

Abstract
Protein engineering by combinatorial site-directed mutagenesis evaluates a portion of the sequence space near a target protein, seeking variants with improved properties (e.g., stability, activity, immunogenicity). In order to improve the hit-rate of beneficial variants in such mutagenesis libraries, we develop methods to select optimal positions and corresponding sets of the mutations that will be used, in all combinations, in constructing a library for experimental evaluation. Our approach, OCoM (Optimization of Combinatorial Mutagenesis), encompasses both degenerate oligonucleotides and specified point mutations, and can be directed accordingly by requirements of experimental cost and library size. It evaluates the quality of the resulting library by one- and two-body sequence potentials, averaged over the variants. To ensure that it is not simply recapitulating extant sequences, it balances the quality of a library with an explicit evaluation of the novelty of its members. We show that, despite dealing with a combinatorial set of variants, in our approach the resulting library optimization problem is actually isomorphic to single-variant optimization. By the same token, this means that the two-body sequence potential results in an NP-hard optimization problem. We present an efficient dynamic programming algorithm for the one-body case and a practically-efficient integer programming approach for the general two-body case. We demonstrate the effectiveness of our approach in designing libraries for three different case study proteins targeted by previous combinatorial libraries—a green fluorescent protein, a cytochrome P450, and a beta lactamase. We found that OCoM worked quite efficiently in practice, requiring only 1 hour even for the massive design problem of selecting 18 mutations to generate 107 variants of a 443-residue P450. We demonstrate the general ability of OCoM in enabling the protein engineer to explore and evaluate trade-offs between quality and novelty as well as library construction technique, and identify optimal libraries for experimental evaluation.
Key words: combinatorial mutagenesis, combinatorial optimization, experiment planning, library diversity, protein engineering
1. Introduction
Biotechnology is harnessing proteins for a wide range of significant applications, from medicine to biofuels (Nelson and Reichert, 2009, la Grange et al., 2010). In order to enable such applications, it is often necessary to modify extant proteins, developing variants with improved properties (e.g., stability, activity, immunogenicity) (Reetz and Carballira, 2007, Fox et al., 2007, Parker et al., 2010) for the task at hand. However, there is a massive space of potential variants to consider. Some protein engineering techniques—e.g., error-prone PCR) (Cadwell and Joyce, 1992), DNA shuffling (Stemmer, 1994), and staggered extension (StEP) (Zhao et al., 1998)—rely primarily on experiment to explore the sequence space, whereas others—e.g., structure-based protein redesign (Bolon and Mayo, 2001, Jiang et al., 2008, Chen et al., 2009)—employ sophisticated models and algorithms in order to identify a small number of variants for experimental evaluation.
Computational design of combinatorial libraries (Voigt et al., 2002, Meyer et al., 2003, Pantazes et al., 2007, Treynor et al., 2007, Ye et al., 2007, Zheng et al., 2009) provides a middle ground between the primarily experimental and primarily computational approaches to development of improved variants. Library-design strategies seek to experimentally evaluate a diverse but focused region of sequence space in order to improve the likelihood of finding a beneficial variant. Such an approach is based on the premise that prior knowledge can inform generalized predictions of protein properties, but may not be sufficient to specify individual, optimal variants (resulting in both false positives and false negatives). Libraries are particularly appropriate when the prior knowledge does not admit detailed, robust modeling of the desired properties, but when experimental techniques are available to rapidly assay a pool of variants. Example scenarios would be instances where a three-dimensional structure is not available (Levin et al., 2007) or cases where definitive decisions regarding specific amino acid substitutions are non-obvious (Reetz and Carballira, 2007).
Nature employs both random mutation and recombination in generating diverse variants, and modern molecular biology has reconstituted these processes as highly controlled in vitro techniques. Here we develop library design methods for mutagenesis, wherein individual residue positions and corresponding mutations are first chosen, and then all possible combinations are constructed and subjected to screening or selection (Fig. 1). Most library optimization work has focused on recombination (i.e., selecting breakpoints), including approaches by Arnold and co-workers (Voigt et al., 2002, Otey et al., 2004, Meyer et al., 2006, Otey et al., 2006), Maranas and co-workers (Moore and Maranas, 2003, Saraf et al., 2004, Saraf et al., 2005), and us (Ye et al., 2007, Zheng et al., 2007, Zheng et al., 2009, Zheng et al., 2010). Mayo and co-workers (Treynor et al., 2007) have extended structure-based variant design to structure-based mutagenic library design, and applied it to the design of a library of green fluorescent proteins. Maranas and co-workers (Pantazes et al., 2007) have developed methods for optimizing both recombination and mutagenesis libraries, and applied them to the design of libraries of cytochrome P450s. LibDesign (Marco and Daugherty, 2005) is another useful tool for combinatorial mutagenesis; however, it requires as input a predesigned library specification (positions and mutations). As we discuss further below, we develop here a more general method that encompasses different forms of computational library evaluation and optimization and experimental library construction, and explicitly optimizes both the quality and the novelty of the variants in the library.
FIG. 1.

Combinatorial mutagenesis libraries. (Left) Specific point mutations at selected positions are introduced and shuffled to generate a library of all combinations of mutations. (Right) Degenerate oligonucleotides (represented here in a regular expression-like notation, rather than IUPAC codes) are incorporated at selected positions, and shuffled to generate a library. Each degenerate oligo can code for a multiset of amino acids; consequently, some mutations may be represented more than others in the resulting library (e.g., in the first position, two codons for E vs. just one for D).
Two techniques are commonly employed to introduce mutations in constructing combinatorial mutagenesis libraries (Fig. 1). When point mutagenesis is employed (Fig. 1, left), an individual oligonucleotide specific to a desired mutation is incorporated; there is a separate oligonucleotide for each such mutation. Combinatorial shuffling techniques (Stutzman-Engwall et al., 2005, van den Beucken et al., 2001) mix and match the mutated genes. When degenerate oligonucleotides are employed (Fig. 1, right), multiple amino acid-level mutations at a position are encoded by a single degenerate 3-mer (Herman and Tawfik, 2007). As with point mutagenesis, a library is generated by combinatorial shuffling. While the degenerate oligo approach is experimentally cheaper (a library costs about the same as a single variant), it can result in redundancy (multiple codons for the same amino acid) and junk (codons for undesired amino acids or stop codons), and is thus more appropriate when a larger library and lower hit rate are acceptable (e.g., when a high-throughput screen is available) (Griswold et al., 2006).
Our method, OCoM (Optimization of Combinatorial Mutagenesis), encompasses both these approaches to experimental library construction. The key question is which mutations to introduce, given that the goal is isolation of functional variants with desired properties. A library-design strategy should therefore assess the predicted quality of prospective library members, e.g., by a sequence potential (Pantazes et al., 2007) or explicit structural evaluation (Treynor et al., 2007). We adopt a general sequence potential based on statistical analysis of a family of homologs to the target. The potential reveals both important residues (single-body conservation) and residue interactions (two-body coupling) for maintenance of protein stability and activity. Importantly, optimizing quality as a sole objective function might well result in libraries composed of sequences that are highly similar or even identical to extant proteins, an undesirable outcome. Thus it is necessary to balance quality assessment with novelty or diversity assessment. While this balance has been explicitly optimized for site-directed recombination (Zheng et al., 2009), previous mutagenic library-design methods have only addressed this issue indirectly, e.g., by controlling factors such as the overall library size and the number of positions being mutated. Here we develop a new metric to explicitly account for the novelty of the variants compared to extant sequences, and we simultaneously optimize libraries for both novelty and quality.
While we have previously characterized the complexity of recombination library design for both quality (Ye et al., 2007) and diversity (Zheng et al., 2007), to our knowledge, mutagenesis library design has never been similarly formalized or characterized. We show that, despite the combinatorial number of variants in the library, the OCoM design of an entire library is equivalent to the design of a single variant. Thus, like single-variant design, library optimization is NP-hard when accounting for a two-body potential. This stands in contrast to the polynomial-time algorithms for combinatorial recombination library design (Ye et al., 2007, Zheng et al., 2007). Consequently, we develop an integer programming approach that works effectively in practice on general OCoM problems, along with a polynomial-time dynamic programming approach that is appropriate for those without the two-body sequence potential.
To summarize the key contributions of OCoM, it supports a general scoring mechanism for variant quality, explicitly evaluates variant novelty, subsumes different approaches to library construction, accounts for bounds on library size and mutational sites, and evaluates the trade-offs between quality and diversity. While we focus on a statistical sequence potential for proposing and assessing mutations, our method is general and could employ a potential based on an initial round of experiments (e.g., from a randomization approach to remove phylogenetic bias) (Jackel et al., 2010) or a list of high-quality results from structure-based design (Chen et al., 2009) from which it is desired to construct a library.
Our results illustrate the effectiveness of our approach. We show library plans for 3 proteins previously examined in combinatorial library experiments: a green florescent protein, a cytochrome P450, and a beta-lactamase. Our results span 6 orders of magnitude of library size, from 102 to 107 members. For each protein, libraries optimized under a range of constraints display distinct trade-offs between quality and novelty, as well as for the choice of library construction method (point mutations or degenerate oligos).
2. Methods
Given a target protein, our goal is to design an optimal combinatorial mutagenesis library, as measured by the overall quality and novelty of its variants.
2.1. Variant evaluation
We start with metrics for assessing variants in a given library. The metrics we present employ position-specific one- and two-body scores, based on a multiple sequence alignment, but our methodology could accept other scoring schemes of a similar form. We first summarize a standard metric for evaluating the quality of a variant (is it likely to be folded and functional?), and then introduce a new metric for evaluating its novelty (how different is it from extant sequences?). While we treat individual variants here, we later show how to use these metrics to evaluate a library as a whole, without enumerating its constituents.
2.1.1. Quality metric ϕ
To evaluate quality, we employ one- and two-body position-specific sequence potentials. Our current implementation uses potential scores derived from statistical analysis of an evolutionarily diverse multiple sequence alignment (MSA) of homologs of the target protein, but the method is generic to any potential of the same form. Details have been previously published (Ye et al., 2007, Parker et al., 2011). Before computing the potentials, we filter the MSA to 90% sequence identity. The one-body term ϕi(a) for amino acid a position i then captures conservation as the negative log frequency of a in the ith column of the MSA. Similarly, the two-body term ϕij(a, b) for amino acid a at i and b at j captures correlated/compensating mutations as the negative log frequency of the pair (a, b) at the ith and jth columns, minus the independent terms ϕi(a) and ϕj(b). By subtracting the independent terms from the pairwise term, ϕij contains only the additional information regarding the correlation between the two positions.
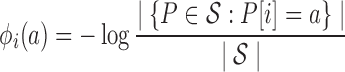 |
(1) |
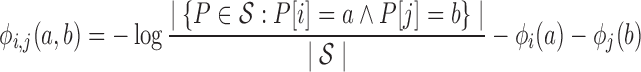 |
(2) |
The quality score of variant S is then 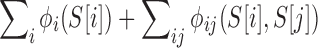 and the total quality score of a library is the sum of the quality scores of its variants. Note that since we subtracted out the one-body terms from the two-body ones, this sum correctly avoids double-counting the contributions from the individual positions. As these scores are based on negative logarithms, smaller is better.
and the total quality score of a library is the sum of the quality scores of its variants. Note that since we subtracted out the one-body terms from the two-body ones, this sum correctly avoids double-counting the contributions from the individual positions. As these scores are based on negative logarithms, smaller is better.
To mitigate overfitting, we restrict ϕij to a relatively small, significant set of residue pairs by a χ2 test of significant correlation. We compute a p-value, subject to a Bonferroni correction for multiple hypothesis testing, dividing the desired p-value by the number of pairs being tested. Alternatively, we could restrict two-body terms to those residues in contact, but we use the χ2 approach here since in previous work (Thomas et al., 2008, 2009a,b) we have found purely statistical models to outperform contact-restricted ones in predictive ability.
2.1.2. Novelty metric ν
Given a whole sequence, we can assess its novelty in terms of how similar it is to the closest homolog (other than the target) in the MSA. That is, compute the minimum percent sequence identity to an extant sequence; the smaller the score, the more novel the variant. Without explicitly accounting for this, a library focused on quality could simply recapitulate natural sequences (which are of course high quality), wasting experimental effort.
To compute the percent sequence identity, we need an entire sequence. However, during the course of optimization, we want to be able to assess the impact on novelty of each mutation under consideration. Thus, we introduce a position-specific novelty score νi(a) for amino acid a at position i, analogous to the quality score discussed above. The novelty contribution νi(a) assesses the sequence space distance between the mutant sequence containing a at i and homologs in the MSA.
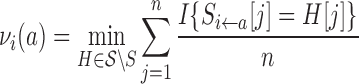 |
(3) |
where S is the target and Si←a is the target with a mutation to amino acid a at position i, 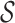 is the MSA, n is the length of S and number of columns of
is the MSA, n is the length of S and number of columns of  , and I{} the indicator function that returns 1 iff the predicate is true. Note that each νi(a) can be precomputed from the target and the MSA.
, and I{} the indicator function that returns 1 iff the predicate is true. Note that each νi(a) can be precomputed from the target and the MSA.
As with quality, the novelty score of variant S is then ∑iνi(S[i]), and the novelty score of a library sums the novelty scores of its variants. (Again, smaller is better.) The value for a variant is much like the percent sequence identity, except that each position does not account for mutations at other positions in computing the identity, and thus could underestimate the contribution. The value for a library is then much like the average percent sequence identity, and reduces the error in the total over the positions, since the library is comprised of the various combinations of mutations. While these thus are only approximations to the overall sequence identity, the error is independent of the actual mutations being made, and thus does not affect the optimization. We find in practice for the case studies presented in the results that the one-body potential is very highly correlated (over 0.99) with the full n-body one. Thus, there is no need to go to a higher-order potential.
2.2. Library representation and evaluation
Since we are optimizing an entire library, rather than individual variants, we need an efficient mechanism for evaluating its overall quality and novelty, without explicitly enumerating all its variants. This section develops a novel “tube” representation compactly encoding the substitutions defining a library, and then shows how to efficiently evaluate quality and novelty of a library represented in that manner. The following section then uses this representation and evaluation to optimize the choices.
2.2.1. “Tube” representation of a library
Recall (Fig. 1) that there are two common molecular biology techniques for generating combinatorial mutagenesis libraries: point mutations and degenerate oligonucleotides. A convenient abstraction subsuming these two methods of library construction is to consider for each position a multiset of amino acids, which we call a tube (as in the experiment). For point mutation, a tube contains a selected set of amino acids to be incorporated at a position. For degenerate oligonucleotides, a tube contains a multiset of amino acids encoded by all codons represented by a degenerate oligonucleotide 3-mer. In this abstraction, we always mean 3-mer. Note that the representation even supports multiple degenerate oligonucleotides (or a degenerate oligonucleotide and a specific one) at a position, which might be desirable to obtain the best balance of library quality, novelty, and size (Herman and Tawfik, 2007).
Given a set of tubes, one per position, the resulting library is defined by the cross-product of the tubes, with separate variants for each instance of an amino acid appearing multiple times in the multiset (Fig. 1). Note that in a multiset, every recurring appearance of an amino acid introduces redundancy, a scenario that is especially undesirable when screening is difficult. In optimizing a library, we select one tube for each position, from a preenumerated set of allowed tubes. These are in turn determined by the amino acids that should be considered as possible substitutions. Our current implementation only allows those appearing at expected uniform frequency 5% or greater in the MSA. This averages to 4 to 5 per position in our case studies, for at most 25 − 1 = 31 tubes when considering all sets of point mutations. For degenerate oligos, we only allow tubes that have a ratio of at least 3:2 between codons for allowed substitutions and those for disallowed ones. We also eliminate tubes that code for the same proportions of amino acids in a larger multiset, for example, we would keep [GC]TC, coding for {L, V} instead of [GC]T[GC], coding equivalently but redundantly for {L, L, V, V}. Finally, we disallow tubes with STOP codons, though recognize that with a very high-throughput screen, those may still be acceptable. All combinations of the 4 nucleotides in each of 3 positions would yield 3375 possible degenerate oligos, but after our global filters there are fewer than 1000, which are further filtered for each position according to allowed substitutions, for an average of 10 in our case studies.
2.2.2. Efficient tube-based library evaluation
Our quality and novelty metrics are expressed in terms of variants in a library. However, in the course of optimizing libraries (next section), we do not want to enumerate all their variants in order to compute these values. In previous work, we showed how to lift one- and two-body position-specific sequence potentials for single variants to corresponding potentials for recombination libraries (Ye et al., 2007, Zheng et al., 2009). We do the same here for combinatorial mutagenesis. For simplicity, consider just the one-body term ϕi; the two-body term ϕij and the novelty νi work similarly.
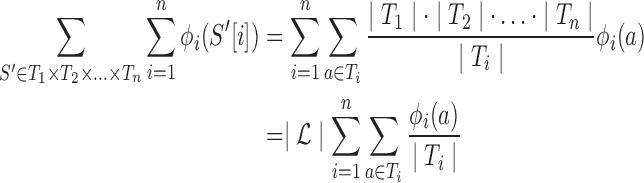 |
(4) |
This follows by recognizing that amino acid type a at position i contributes ϕi(a) to each variant, i.e., each choice of amino acid types for the other positions.
Thus, we develop a tube-based library potential by averaging over the set of amino acids in the tube, taking a convex combination of quality and novelty under a weighting factor α:
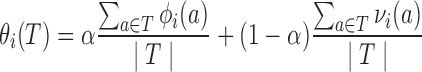 |
(5) |
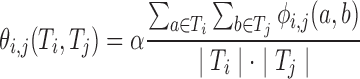 |
(6) |
For simplicity of subsequent formulas, we assume that α is fixed before computing θi; recall that we do not have a two-body novelty term.
Note that our tube-based scores avoid a potential pitfall by automatically accounting for the relative frequencies of amino acids at a position, and their relative contribution to the library. That is, if one position has three amino acid types and another two (Fig. 1), then the contributions of the constituent amino acids are weighted by 1/3 and 1/2, respectively.
2.3. Library optimization
With the pieces in place, we can now formally define our problem.
Problem 2.1 (OCoM)
Given a protein sequence S of length n and, for each position i a set
 of allowed tubes, optimize a library
of allowed tubes, optimize a library
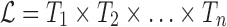 where for each i,
where for each i,
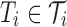 , so as to minimize
, so as to minimize
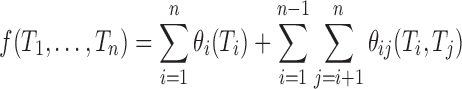 |
(7) |
The experimental cost can be constrained by the number of sites being substituted, the number of amino acids (including duplicates) in each tube, and the size of the library.
Recall that θi is defined in terms of a parameter α that controls the relative trade off between quality and novelty. For the results, we try a range of values, recognizing that in the future it is desirable to consider all trade-offs and select plans that are Pareto optimal (He et al., 2010).
2.3.1. Complexity
As Eq. 7 makes clear, once we have normalized tube scores, library optimization looks just like single-variant optimization, though over an “alphabet” of tubes rather than amino acids or rotamers. It immediately follows from the NP-hardness of protein design with a two-body potential (Pierce and Winfree, 2002) that OCoM-based combinatorial mutagenesis library design is NP-hard.
2.3.2. Dynamic programming
Without the two-body sequence potential, we can readily develop an efficient dynamic programming algorithm. Let M(i, T) be the best score of a library optimized through position i, with tube T at position i. Because the one-body score allows for the choice of the optimal T at each position without consideration of any other position, the optimal library determined by the additional choice of T at i depends only on the library through i − 1. Thus
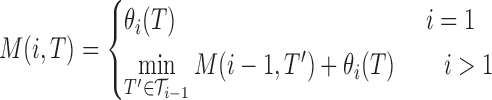 |
(8) |
The time and space complexity is quadratic in the size of the input: O(nm) for n the length of the sequence and m the maximum number of allowable tubes at any position. We can easily add a dimension to the DP matrix to count total mutational sites (up to M), for a total complexity of O(nmM).
2.3.3. Integer programming
In order to solve the full library design problem, including the two-body potential, we develop an integer programming formulation that works well in practice using the IBM ILOG CPLEX solver.
Define singleton binary variable si,t to indicate whether or not tube t is at position i. Similarly, define pairwise binary variable pi,j,t,u to indicate whether or not the tubes t,u are at i, j respectively.
We rewrite our objective function (Eq. 7) in terms of these binary variables:
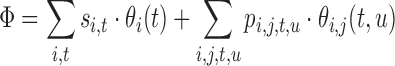 |
(9) |
In order to guarantee that the variable assignments yield a valid combinatorial library, we impose the following constraints:
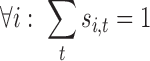 |
(10) |
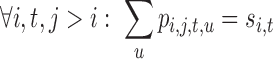 |
(11) |
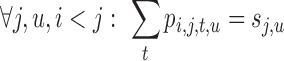 |
(12) |
Eq. 10 ensures that exactly one tube is chosen at each position i. Eq. 11 and Eq. 12 maintain consistency between singleton and pairwise variables.
In order to specify desired properties of the mutated sites and library size, we impose the following additional constraints.
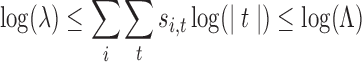 |
(13) |
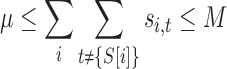 |
(14) |
The bounds on the library size (Eq. 13) and number of mutations per position (Eq. 14) may be set by the technology and resources available for library construction and screening. The expression t ≠ {S[i]} determines whether or not the tube has only the wild-type amino acid, and thereby whether or not that is a mutated position. We could likewise incorporate additional constraints on the number of mutated positions. We use these as constraints instead of terms in the objective function because there are likely to be a relatively small number of values to try, and the results can be compared and contrasted. Furthermore, our objective function incorporates an explicit novelty score; these terms somewhat implicitly affect diversity. A larger λ means more variants, which must be different from each other in some way, except in the case of redundant codons. A larger μ allows, but does not guarantee, greater site diversity.
2.3.4. Implementation
The integer program is solved by the IBM ILOG optimization software. The code to generate the problem formulation and read the solution is implemented in Java. We have placed a limited-capability demonstration at http://www.cs.dartmouth.edu/∼cbk/ocom/; our Java code is available for academic use by contacting the authors.
3. Results
We applied OCoM to optimize libraries for three different proteins for which combinatorial libraries had previously been developed. We found that OCoM worked quite efficiently in practice, requiring only 1 hour even for the massive design problem of selecting 18 mutations to generate 107 variants for a 443-residue sequence. We demonstrate the general ability of OCoM in enabling the protein engineer to explore and evaluate trade-offs between quality and novelty as well as library construction technique, and identify optimal libraries for experimental evaluation.
3.1. Green fluorescent protein (GFP)
GFP presents a valuable engineering target due to its widespread use in imaging experiments; the availability of distinct colors, some engineered, enables in vivo visualization of differential gene expression and protein localization and measurement of protein association by fluorescence resonance energy transfer (Huh et al., 2003, Heim et al., 1994, Soboleski et al., 2005, Zhang et al., 2002). Following the work of Mayo and colleagues, we targeted the wild type 238-residue GFP from Aequorea victoria (uniprot entry name GFP_AEQVI) with mutation S65T (Treynor et al., 2007). The sequence potential is derived from the 243 homologs in Pfam PF01353.
Figure 2 (left) illustrates the trade-offs between library quality and novelty scores for fixed library size bounds and library construction techniques, over a range of α values (recall that higher α places more focus on quality). While we targeted 100- and 1000-member libraries, depending on the input and choice of parameters, not every exact library size is possible. Thus these numbers represent lower bounds on the library sizes; the upper bounds are slightly relaxed. The curves are fairly smooth but sometimes steep as a swift change in one property is made at relatively little cost to the other. Interestingly, the ≈100-member library curves intersect the ≈1000-member library curves. To the left of that point, the ≈100-member libraries yield better quality for a given novelty, while to the right, the ≈1000-member libraries yield a better novelty for a given quality, and thus would be preferred if that screening capacity is available. The curves intersect where the larger library approaches its maximum quality and the smaller library reaches its maximum novelty; thus adjusting α only sends library plans along the vertical or horizontal.
FIG. 2.

GFP plans under varying quality-novelty trade-offs, at fixed library size bounds, with two library construction techniques. Smaller scores are better. The left panels plot the scores of plans (one per point) for libraries of ≈100 members (red diamond solid) and ≈1000 members (blue square dash). The right panels detail the ≈100-member library plans, with selected positions and their wild-type amino acid types (underlined) and mutations.
The right panels of Figure 2 summarize the mutations comprising each library. Within the degenerate oligo plans we notice single substitutions at each site, while within the point mutation plans we notice a set of different substitutions at the same site, including some that fall outside the natural degeneracy in the genetic code. We also notice that a number of mutations are attractive across a range of α values, and under both construction techniques. Several times both construction methods identify the same site and same mutation. And in both cases, we see concentration of mutations on less conserved sites (e.g., 124[EK] where Lysine is the consensus residue at 31%) for better quality, and spreading mutations over the sequence for better novelty.
Figure 3 (left) illustrates trends in planning GFP libraries of a wide range of sizes. The y-axis gives the total quality score summed over the unique variants in the library (lower is better). Compared to the number of variants in the library to be screened, this is a measure of the library efficiency. The point mutation libraries remain linear at an approximate slope of 1 on this log-log plot; essentially, each mutation is picking up a constant “penalty” against quality. While, as we also see in Figure 2, degenerate oligo libraries tend to have better quality scores due to their multiset nature, the redundancy leads to fewer unique variants and thus fewer expected “hits” for the same screening effort. Consequently, up to a factor of 103 more degenerate oligo variants than point mutation variants need to be screened to achieve the unique library size, consistent with trends in other studies (Reetz et al., 2008). On the other hand, degenerate oligo libraries are also cheaper to construct. These curves help elucidate the trade-offs. The degenerate oligo curve flattens out at 106 to 107 largely because the algorithm has reduced capacity to find more unique reasonable quality variants on this particular and relatively smaller protein.
FIG. 3.

Efficiency evaluation of plans for different GFP (left) and P450 (right) library sizes, under degenerate oligos (blue solid squares) and point mutations (red dashed diamonds). The y-axis plots the total quality score (ϕ; lower is better) of the unique variants in the library (i.e., removing duplicates from degenerate oligos). The degenerate oligo curve is labeled with the number of unique variants.
To further study the use of degeneracy in library generation, we compared libraries using selected degenerate oligos with those using saturation mutagenesis, either with the NNK degenerate codon (coding all 20 amino acids) or the NDT degenerate codon (12 diverse amino acids). Reetz et al. (2008) have studied the relative efficiency of the two saturation mutagenesis techniques, in the context of directed evolution. Using OCoM, we can further compare and contrast the selection of positions to mutate, at different levels of degeneracy. We separately optimized relatively conserved core residues (positions 57–72) (Treynor et al., 2007) and relatively less conserved surface ones. Figure 4 shows the efficiency of libraries (using the total quality metric of the preceding paragraph) for different number of sites to mutate. As in our above library studies, there are sufficient degrees of freedom in any method, and both in the core and on the surface, to continue taking mutations at roughly the same penalty. Strikingly, the relative efficiency (ratio) of saturation, half saturation, and any choice is about the same in the core or on the surface, across the number of sites mutated. We also evaluated the use of “double-degenerate” oligos, combining two different degenerate oligos in a single tube. However, for these studies they yielded exactly the same plans as did the regular degenerate oligos. There was apparently insufficient motivation to select amino acids sufficiently different not to be naturally covered by the degeneracy in the genetic code.
FIG. 4.

Efficiency evaluation (as in Fig. 3) for GFP libraries optimized at different levels of degeneracy, for core or surface, at different numbers of mutated sites.
Notably, all of our full length GFP plans in Figure 2 (right) avoid the conserved core region when constructed with degenerate oligos or point mutations under α favoring quality or novelty. However, all plans consistently choose the immediately adjacent mutation, 73[KR]. This is largely due to the fact that 73 is less conserved than the core positions, and Lys is the consensus residue at 57%. While these plans were designed by global optimization of our scoring metrics over the entire protein, it is possible to incorporate addition constraints restricting the mutated positions and thereby targeting specific regions.
In order to compare with previous library studies (Treynor et al., 2007, Pantazes et al., 2007), which both exclusively focused on the GFP core, we restricted OCoM to the core residues, 57–72. OCoM proposed a 29 plan that is highly similar to the OPTOLIGO one (Pantazes et al., 2007) and shares some similarity with the DBISORBIT plan (Treynor et al., 2007). The plans are summarized in Table 1. OCoM and OPTOLIGO choose 8 of the same 9 positions to mutate, and 5 of the same substitutions at those 8 positions. Interestingly, for one mutation selected by OCoM but not OPTOLIGO, 68[AV], OCoM agrees with the DBISORBIT library, which experimentally gave the best preservation of function and introduction of diversity among their designed libraries, and was the only one to include 68[AV]. However, as Pantazes et al. noted, the very best Treynor et al. libraries score particularly poorly using a sequence potential because the 61[LV] and 65[AT] substitutions are not well represented in the sequence record. All three libraries reach consensus at two positions: 62 and 72.
Table 1.
GFP Libraries for the Conserved Core 57–72 Positions, by Our Method OCoM, along with OPTOLIGO by Pantazes et al. (2007) and DBISORBIT by Treynor et al. (2007)
| Method | Library | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OCoM | 59[IT] | 61[AV] | 62[AT] | 63[ST] | 64[FL] | 65[ST] | 68[AV] | 69[RQ] | 72[AS] | ||
| OPTOLIGO | 58[DP] | 59[IT] | 62[AT] | 63[AT] | 64[FL] | 65[ST] | 68[IV] | 69[LQ] | 72[AS] | ||
| DBIS ORBIT | 58[AP] | 59[ST] | 61[LV] | 62[AT] | 65[AT] | 68[AV] | 69[LQ] | 71[FL] | 72[AS] | ||
Underlined residue: wild type.
3.2. Cytochrome P450
Cytochrome P450 is an essential enzyme at all levels of cellular life and thus extensively studied, especially given its significant engineering applications in biofuels (Fukuda et al., 1994). We chose as a target a P450 from Bacillus subtilis, CYP102A2 (uniprot gene synonym cypD), used in previous library studies (Otey et al., 2004, Pantazes et al., 2007). The P450 family is very diverse, so we identified a set of 194 homologs to our target by running PSI-BLAST for 3 iterations, and then multiply aligned them with ClustalW using default parameters on the EBI portal. As in the earlier studies, we focused on residues 6–449 because the remaining portions of the MSA were too sparse for meaningful statistics.
The trade-off curves (Fig. 5) are more distinct than those for GFP, and are quite sharp and sparse. This may be a result of looking here at a small library size with relatively few mutations, relative to the much larger size of this protein. The degenerate oligo plans focus on a few positions (an average of 4), while the point mutation plans are more spread over the sequence (an average of 7).
FIG. 5.

P450 plans under varying quality-novelty trade-offs (see Fig. 2 for description).
With increasing library size (Fig. 3, right), we see similar trends as for GFP. As the library size increases, more and more screening effort (up to three orders of magnitude) is required to find fewer good unique variants in the degenerate oligo libraries. This illustrates a fundamental difference between the two library construction methods, and highlights a key advantage: using discrete oligos for each individual point mutation can always specifically target beneficial amino acids, even with increasing library size.
While we can compare our designs with the OPTOLIGO ones by Pantazes et al. (2007), our plans are from global optimization over the entire 400-residue protein, while theirs were limited to the 10 most variable positions as determined by their sequence analysis. Remarkably, there is one design site in common, Asn161. Our point mutation plans make 161[GN] or in one case 161[DGN]. However, OPOLIGO makes 161[NS] and rules out substituting pairs Ser and Gly or Ser and Asp at the same position. They eliminated a number of pairs of amino acids with similar qualities which therefore scored similarly under their metrics. These expert rules precluded the two systems from adopting the same substitutions at site 161.
For a more direct comparison, we targeted OCoM to the ten residue positions in the OPTOLIGO experiment, and employed only point mutations, the same as OPTOLIGO. Of the ten targeted positions, the OPTOLIGO plans were available for four in Figure 9 of Pantazes et al. (2007). OCoM plans incorporated three of those four positions (25, 161, and 191, leaving out 441). We found the OCoM and OPTOLIGO plans to vary substantially (Table 2). This can be attributed to the differences in the MSA and the subsequent sequence analysis. OPTOLIGO uses a consensus sequence as a reference and does not always include a wild-type in the library. Furthermore, our MSA shows some residues to be quite conserved, such that OCoM has very few choices at these sites. There is some overlap when both methods consider a position variable, especially at the larger library sizes. For example, at the 105 library size both methods employ Ala, Glu, and Ser at 25; Asn and Ser at 161; and Asn, Arg, Ser, and The at 191. In general, we also notice the same phenomenon reported by Pantazes et al., that substitutions chosen at smaller library sizes are largely retained at larger library sizes.
Table 2.
P450 Plan Details for Three Residue Positions, under OCoM and OPTOLIGO
| D25 | N161 | Q191 | ||||
|---|---|---|---|---|---|---|
| Library size | OCoM | OPTOLIGO | OCoM | OPTOLIGO | OCoM | OPTOLIGO |
| 100 | ADEKRS | AL | DGN | LNS | Q | MPT |
| 1000 | ADEKRS | ALS | DGNS | LNS | KQT | MPRT |
| 10000 | ADEKRS | ALS | DGNS | ELNSY | KNQRST | MNPRST |
| 100000 | ADEKRS | AELS | DGNS | AELNSY | KNQRST | MNPRST |
The wild-type residue is underlined in the OCoM plans; OPTOLIGO does not restrict the library to include wild-type.
3.3. Beta lactamase
The beta lactamase enzyme family hydrolyzes the beta lactam ring of penicillin-like drugs thereby conferring resistance to bacteria and presenting a potential drug target (Harding et al., 2005). As it supports easy and inexpensive activity screening, beta lactamase is an ideal candidate for testing combinatorial library methods (Meyer et al., 2003, 2006; Hiraga and Arnold, 2003, Ye et al., 2007, Zheng et al., 2009). However, these previous studies use recombination, while OCoM uses mutagenesis. The assumptions underlying the two techniques are quite different: recombination takes coarser-grained steps through sequence space, interpolating parental genes by mixing-and-matching, while mutagenesis takes finer-grained steps, moving away from a wild-type. Ultimately, a combination of the two methods may be most helpful.
We took as target the TEM-1 beta lactamase from E. coli, and developed the sequence potential from an MSA of 149 homologs aligned to 263 residues used in our previous combinatorial recombination work (Ye et al., 2007). We found the trends too similar to our other case studies to merit repetition of detail here, but we note that in contrast to P450, but like GFP (of a more similar size), the trade-off curves are less sharp and more full (Fig. 6). The efficiency trends (Fig. 7) are quite similar to those of both GFP and P450 (Fig. 3). Like GFP and P450, the targeted mutation sites are similar, but the repertoire of substitutions can differ. For example, at Lys261 the degenerate oligo plans make D,E,N substitutions, while the point mutations make A,D,E,Q,V substitutions.
FIG. 6.

Beta lactamase plans under varying quality-novelty trade-offs (see Fig. 2 for description).
FIG. 7.

Efficiency evaluation (as in Fig. 3) of plans for different beta lactamase library sizes, under degenerate oligos (blue solid squares) and point mutations (red dashed diamonds). The y-axis plots the total quality score (ϕ; lower is better) of the unique variants in the library (i.e., removing duplicates from degenerate oligos). The degenerate oligo curve is labeled with the number of unique variants.
4. Discussion
OCoM provides a powerful and general mechanism to optimize combinatorial mutagenesis libraries so as to improve the “hit-rate” of novel variants with properties of interest. It enables protein engineers to study the trade-offs among predicted quality and novelty, library size, and expected success over two different approaches to library construction. While it readily allows effort to be focused on residues or regions of interest, that is not required; OCoM supports global design of a protein, accounting for interrelated effects of mutations. While the design problem is NP-hard in theory and clearly combinatorial in practice, our encoding of the constraints and homology-based filtering of poor choices, along with the power of the IBM ILOG solver, yielded an implementation that was able to compute the optimal 107 size library for each test case in under an hour.
As we have implemented here, 2-body quality scores are considered state-of-the art, and necessary for evaluation of stability and activity of new proteins (Russ et al., 2005, Socolich et al., 2005). However, there may be cases, such as large proteins (or complexes) with high degrees of sequence variability (and thus large tube sets), where only a 1-body potential will be practical because of the combinatorial explosion. In such cases, our dynamic programming formulation will still enable the optimization of libraries based on conservation statistics.
Since OCoM is modular, it is easily extensible to additional forms of variant and library evaluation and constraint, and those are key steps for our future work. For example, rather than a general sequence potential and global design, it could be targeted to exploration of sequence space most affecting activity or stability, or it could be extended to incorporate evaluation of immunogenicity (Parker et al., 2010, 2011). And as mentioned in the introduction, the potential could be derived from initial experiments or from structure-based analysis. Although beyond the scope of this article, prospective application of OCoM in designing libraries for targets of engineering interest is of course the whole motivation of the work.
Acknowledgments
We thank Alan Friedman (Purdue) for helpful discussions on library design. This work was funded in part by the NSF (grant CCF-0915388 to C.B.K.).
Disclosure Statement
No competing financial interests exist.
References
- Bolon D. Mayo S.L. Enzyme-like proteins by computational design. Proc. Natl. Acad. Sci. USA. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell R.C. Joyce G.F. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- Chen C.-Y. Georgiev I. Anderson A.C., et al. Computational structure-based redesign of enzyme activity. Proc. Natl. Acad. Sci. USA. 2009;106:3764–3769. doi: 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox R.J. Davis S.C. Mundorff E.C., et al. Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol. 2007;25:338–344. doi: 10.1038/nbt1286. [DOI] [PubMed] [Google Scholar]
- Fukuda H. Fuiji T. Sukita E., et al. Reconstitution of the isobitene-forming reaction catalyzed by cytochrome p450 and p450 reductase from Rhodotorula minuta: decarboxylation with the formation of isobutene. Biochem. Biophys. Res. Commun. 1994;201:516–522. doi: 10.1006/bbrc.1994.1732. [DOI] [PubMed] [Google Scholar]
- Griswold K.E. Aiyappan N.S. Iverson B. L., et al. The evolution of catalytic efficiency and substrate promiscuity in human theta class 1-1 glutathione transferase. J. Mol. Biol. 2006;364:400–410. doi: 10.1016/j.jmb.2006.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding F.A. Liu A.D. Stickler M., et al. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol. Cancer Ther. 2005;4:1791–1800. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- He L. Friedman A.M. Bailey-Kellogg C. Pareto optimal protein design. Proc. 3dsig Struct. Bioinform. Comput. Biophys. 2010:69–70. [Google Scholar]
- Heim R. Prasher D.C. Tsien R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman A. Tawfik D.S. Incorporating synthetic oligonucleotides via gene reassembly (ISOR): a versatile tool for generating targeted libraries. Protein Eng. Des. Sel. 2007;20:219–226. doi: 10.1093/protein/gzm014. [DOI] [PubMed] [Google Scholar]
- Hiraga K. Arnold F.H. General method for sequence-independent site-directed chimeragenesis. J. Mol. Biol. 2003;330:287–296. doi: 10.1016/s0022-2836(03)00590-4. [DOI] [PubMed] [Google Scholar]
- Huh W. Falvo J.V. Gerke L.C., et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Jackel C. Bloom J.D. Kast P., et al. Consensus protein design without phylogenetic bias. J. Mol. Biol. 2010;399:541–546. doi: 10.1016/j.jmb.2010.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L. Althoff E.A. Clemente F.R., et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Grange D.C. den Haan R. van Zyl W.H. Engineering cellulolytic ability into bioprocessing organisms. Appl. Microbiol. Biotechnol. 2010;87:1195–1208. doi: 10.1007/s00253-010-2660-x. [DOI] [PubMed] [Google Scholar]
- Levin A.M. Murase K. Jackson P.J., et al. Double barrel shotgun scanning of the Caveolin-1 scaffolding domain. ACS Chem. Biol. 2007;2:493–500. doi: 10.1021/cb700055t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A.M. Daugherty P.S. Automated design of degenerate codon libraries. Protein Eng. Des. Sel. 2005;18:559–561. doi: 10.1093/protein/gzi061. [DOI] [PubMed] [Google Scholar]
- Meyer M.M. Hochrein L. Arnold F.H. Structure-guided SCHEMA recombination of distantly related beta-lactamases. Protein Eng. Des. Sel. 2006;19:563–570. doi: 10.1093/protein/gzl045. [DOI] [PubMed] [Google Scholar]
- Meyer M.M. Silberg J.J. Voigt C.A., et al. Library analysis of SCHEMA-guided protein recombination. Protein Sci. 2003;12:1686–1693. doi: 10.1110/ps.0306603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore G.L. Maranas C.D. Identifying residue-residue clashes in protein hybrids by using a second-order mean-field approach. Proc. Natl. Acad. Sci. USA. 2003;100:5091–5096. doi: 10.1073/pnas.0831190100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson A.L. Reichert J.M. Development trends for therapeutic antibody fragments. Nat. Biotech. 2009;27:331–337. doi: 10.1038/nbt0409-331. [DOI] [PubMed] [Google Scholar]
- Otey C.R. Landwehr M. Endelman J.B., et al. Structure-guided recombination creates an artificial family of cytochromes P450. PLoS Biol. 2006;4:e112. doi: 10.1371/journal.pbio.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey C.R. Silberg J.J. Voigt C.A., et al. Functional evolution and structural conservation in chimeric cytochromes P450: calibrating a structure-guided approach. Chem. Biol. 2004;11:309–318. doi: 10.1016/j.chembiol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Pantazes R.J. Saraf M.C. Maranas C.D. Optimal protein library design using recombination or point mutations based on sequence-based scoring functions. Protein Eng. Des. Sel. 2007;20:361–373. doi: 10.1093/protein/gzm030. [DOI] [PubMed] [Google Scholar]
- Parker A.S. Griswold K. Bailey-Kellogg C. Optimization of therapeutic proteins to delete T-cell epitopes while maintaining beneficial residue interactions. J. Bioinform. Comput. Biol. 2011:207–229. doi: 10.1142/s0219720011005471. [DOI] [PubMed] [Google Scholar]
- Parker A.S. Zheng W. Griswold K., et al. Optimization algorithms for functional deimmunization of therapeutic proteins. BMC Bioinform. 2010;11:180. doi: 10.1186/1471-2105-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce N. Winfree E. Protein design is NP-hard. Protein Eng. 2002;15:779–782. doi: 10.1093/protein/15.10.779. [DOI] [PubMed] [Google Scholar]
- Reetz M.T. Carballira J.D. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protocols. 2007;2:891–903. doi: 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- Reetz M.T. Kahakeaw D. Lohmer R. Addressing the numbers problem in directed evolution. ChemBioChem. 2008;9:1797–1804. doi: 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- Russ W.P. Lowery D.M. Mishra P., et al. Natural-like function in artificial WW domains. Nature. 2005;437:579–583. doi: 10.1038/nature03990. [DOI] [PubMed] [Google Scholar]
- Saraf M.C. Gupta A. Maranas C.D. Design of combinatorial protein libraries of optimal size. Proteins. 2005;60:769–777. doi: 10.1002/prot.20490. [DOI] [PubMed] [Google Scholar]
- Saraf M.C. Horswill A.R. Benkovic S.J., et al. FamClash: a method for ranking the activity of engineered enzymes. Proc. Natl. Acad. Sci. USA. 2004;12:4142–4147. doi: 10.1073/pnas.0400065101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboleski M.R. Oaks J. Halford W.P. Green fluorescent protein is a quantitative reporter of gene expression in individual eukaryotic cells. FASEB J. 2005;19:440–442. doi: 10.1096/fj.04-3180fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socolich M. Lockless S.W. Russ W.P., et al. Evolutionary information for specifying a protein fold. Nature. 2005;437:512–518. doi: 10.1038/nature03991. [DOI] [PubMed] [Google Scholar]
- Stemmer W.P.C. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. USA. 1994;91:10747–10751. doi: 10.1073/pnas.91.22.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzman-Engwall K. Conlon S. Fedechko R., et al. Semi-synthetic DNA shuffling of aveC leads to improved industrial scale production of doramectin by Streptomyces avermitilis. Metab. Eng. 2005;7:27–37. doi: 10.1016/j.ymben.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Thomas J. Ramakrishnan N. Bailey-Kellogg C. Graphical models of residue coupling in protein families. IEEE/ACM Trans. Comput. Biol. Bioinform. 2008;5:183–197. doi: 10.1109/TCBB.2007.70225. [DOI] [PubMed] [Google Scholar]
- Thomas J. Ramakrishnan N. Bailey-Kellogg C. Graphical models of protein-protein interaction specificity from correlated mutations and interaction data. Proteins. 2009a;76:911–929. doi: 10.1002/prot.22398. [DOI] [PubMed] [Google Scholar]
- Thomas J. Ramakrishnan N. Bailey-Kellogg C. Protein design by sampling an undirected graphical model of residue constraints. IEEE/ACM Trans. Comput. Biol. Bioinform. 2009b;6:506–516. doi: 10.1109/TCBB.2008.124. [DOI] [PubMed] [Google Scholar]
- Treynor T.P. Vizcarra C.L. Nedelcu D., et al. Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function. Proc. Natl. Acad. Sci. USA. 2007;104:48–53. doi: 10.1073/pnas.0609647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Beucken T. van Neer N. Sablon E., et al. Building novel binding ligands to B7.1 and B7.2 based on human antibody single variable light chain domains. J. Mol. Biol. 2001;310:591–601. doi: 10.1006/jmbi.2001.4703. [DOI] [PubMed] [Google Scholar]
- Voigt C.A. Martinez C. Wang Z.G., et al. Protein building blocks preserved by recombination. Nat. Struct. Biol. 2002;9:553–558. doi: 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- Ye X. Friedman A.M. Bailey-Kellogg C. Hypergraph model of multi-residue interactions in proteins: sequentially-constrained partitioning algorithms for optimization of site-directed protein recombination. J. Comput. Biol. 2007;14:777–790. doi: 10.1089/cmb.2007.R016. [DOI] [PubMed] [Google Scholar]
- Zhang J. Campbell R. Ting A., et al. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- Zhao H. Giver L. Shao Z., et al. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotech. 1998;16:258–261. doi: 10.1038/nbt0398-258. [DOI] [PubMed] [Google Scholar]
- Zheng W. Friedman A.M. Bailey-Kellogg C. Algorithms for joint optimization of stability and diversity in planning combinatorial libraries of chimeric proteins. J. Comput. Biol. 2009;16:1151–1168. doi: 10.1089/cmb.2009.0090. [DOI] [PubMed] [Google Scholar]
- Zheng W. Griswold K.E. Bailey-Kellogg C. Protein fragment swapping: a method for asymmetric, selective site-directed recombination. J. Comput. Biol. 2010;17:459–475. doi: 10.1089/cmb.2009.0189. [DOI] [PubMed] [Google Scholar]
- Zheng W. Ye X. Friedman A.M., et al. Algorithms for selecting breakpoint locations to optimize diversity in protein engineering by site-directed protein recombination. Proc. CSB. 2007:31–40. [PubMed] [Google Scholar]