splice, and Alt First Exon were the major AS in the two breeds. There were more Skipped Exon and Alt First Exon in the DPL than the Landrace piglets. However, other AS were similar in the two breeds. The distribution of the AS events was shown in Table 4.
Table 4.
The distribution of AS in the two breeds.
| Animal | Skipped Exon | Intron Retention | Alternative 5′ Splice Sites | Alt 3′ Splice Sites | Alt Last Exon | Alt First Exon |
|---|---|---|---|---|---|---|
| L1 | 1,782 | 477 | 270 | 575 | 208 | 678 |
| L2 | 1,049 | 440 | 199 | 373 | 143 | 456 |
| L3 | 1,476 | 483 | 221 | 508 | 166 | 560 |
| D1 | 2,048 | 623 | 286 | 653 | 215 | 788 |
| D2 | 2,519 | 598 | 359 | 780 | 266 | 1,068 |
3.7. qRT-PCR Confirmation of the Gene Expression Data from RNA-seq
To verify the gene expression data by RNA-seq, qRT-PCR was performed for six genes according to their biological function, including two genes with lower expression levels and four genes with higher expression levels in the Landrace piglets compared to the DPL piglets. The expression patterns of the six genes were generally consistent with the RNA-seq results (Figure 3), suggesting that the results of the RNA-seq experiments were accurate and reliable.
Figure 3.
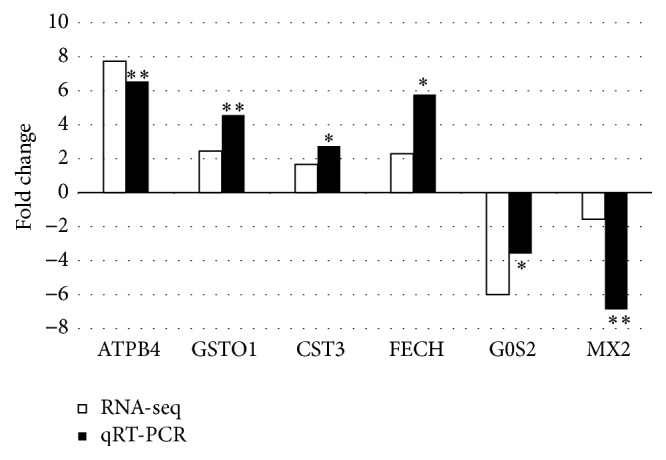
Validation of the 6 DEGs from RNA-seq analysis by qRT-PCR. The direction and magnitude of the fold changes obtained using the qRT-PCR technique were similar to those of the RNA-seq data (∗ p < 0.05; ∗∗ p < 0.01).
4. Discussion
In this study, we compared the gene expression differences of the two genetically different pig breeds, the DPL, and the Landrace pigs. To the best of our knowledge, our study was the first report on transcriptome gene expression profiling of whole blood in DPL and Landrace piglets using RNA-seq technology. The current study generated 30.2 Gb clean data, more than 10-fold the pig genome size. Moreover, the map ration to reference genome ranged from 77.35% to 79.4%, meeting the requirements for the subsequent analysis. In this study, we used Sus scrofa as reference sequence; the low mapping rates could be related to the difference between the sample genome and the reference genome. Moreover, compared with human and mouse genomic sequence, pig genomic sequence is incomplete. Therefore, the mapping rate of pigs is generally less than 80% [30]. The identified DEGs, SNPs, and AS may help to illustrate the underlying differences in the whole blood transcriptome between these two breeds and will be valuable for future studies on the identification of major genes that may affect pig growth performance, immune capacity, and meat quality.
In this study, totally 83 genes were shown to be significantly differentially expressed between the two breeds. The GO annotation and KEGG pathways analysis showed that the DEGs were mainly involved in single-organism process, cellular process and metabolic process of biological process, the cell part, cell and organelle of a cellular component, and the binding and catalytic activity of molecular function. Our finding was in accordance with the transcriptome analysis of other studies [10, 31]. It is well known that the molecular regulation of animal traits is very complicated and the relationship between genes and traits can be one-to-many or many-to-one. Therefore, it was not surprising that our identified DEGs were enriched not only in immune-related pathways but also in those involved in metabolic process. The immune-related and metabolic pathways are important for improving pig traits such as disease resistance and growth. Based on our data, further functional studies with these DEGs are warranted to identify key genes influencing growth performance and immune capacity of swine.
Our current study has identified several differentially expressed genes between DPL and Landrace that are important in disease resistance, such as MX2 and CEBPE. The MX2 gene was identified as a novel IFN-induced gene, which inhibited a variety of virus infection [32–34]. Studies have shown that the expression levels of MX2 was upregulated in piglets after coinfection of porcine reproductive and respiratory syndrome virus (PRRSV) and Mycoplasma hyopneumoniae [35]; it was also increased with the sudden change of feeding regime after weaning in piglet gut [36]. Moreover, MX2 also showed antiviral ability against influenza virus [37], vesicular stomatitis virus [33], and PRRSV in swine [38]. Additionally, previous studies have shown that DPL piglets exhibited strong resistance to PRRSV [39, 40]. In the current study, the expression level of MX2 gene was higher in the DPL piglets than the Landrace piglets. Therefore, higher transcription level of MX2 in DPL could be an indicator for resistance to PRRSV. CEBPE, which expressed only in myeloid cells including monocytes and macrophages, has been shown to be required not only for hematopoietic cell development, but also for cytokine expression [41–43]. Studies have shown that CEBPE-deficient mice developed normally but failed to generate functional neutrophils [44]. Furthermore, the phagocytic function of CEBPE-deficient macrophages was also impaired [45]. The higher expression level of CEBPE in the DPL piglets may also suggest CEBPE as an indicator for disease resistance. In addition, other immune-related genes including GNLY, GZMA, CSF1R, ADAM8, and CD59 were also differentially expressed in the two breeds. The expression of the immune relevant DEGs in DPL and Landrace pigs could be related to the differences of the breed and the immune system. Our data suggest that these genes might play important roles in disease resistance after birth.
Our study also identified DEGs between DPL and Landrace that are important for body growth, including KIT, PHOSPHO1, and CST3. Compared with other industrial pig breeds, Chinese indigenous pigs have the disadvantage of slow growth rate and high fat content, but with the advantage of high fertility, special meat flavor, and strong adaptability [1]. Our data showed that KIT gene had higher expression in the Landrace piglets compared to DPL. It was reported that the KIT genotypes affected piglet birth weight and body weight gain until weaning [46]. Two other important candidate genes for body growth, namely, PHOSPHO1 and CST3, had higher expression in the Landrace piglets compared to the DPL. PHOSPHO1 is a member of the haloacid dehalogenase superfamily and it plays an essential for the initiation of skeletal mineralization [47]. Recently, PHOSPHO1 was found to have an important function during growth and skeletal development in mouse, which could avoid low bone mineral density, spontaneous greenstick fractures, osteomalacia, and prominent scoliosis [47, 48]. Moreover, PHOSPHO1 was shown to play an important role in activating inside chondrocyte- and osteoblast-derived matrix vesicles [48]. CST3 serves an important physiological role as the controlling inhibitor of extracellular cysteine proteinase activity [49]. Previous studies also suggested it as an excellent candidate gene to influence fetal development and growth [50]. The current study has revealed highly enriched DEGs in piglets that are involved in organ morphogenesis, skeletal system development, and immune. These data provide a basis for future functional research that may aid in the discovery of genetic mechanism of body growth in pigs.
Genes that are important in adiposity were also found to be differently expressed between the DPL and Landrace piglets, including G0S2 and CEBPE. Adiposity can increase the cost for pig farming because it reduces feed efficiency and carcass yield. Lipids and their metabolites can regulate gene expression and therefore affect many physiological processes. Our data showed that G0S2 and CEBPE were highly expressed in the DPL piglets compared to Landrace. G0/G1 switch gene 2 (G0S2) was initially found to be differentially expressed in lymphocytes during lectin-induced switch from the G0 to G1 phase of the cell cycle [51, 52]. Moreover, G0S2 was abundantly expressed in porcine adipose tissue and liver among various tissues [53]. Other data also showed that G0S2 was associated with fat content in bovine species [54]. Fatty acid composition is a critical aspect of meat quality, and its variation affects flavor, color, firmness, and softness of the fat in meat [55]. Since DPL pigs have higher fat content than Landrace, G0S2 may be a promising candidate gene for meat quality. CEBPE not only associates with immunity but also is involved in systemic lipid metabolism [56]. Macrophages from CEBPE-deficient mice showed less lipid accumulation than control mice [57]. At present, limited information is available about the roles of G0S2 and CEBPE in pigs; therefore, further investigations are needed to explain the role of these genes in porcine lipid metabolic. In addition, other genes unrelated to growth performance, immune capacity, or meat quality were found to be differently expressed in the two breeds. These genes included HRH4, REXO2, NTAN1, CA2, REXO2, and FAM213A. However, there are very few studies that have been performed to study these genes' functions. Further studies are needed to gain more insights into the function of these genes.
NGS has been widely used to analyze SNPs in eukaryotes [31, 58]. The DPL piglets have twofold more SNPs than the Landrace piglets. These different SNPs in the two breeds will be valuable molecular markers for future study. SNPs identification may be affected due to RNA editing, sequencing errors (in spite of sequencing quality filtration), mapping error, or the reference sequencing errors [59]. Therefore, further studies are needed to validate the identified SNPs.
Another important advantage of RNA-seq is its ability to detect AS events [22]. AS is an important model of gene expression regulation and is not generally assessable using microarray method. We found that Skipped Exon was the most common type of AS in pigs, which is similar to human [24] and yeast [60]. Our data showed that DPL had more AS than Landrace, which is consistent with the SNPs result. The agreement of SNPs and AS result may suggest that many genes undergo the same transcriptional and translational regulation in both breeds. Further research will be able to determine the detailed regulation in each breed, and theses observed differences will be highly useful in future studies.
In summary, this study was conducted to screen DEGs from the whole blood of DPL and Landrace piglets using RNA-seq technology. We found that 83 genes were significantly differentially expressed between the two breeds. Bioinformatics analyses identified significantly enriched pathways that are important for disease resistance and growth performance. The identification of specialized biological functions and key genes will be very valuable for further studies for breed improvement programs. Additionally, more SNPs and AS were found in the DPL than Landrace pigs. These findings provide new insights into the postnatal status of the piglets and will facilitate future genomic and gene function research on the different pig breeds.
Supplementary Material
Detailed information of the 83 DEGs in the whole blood of the DPL and Landrace piglets.
Acknowledgments
This study was supported by the National Animal Breed Resource Preservation Project of China (no. 2130135), the National Project for Breeding Transgenic Pigs of China (no. 2013ZX08006-002), Shandong Province Modern Pig Technology and Industry System Project (no. SDAIT-08-02), and Shandong Province Agricultural Animal Breeding Project of China (nos. 2013LZ02-015 and 2014LZ03-016).
Competing Interests
There is no conflict of interests.
Authors' Contributions
Jiaqing Hu and Dandan Yang contributed equally to this work.
References
- 1.Yang H. Livestock development in China: animal production, consumption and genetic resources. Journal of Animal Breeding and Genetics. 2013;130(4):249–251. doi: 10.1111/jbg.12045. [DOI] [PubMed] [Google Scholar]
- 2.Wang J., Guo J., Zhang Q., et al. Genetic diversity of Chinese indigenous pig breeds in shandong province using microsatellite markers. Asian-Australasian Journal of Animal Sciences. 2011;24(1):28–36. [Google Scholar]
- 3.Chaussabel D., Pascual V., Banchereau J. Assessing the human immune system through blood transcriptomics. BMC Biology. 2010;8, article 84 doi: 10.1186/1741-7007-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohr S., Liew C.-C. The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends in Molecular Medicine. 2007;13(10):422–432. doi: 10.1016/j.molmed.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Wilhelm B. T., Landry J.-R. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods. 2009;48(3):249–257. doi: 10.1016/j.ymeth.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh M., Sodhi S. S., Song K.-D., et al. Evaluation of body growth and immunity-related differentially expressed genes through deep RNA sequencing in the piglets of Jeju native pig and Berkshire. Animal Genetics. 2015;46(3):255–264. doi: 10.1111/age.12281. [DOI] [PubMed] [Google Scholar]
- 7.Xiao J., Zhong H., Liu Z., et al. Transcriptome analysis revealed positive selection of immune-related genes in tilapia. Fish & Shellfish Immunology. 2015;44(1):60–65. doi: 10.1016/j.fsi.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Mu Y., Li M., Ding F., et al. De novo characterization of the spleen transcriptome of the large yellow croaker (Pseudosciaena crocea) and analysis of the immune relevant genes and pathways involved in the antiviral response. PLOS ONE. 2014;9(5) doi: 10.1371/journal.pone.0097471.e97471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sodhi S. S., Park W. C., Ghosh M., et al. Comparative transcriptomic analysis to identify differentially expressed genes in fat tissue of adult Berkshire and Jeju Native Pig using RNA-seq. Molecular Biology Reports. 2014;41(9):6305–6315. doi: 10.1007/s11033-014-3513-y. [DOI] [PubMed] [Google Scholar]
- 10.Shan L., Wu Q., Li Y., et al. Transcriptome profiling identifies differentially expressed genes in postnatal developing pituitary gland of miniature pig. DNA Research. 2014;21(2):207–216. doi: 10.1093/dnares/dst051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroyen M., Tuggle C. K. Current transcriptomics in pig immunity research. Mammalian Genome. 2015;26(1-2):1–20. doi: 10.1007/s00335-014-9549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan Q., Shai O., Lee L. J., Frey B. J., Blencowe B. J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 13.Marquez Y., Brown J. W. S., Simpson C., Barta A., Kalyna M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Research. 2012;22(6):1184–1195. doi: 10.1101/gr.134106.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graveley B. R., Brooks A. N., Carlson J. W., et al. The developmental transcriptome of Drosophila melanogaster . Nature. 2011;471(7339):473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trapnell C., Pachter L., Salzberg S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chowdhury S. R., King D. E., Willing B. P., et al. Transcriptome profiling of the small intestinal epithelium in germfree versus conventional piglets. BMC Genomics. 2007;8, article 215:16. doi: 10.1186/1471-2164-8-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canario L., Voillet V., Iannuccelli N., et al. Transcriptomic prediction of piglet vitality from umbilical cord blood of purebreds and crossbreds born in the same litter—comparison of Meishan and Large White Sows. Proceedings of the 10th World Congress on Genetics Applied to Livestock Production. Presented at 10. World Congress on Genetics Applied to Livestock Production (WCGALP '14); 2014; Vancouver, Canada. American Society of Animal Science; [Google Scholar]
- 18.Muns R., Nuntapaitoon M., Tummaruk P. Non-infectious causes of pre-weaning mortality in piglets. Livestock Science. 2016;184:46–57. doi: 10.1016/j.livsci.2015.11.025. [DOI] [Google Scholar]
- 19.Jiang Y., Cen W., Xing S., et al. Tissue expression pattern and polymorphism of G0S2 gene in porcine. Gene. 2014;539(1):173–179. doi: 10.1016/j.gene.2014.01.058. [DOI] [PubMed] [Google Scholar]
- 20.Martino A., Cabiati M., Campan M., et al. Selection of reference genes for normalization of real-time PCR data in minipig heart failure model and evaluation of TNF-α mRNA expression. Journal of Biotechnology. 2011;153(3-4):92–99. doi: 10.1016/j.jbiotec.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Cinar M. U., Islam M. A., Pröll M., et al. Evaluation of suitable reference genes for gene expression studies in porcine PBMCs in response to LPS and LTA. BMC Research Notes. 2013;6(1, article 56) doi: 10.1186/1756-0500-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C., Williams B. A., Pertea G., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11, article R106 doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Handsaker B., Wysoker A., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Florea L., Li S. CLASS and ASprofile: resources for alternative splicing annotation from RNA-seq data. Proceedings of the 21th International Plant and Animal Genome Conference; 2013. [Google Scholar]
- 26.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Bioinformatics Methods and Protocols. 1999:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 27.Wang J., Wang Y., Guo J., et al. Slection of reference genes and determination of cytokines and receptor mRNA expression in peripheral blood of piglets. Scientia Agricultura Sinica. 2015;48:1437–1444. [Google Scholar]
- 28.Livak K. J., Schmittgen T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 30.Wang J., Wang Y., Wang H., et al. Transcriptomic analysis identifies candidate genes and gene sets controlling the response of porcine peripheral blood mononuclear cells to poly I:C stimulation. G3-Genes Genomes Genetics. 2016;6(5):1267–1275. doi: 10.1534/g3.116.028290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nie Q., Fang M., Jia X., et al. Analysis of muscle and ovary transcriptome of sus scrofa: assembly, annotation and marker discovery. DNA Research. 2011;18(5):343–351. doi: 10.1093/dnares/dsr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kane M., Yadav S. S., Bitzegeio J., et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 2013;502(7472):563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki K., Tungtrakoolsub P., Morozumi T., Uenishi H., Kawahara M., Watanabe T. A single nucleotide polymorphism of porcine MX2 gene provides antiviral activity against vesicular stomatitis virus. Immunogenetics. 2014;66(1):25–32. doi: 10.1007/s00251-013-0745-2. [DOI] [PubMed] [Google Scholar]
- 34.Jin H. K., Takada A., Kon Y., Haller O., Watanabe T. Identification of the murine Mx2 gene: interferon-induced expression of the Mx2 protein from the feral mouse gene confers resistance to vesicular stomatitis virus. Journal of Virology. 1999;73(6):4925–4930. doi: 10.1128/jvi.73.6.4925-4930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li B., Du L., Xu X., et al. Transcription analysis on response of porcine alveolar macrophages to co-infection of the highly pathogenic porcine reproductive and respiratory syndrome virus and Mycoplasma hyopneumoniae . Virus Research. 2015;196:60–69. doi: 10.1016/j.virusres.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Bomba L., Minuti A., Moisá S. J., et al. Gut response induced by weaning in piglet features marked changes in immune and inflammatory response. Functional & Integrative Genomics. 2014;14(4):657–671. doi: 10.1007/s10142-014-0396-x. [DOI] [PubMed] [Google Scholar]
- 37.Morozumi T., Naito T., Lan P. D., et al. Molecular cloning and characterization of porcine Mx2 gene. Molecular Immunology. 2009;46(5):858–865. doi: 10.1016/j.molimm.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 38.Wang H., Bai J., Fan B., Li Y., Zhang Q., Jiang P. The interferon-induced Mx2 inhibits porcine reproductive and respiratory syndrome virus replication. Journal of Interferon & Cytokine Research. 2016;36(2):129–139. doi: 10.1089/jir.2015.0077. [DOI] [PubMed] [Google Scholar]
- 39.Xing J., Xing F., Zhang C., et al. Genome-wide gene expression profiles in lung tissues of pig breeds differing in resistance to porcine reproductive and respiratory syndrome virus. PLoS ONE. 2014;9(1) doi: 10.1371/journal.pone.0086101.e86101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang C., Xing F., Xing J., Jiang Y., Zhou E. Different expression patterns of PRRSV mediator genes in the lung tissues of PRRSV resistant and susceptible pigs. Developmental & Comparative Immunology. 2013;39(1-2):127–131. doi: 10.1016/j.dci.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Akagi T., Thoennissen N. H., George A., et al. In vivo deficiency of both C/EBPβ and C/EBPε results in highly defective myeloid differentiation and lack of cytokine response. PLoS ONE. 2010;5(11) doi: 10.1371/journal.pone.0015419.e15419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C., Chen J., Sun H., Sun L., Liu Y. CEBPE polymorphism confers an increased risk of childhood acute lymphoblastic leukemia: a meta-analysis of 11 case-control studies with 5,639 cases and 10,036 controls. Annals of Hematology. 2015;94(2):181–185. doi: 10.1007/s00277-014-2186-x. [DOI] [PubMed] [Google Scholar]
- 43.Gombart A. F., Kwok S. H., Anderson K. L., Yamaguchi Y., Torbett B. E., Koeffler H. P. Regulation of neutrophil and eosinophil secondary granule gene expression by transcription factors C/EBPε and PU.1. Blood. 2003;101(8):3265–3273. doi: 10.1182/blood-2002-04-1039. [DOI] [PubMed] [Google Scholar]
- 44.Lekstrom-Himes J., Xanthopoulos K. G. CCAAT/enhancer binding protein ε is critical for effective neutrophil-mediated response to inflammatory challenge. Blood. 1999;93:3096–3105. [PubMed] [Google Scholar]
- 45.Tavor S., Vuong P. T., Park D. J., Gombart A. F., Cohen A. H., Koeffler H. P. Macrophage functional maturation and cytokine production are impaired in C/EBPε-deficient mice. Blood. 2002;99(5):1794–1801. doi: 10.1182/blood.v99.5.1794. [DOI] [PubMed] [Google Scholar]
- 46.Fésüs L., Zsolnai A., Komlósi I. Influence of porcine coat colour genotypes on haematological parameters, piglet birth weight and body weight gain until weaning. Journal of Animal Breeding and Genetics. 2005;122(2):127–130. doi: 10.1111/j.1439-0388.2005.00504.x. [DOI] [PubMed] [Google Scholar]
- 47.MacRae V. E., Davey M. G., McTeir L., et al. Inhibition of PHOSPHO1 activity results in impaired skeletal mineralization during limb development of the chick. Bone. 2010;46(4):1146–1155. doi: 10.1016/j.bone.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huesa C., Yadav M., Finnila M., et al. PHOSPHO1 is essential for mechanically competent mineralization and the avoidance of spontaneous fractures. Bone. 2010;47:S84–S85. doi: 10.1016/j.bone.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abrahamson M., Barrett A. J., Salvesen G., Grubb A. Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. The Journal of Biological Chemistry. 1986;261(24):11282–11289. [PubMed] [Google Scholar]
- 50.Song G., Bailey D. W., Dunlap K. A., et al. Cathepsin B, cathepsin L, and cystatin C in the porcine uterus and placenta: potential roles in endometrial/placental remodeling and in fluid-phase transport of proteins secreted by uterine epithelia across placental areolae. Biology of Reproduction. 2010;82(5):854–864. doi: 10.1095/biolreprod.109.080929. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura N., Shimaoka Y., Tougan T., et al. Isolation and expression profiling of genes upregulated in bone marrow-derived mononuclear cells of rheumatoid arthritis patients. DNA Research. 2006;13(4):169–183. doi: 10.1093/dnares/dsl006. [DOI] [PubMed] [Google Scholar]
- 52.Russell L., Forsdyke D. R. A human putative lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a small basic protein with the potential to be phosphorylated. DNA and Cell Biology. 1991;10(8):581–591. doi: 10.1089/dna.1991.10.581. [DOI] [PubMed] [Google Scholar]
- 53.Ahn J., Oh S.-A., Suh Y., Moeller S. J., Lee K. Porcine G0/G1 switch gene 2 (G0S2) expression is regulated during adipogenesis and short-term in-vivo nutritional interventions. Lipids. 2013;48(3):209–218. doi: 10.1007/s11745-013-3756-8. [DOI] [PubMed] [Google Scholar]
- 54.Ahn J., Li X., Choi Y. M., et al. Differential expressions of G0/G1 switch gene 2 and comparative gene identification-58 are associated with fat content in bovine muscle. Lipids. 2014;49(1):1–14. doi: 10.1007/s11745-013-3866-3. [DOI] [PubMed] [Google Scholar]
- 55.Wood J. D., Richardson R. I., Nute G. R., et al. Effects of fatty acids on meat quality: a review. Meat Science. 2004;66(1):21–32. doi: 10.1016/s0309-1740(03)00022-6. [DOI] [PubMed] [Google Scholar]
- 56.Okamoto R., Gery S., Gombart A. F., et al. Deficiency of CCAAT/enhancer binding protein-epsilon reduces atherosclerotic lesions in LDLR−/− mice. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0085341.e85341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gombart A. F., Krug U., O'Kelly J., An E., Vegesna V., Koeffler H. P. Aberrant expression of neutrophil and macrophage-related genes in a murine model for human neutrophil-specific granule deficiency. Journal of Leukocyte Biology. 2005;78(5):1153–1165. doi: 10.1189/jlb.0504286. [DOI] [PubMed] [Google Scholar]
- 58.Cánovas A., Rincon G., Islas-Trejo A., Wickramasinghe S., Medrano J. F. SNP discovery in the bovine milk transcriptome using RNA-Seq technology. Mammalian Genome. 2010;21(11-12):592–598. doi: 10.1007/s00335-010-9297-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu T., Lu G., Fan D., et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Research. 2010;20(9):1238–1249. doi: 10.1101/gr.106120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagalakshmi U., Wang Z., Waern K., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed information of the 83 DEGs in the whole blood of the DPL and Landrace piglets.