

Abstract
Background
Besides being a major regulator of the response to acetic acid in Saccharomyces cerevisiae, the transcription factor Haa1 is an important determinant of the tolerance to this acid. The engineering of Haa1 either by overexpression or mutagenesis has therefore been considered to be a promising avenue towards the construction of more robust strains with improved acetic acid tolerance.
Results
By applying the concept of global transcription machinery engineering to the regulon-specific transcription factor Haa1, a mutant allele containing two point mutations could be selected that resulted in a significantly higher acetic acid tolerance as compared to the wild-type allele. The level of improvement obtained was comparable to the level obtained by overexpression of HAA1, which was achieved by introduction of a second copy of the native HAA1 gene. Dissection of the contribution of the two point mutations to the phenotype showed that the major improvement was caused by an amino acid exchange at position 135 (serine to phenylalanine). In order to further study the mechanisms underlying the tolerance phenotype, Haa1 translocation and transcriptional activation of Haa1 target genes was compared between Haa1 mutant, overproduction and wild-type strains. While the rapid Haa1 translocation from the cytosol to the nucleus in response to acetic acid was not affected in the Haa1S135F mutant strain, the levels of transcriptional activation of four selected Haa1-target genes by acetic acid were significantly higher in cells of the mutant strain as compared to cells of the wild-type strain. Interestingly, the time-course of transcriptional activation in response to acetic acid was comparable for the mutant and wild-type strain whereas the maximum mRNA levels obtained correlate with each strain’s tolerance level.
Conclusion
Our data confirms that engineering of the regulon-specific transcription factor Haa1 allows the improvement of acetic acid tolerance in S. cerevisiae. It was also shown that the beneficial S135F mutation identified in the current work did not lead to an increase of HAA1 transcript level, suggesting that an altered protein structure of the Haa1S135F mutant protein led to an increased recruitment of the transcription machinery to Haa1 target genes.
Keywords: Saccharomyces cerevisiae, Acetic acid tolerance, Response and adaptation to acetic acid, Haa1, Transcription factor engineering
Background
The yeast Saccharomyces cerevisiae is a favorite microorganism in industrial biotechnology, mainly due to its robustness under real process conditions, its tolerance to low pH, and its impressive accessibility and versatility for metabolic engineering. Although bioethanol is still the largest-scale product in industrial biotechnology, research and development programs on engineering S. cerevisiae for the production of other valuable compounds are underway, and production processes for a few biofuels, bulk and fine chemicals have already been commercialized. Examples of the latter are isobutanol, farnesene, succinic acid and resveratrol [1].
High tolerance to acetic acid is a favorable phenotype for microorganisms used in industrial biotechnology, in particular in the light of using renewable feedstocks such as lignocellulosic biomass. In fact, acetic acid in lignocellulosic hydrolysates originates from the acetyl groups in hemicelluloses, which are released after the essential pre-treatment step and the subsequent enzymatic breakdown of the polysaccharides into fermentable sugars [2, 3]. At the low pH values usually used in fermentation processes, acetic acid is mainly present in its protonated form, which can freely diffuse across the plasma membrane into the cell. Once inside the cytosol at near-neutral pH, acetic acid dissociates into a proton and the acetate counterion, both accumulating in the cytosol (Fig. 1). The cytosolic acidification together with the induced permeabilization of the plasma membrane is assumed to lead to the dissipation of the proton gradient across the plasma membrane required for secondary transport, thereby inhibiting or completely blocking metabolic activity depending on the level of the stress [4]. Swinnen et al. [5] and Fernandez-Nino et al. [6] have recently shown that only a fraction of cells in a population is able to recover from the stressful conditions evoked by acetic acid and resume proliferation; the size of this fraction depends both on the concentration of the acid and the genetic background of the strain. Notably, most previous studies (including those mentioned in the next paragraphs) have been conducted in comparably low acetic acid concentrations that allowed the majority of cells to resume proliferation after acetic acid exposure but still significantly influenced the duration of the latency phase.
Fig. 1.
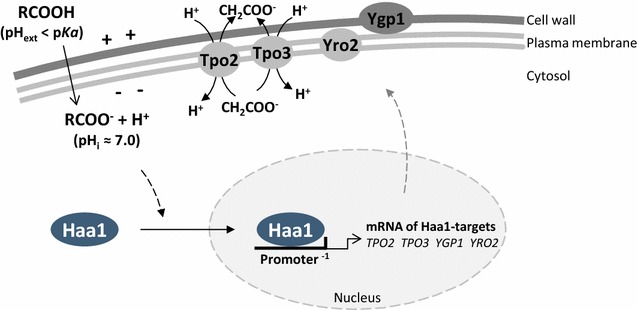
Haa1-induced activation of the four selected target genes within the Haa1-regulon that are relevant in the current study and involved in the adaptation to acetic acid stress. Upon exposure of cells to acetic acid, Haa1 binds to the promoter region of its target genes and thereby regulates their expression. We refer to the background section for more detailed information regarding the function of the depicted target genes
In order to investigate the complex global response of S. cerevisiae to acetic acid exposure, the S. cerevisiae deletion strain collection has been screened to identify single-gene deletions that alter susceptibility to acetic acid [4], and a number of transcriptomic analyses have been conducted in order to study the transcriptional response of this organism to acetic acid exposure [7–13]. In this way, the transcription factor Haa1 has been found to be a major regulator of the yeast’s response to acetic acid. It is involved in the activation of approximately 80% of the acetic acid-responsive genes, which include genes encoding protein kinases, multidrug resistance transporters, proteins involved in lipid metabolism and nucleic acid processing, and proteins of unknown function [7]. Haa1 seems to be also an important determinant of the yeast’s tolerance to acetic acid stress, as the deletion of a significant number of genes of the Haa1-regulon was shown to result in increased susceptibility to acetic acid [7]. Moreover, several recent studies have shown that overexpression of HAA1 improves yeast’s tolerance to high concentrations of acetic and lactic acid [14–17]. Given that the deletion of the HAA1 gene was found to lead to a higher accumulation of labeled acetic acid in stressed cells [18], the role of Haa1 is, at least partially, related with its involvement in the reduction of the intracellular acetate concentration. It is thought that the latter is mediated by Haa1-induced activation of TPO2 and TPO3 genes encoding two drug/H+-antiporters that presumably mediate the active efflux of the acetic acid counterion [18] (Fig. 1).
Haa1 binds the promoter region of its target genes through the recognition of the minimal functional binding motif Haa1-responsive element (HRE) 5′-(G/C)(A/C)GG(G/C)G-3′ [19]. The Haa1/HRE recognition process was examined using a new Quartz Crystal Microbalance (QCM) analytical method and a transmission line model (TLM) algorithm to characterize the mechanical properties of the Haa1/DNA complex and the effect of single point mutations on this recognition process [20, 21]. Among all genes upregulated by acetic acid exposure that are dependent on Haa1 expression, 55% contain the HRE motif in the promoter region, and are considered to be direct targets of Haa1. Among the latter genes are the above-mentioned TPO2 and TPO3 genes encoding two major facilitator superfamily transporters required for multidrug resistance (MFS-MDR), previously involved in polyamine resistance and efflux [22]; the YGP1 gene encoding a cell-wall glycoprotein, expressed under nutrient starvation conditions [23]; and the YRO2 gene encoding a protein homolog to Hsp30 up-regulated upon potassium starvation and required for yeast tolerance to acetic acid stress [24] (Fig. 1). The remaining genes that do not contain the HRE motif in the promoter region are presumed to be indirect targets of Haa1 [7]. In this respect, Haa1 directly regulates the transcription factor encoding genes MSN4, NRG1, FKH2, STP4 and COM2 in the presence of acetic acid, while their respective gene products regulate the expression of several other genes, which are thus Haa1-indirect targets via a complex regulatory network [7].
In response to inhibitory concentrations of lactic acid, Haa1 was found to rapidly migrate from the cytoplasm to the nucleus suggesting that the biological activity of the transcription factor is dependent on its subcellular localization [14]. The Haa1 translocation is accompanied by a decrease in the phosphorylation level of the protein indicating that the transcription factor is activated upon dephosphorylation [14]. The mediated export of Haa1 by the exportin Msn5, found to directly interact with Haa1 in a large-scale yeast two-hybrid screening [25], was confirmed in msn5Δ cells harboring GFP-fused Haa1 [14]. The cells accumulated Haa1 in the nucleus even in the absence of lactic acid, suggesting that Haa1 constantly shuffles between the cytoplasm and the nucleus, being retained in the nucleus upon lactic acid stress [14]. No equivalent information is currently available for Haa1 translocation in response to acetic acid stress.
Inspired by the success of global transcription machinery engineering (gTME) in improving industrially-relevant multifactorial properties of microorganisms [26], we thought about a similar approach for improving acetic acid tolerance of S. cerevisiae but focusing on the regulon-specific transcription factor Haa1 rather than on a global transcription factor. After error-prone PCR of the HAA1 coding sequence, a highly tolerant mutant allele carrying two point mutations was selected for further study of the mechanisms underlying the tolerance phenotype. A strain harboring a second copy of the native HAA1 expression cassette was also constructed and included in the study. We investigated the impact of the modifications on the fraction of cells resuming proliferation and on the mRNA levels from selected Haa1-target genes. We also studied the subcellular translocation of Haa1 wild-type and mutant proteins during early response to acetic acid stress.
Methods
Strains and cultivation conditions
All S. cerevisiae strains used in this study are listed in Table 1. S. cerevisiae cells were routinely maintained on solid YPD medium containing 10 g L−1 yeast extract, 20 g L−1 peptone, 20 g L−1 glucose and 15 g L−1 agar, or on solid synthetic medium containing 5 g L−1 (NH4)2SO4, 3 g L−1 KH2PO4, 0.5 g L−1 MgSO4·7H2O, 20 g L−1 glucose, 20 g L−1 agar and appropriate amounts of trace elements and vitamins according to Verduyn et al. [27]. Acetic acid tolerance assays were performed in synthetic medium containing acetic acid at the indicated concentrations, and pH was adjusted to 4.5 with 4 M KOH. S. cerevisiae cells were routinely cultivated in a static incubator at 30 °C, or in an orbital shaker at 200 rpm and 30 °C.
Table 1.
S. cerevisiae strains used in this study
| Strain name | Genotype | References |
|---|---|---|
| BY4741 haa1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 HAA1::kanMX | [43] |
| CEN.PK113-13D | MATα MAL2-8 c SUC2 ura3-52 | [44] |
| CEN.PK113-13D haa1Δ | MATα MAL2-8 c SUC2 ura3-52 HAA1::kanMX | This study |
| CEN.PK113-7D | MATa MAL2-8 c SUC2 | [44] |
| CEN.PK113-7D Haa1S135F | MATa MAL2-8 c SUC2 HAA1::HAA1 C404T | This study |
| CEN.PK113-7D Haa1Truncated | MATa MAL2-8 c SUC2 HAA1::HAA1 G1447T | This study |
| CEN.PK113-7D Haa1S135F_Truncated | MATa MAL2-8 c SUC2 HAA1::HAA1 C404T_G1447T | This study |
| CEN.PK113-7D Haa1OE | MATa MAL2-8 c SUC2 YGLCτ3::HAA1 | This study |
Escherichia coli cells (DH5α) were cultivated in lysogeny broth (LB) medium containing 10 g L−1 yeast extract, 20 g L−1 peptone and 10 g L−1 NaCl. For solid medium, 15 g L−1 agar was added. E. coli cells were cultivated in a static incubator at 37 °C, or in an orbital shaker at 250 rpm and 37 °C.
General molecular biology methods
Saccharomyces cerevisiae transformations were performed using the lithium acetate method described by Gietz et al. [28], and E. coli transformations using the CaCl2 and heat shock method described by Sambrook and Russell [29]. Genomic DNA was extracted from S. cerevisiae cells using a mixture of phenol, chloroform and isoamyl-alcohol according to Hoffman and Winston [30]. Polymerase chain reaction (PCR) was performed with TaKaRa Ex Taq Polymerase (Merck KGaA, Darmstadt, Germany) for diagnostic purposes, and Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, MA, USA) for genetic manipulation and sequencing purposes. Sequencing was carried out using the dideoxy chain-termination method [31] at GATC Biotech AG (Konstanz, Germany). Sequences were analyzed with Sequencher® version 5.3 sequence analysis software (Gene Codes Corporation, Ann Arbor, USA; available at http://genecodes.com) and SnapGene® software (GSL Biotech; available at http://www.snapgene.com/).
Deletion of HAA1 in strain CEN.PK113-13D
The HAA1 gene was deleted in strain CEN.PK113-13D by using a cassette conferring resistance to G418 as a selectable trait. The deletion cassette was PCR-amplified from genomic DNA of strain BY4741 haa1Δ with primers HAA1_del_fw and HAA1_del_rv (Table 2). The deletion cassette was purified from the reaction mixture by using a PCR purification kit (Qiagen, Hilden, Germany), and subsequently used for transformation of the strain CEN.PK113-13D. Transformants were selected on YD containing 100 mg L−1 G418. The correct integration of the deletion cassette was checked by PCR with primers HAA1_del_control_fw and HAA1_del_control_rv (Table 2).
Table 2.
Primers used in this study
| Primer name | Sequence (5′–3′) |
|---|---|
| HAA1_del_fw | ACAGCACCAGCACTTGATTG |
| HAA1_del_rv | ATGGTCTTACTCTCTGATACC |
| HAA1_del_control_fw | ACAGAGTCGTGCATTTCCAC |
| HAA1_del_control_rv | CCATGAGTGACGACTGAATC |
| HAA1_BamHI_fw | CAGTGGATCCCTCTATGAGAAGAACCCACG |
| HAA1_BamHI_rv | CAGTGGATCCTACACAACAAACTACGCAAGG |
| HAA1_epPCR_fw | AAAAAGGAAACAAAAGTATAGAAAAAAAAAACCTAAAAAATAATG |
| HAA1_epPCR_rv | AAACTACAGTTACAGAGAAGCAAGAGACGAAAAGCAAATTTATCA |
| HAA1_C404T_GIN11_fw | CTGGCACAGAAAGCCAAAGAAGAAGCAAGAGCTAAAGCCAATCGGAACCCTAAAGGGAGC |
| HAA1_G1447T_GIN11_fw | TTTACAGATTCATCGTCGATTTCAACGCTTTCCCGTGCAAATCGGAACCCTAAAGGGAGC |
| HAA1_GIN11_rv | GGTACCAGGAAATGAAAGCG |
| HAA1_kanMX_fw | GGGCTGCAGGAATTCGATATCAAGCTTATCGATACCGTCGCAGCTGAAGCTTCGTACGC |
| HAA1_C404T_kanMX_rv | TCAGCTTTCGCTGGCAAGCTTACCGAACTATCTTGCCAGTGCATAGGCCACTAGTGGATCTG |
| HAA1_G1447T_kanMX_rv | CAGCTAGGTTTGAAGGGTCCATCATCATATTTGCTATCGAGCATAGGCCACTAGTGGATCTG |
| HAA1_control_fw | ACAGCACCAGCACTTGATTG |
| GIN11_rv | CTGAAACGCAGATGTGCCTC |
| HAA1_C404T_fw | TGTGAGAGGTGCATAAGAGG |
| HAA1_C404T_rv | CCAATCGTAGACCAAAGAGC |
| HAA1_G1447T_fw | GCTCTTTGGTCTACGATTGG |
| HAA1_G1447T_rv | TGGTTCATCTCGTCTGGTAC |
| HAA1_OE_GIN11_fw | AGCTTCTTTCCACGAAAGAAATAGTGTAAGTTAGAGGTACATCGGAACCCTAAAGGGAGC |
| HAA1_OE_kanMX_rv | ATTAAGAAGCAAATACCTTCCTGTTGCTTGATTTGCCCTGGCATAGGCCACTAGTGGATCTG |
| HAA1_OE_fw | ATGTAGTTCAACTTCTATGAATGCTCGGCGATACGATATGCTCTATGAGAAGAACCCACG |
| HAA1_OE_rv | AAGGCTCATTTCCATGATGGGGTCACAATTATTATCGCACTACACAACAAACTACGCAAGG |
| HAA1_OE_control_fw | GCAATCTCGACGATCAACTG |
| HAA1_OE_control_rv | AAGAACCAGAATGGCAGGAC |
| HAA1_GFP-NAT_fw | CGATCAAGGATTTGCGGATTTGGATAATTTCATGTCTTCGTTA CGG ATC CCC GGG TTA ATT AA |
| HAA1_GFP-NAT_rv | CTACAGTTACAGAGAAGCAAGAGACGAAAAGCAAATTTA TCAGAATTCGAGCTCGTTTAAAC |
| HAA1_STOP483_GFP-NAT_fw | CTTTGACACCGAGTTTTATGGATATTCCCGAAAAAGAAAGA CGG ATC CCC GGG TTA ATT AA |
| HAA1_STOP483_GFP-NAT_rv | CTGTCAGTAATGTAATTGGATGATGGCGATCTTTCCGTTTA TCA GAATTCGAGCTCGTTTAAAC |
| ACT1_RT_fw | CTCCACCACTGCTGAAAGAGAA |
| ACT1_RT_rv | CCAAGGCGACGTAACATAGTTTT |
| HAA1_RT_fw | TCGTGTGGGCGAAGTTAGC |
| HAA1_RT_rv | CCAACCCCATCAATGTCAGAA |
| TPO2_RT_fw | TGAGTGATCAAGAATCTGTTG |
| TPO2_RT_rv | CGGTACGGTTCAATTGCTTT |
| TPO3_RT_fw | TTGTGACTGGCGATCCAGAA |
| TPO3_RT_rv | ACTCCAACGGATCCATGCA |
| YGP1_RT_fw | TGTACAATGTTGCCCGTGTTG |
| YGP1_RT_rv | GGCACCGGCGGATGA |
| YRO2_RT_fw | TGGATCCCAGTCAGAGCAAAGT |
| YRO2_RT_rv | ACCTGGGTGCTCCTTTTGG |
| FPS1_RT_fw | ATCTGGGCCCACGTCTTG |
| FPS1_RT_rv | ATGCACCCAAAGCATTTTATGA |
Construction of plasmid pRS416-HAA1
The HAA1 gene (containing the HAA1 coding sequence together with 1086 bp upstream and 412 bp downstream of the start and stop codon, respectively) was PCR-amplified from genomic DNA of strain CEN.PK113-13D with primers HAA1_BamHI_fw and HAA1_BamHI_rv (Table 2). Each of the two primers contained at its 3′ terminal end a sequence that is complementary to a region flanking the HAA1 gene, and at its 5′ terminal end the recognition site for the restriction enzyme BamHI. The amplified fragment was purified from the reaction mixture by using a PCR purification kit, and subsequently digested with BamHI (Thermo Fisher Scientific, MA, USA). The HAA1 gene was then ligated into the BamHI site of the dephosphorylated low copy (CEN/ARS) plasmid pRS416 [32] using T4 DNA ligase (Thermo Fisher Scientific, MA, USA). The ligation mixture was used to transform chemically competent E. coli cells, after which transformants were selected on LB medium containing 100 mg L−1 ampicilin. Plasmids were subsequently extracted from E. coli cells by using a commercial miniprep kit (Qiagen, Hilden, Germany). The correct integration of the HAA1 gene into the plasmid pRS416 was checked by restriction analysis, and the HAA1 gene sequence was verified by sequencing.
Construction of the HAA1 mutant library
A library of mutant versions of the HAA1 coding sequence was constructed using the GeneMorph II Random Mutagenesis kit from Stratagene (California, US). At first, mutant alleles of the HAA1 coding sequence were created via error-prone PCR using plasmid pRS416-HAA1 as a template. The forward and reverse primers (HAA1_epPCR_fw and HAA1_epPCR_rv; Table 2) contained at their very 3′ terminal end the start or stop codon of the HAA1 gene, respectively, which implies that mutations were only introduced in the sequences between start and stop codon. The 50-µL PCR mixtures contained 5 µL of Mutazyme II reaction buffer (10×), 1 µL of Mutazyme II DNA polymerase (2.5 U µL−1), 1 µL of both forward and reverse primer (10 pmol µL−1 each primer), 1 µL of dNTP mix (10 mM each dNTP) and 1 µL of template DNA. In total nine PCR mixtures were prepared containing three different amounts of the HAA1 coding sequence (50, 250, and 750 ng) according to the kit’s guidelines. The following cycling parameters were used: 2 min of initial denaturation at 95 °C, and 30 cycles comprising a 30-s denaturation step at 95 °C, a 30-s annealing step at 66 °C, and a 3-min elongation step at 72 °C. The final elongation step was performed at 72 °C for 10 min. After PCR, the amplified fragments were isolated from each of the nine PCR mixtures by extraction from an agarose gel using a gel extraction kit (Qiagen, Hilden, Germany). DNA concentrations were determined using the NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Delaware, USA), after which equal amounts of the individually obtained PCR products were combined.
In order to replace the wild-type HAA1 coding sequence in the plasmid pRS416-HAA1 by the mutant HAA1 alleles, the plasmid was first digested with restriction enzymes EcoNI and BspEI (Thermo Fisher Scientific, MA, USA), which removed most part of the HAA1 coding sequence (EcoNI cuts at 33 bp downstream of the start codon, and BspEI at 122 bp upstream of the stop codon), and subsequently recovered from the restriction mixture by extraction from an agarose gel. Appropriate amounts of the digested pRS416-HAA1 plasmid and PCR products of the mutant HAA1 alleles in a molar ratio of 1–5 (100 and 147 ng of DNA, respectively) were then used to transform strain CEN.PK113-13D haa1Δ. The flanking sequences of the PCR products homologous to the HAA1 promoter and terminator allowed in vivo recombinatorial cloning. The transformation mixtures were afterwards spread on 23 square Petri dishes (120 × 120 mm) with solid synthetic medium. After 4 days of incubation at 30 °C, 2000–5000 single cell colonies were obtained on each plate. A parallel transformation only using the digested plasmid resulted in about 200–300 colonies indicating that less than 1 in 10 colonies reflecting the HAA1 mutant library might contain an insert-less plasmid. To obtain the total HAA1 mutant library the cells from each of the 23 plates were washed off with 1 mL of sterile water, and subsequently mixed together in one tube. Aliquots of 1 mL were mixed with 200 µL of glycerol and stored at −80 °C.
Enrichment of the HAA1 mutant library for alleles that confer improved acetic acid tolerance
For pre-culture, 5 mL of synthetic medium in a glass tube were inoculated with 50 µL of the HAA1 mutant library glycerol stock. The cells were cultivated overnight in an orbital shaker at 200 rpm and 30 °C. The pre-culture was used to inoculate 50 mL of fresh synthetic medium in a shake flask to an optical density (OD600) of 0.2. This culture was cultivated under the same conditions as the pre-culture for 6–8 h until mid-exponential phase was reached (i.e. OD600 between 1.0 and 1.5). An appropriate amount of cells to obtain an OD600 of 0.2 in 50 mL was pelleted by centrifugation (800g for 5 min), and resuspended in 50 mL of synthetic medium containing 200 mM acetic acid at pH 4.5. This culture was cultivated until early stationary phase was reached. At this time point, a glycerol stock of the culture, which is supposed to be enriched in acetic acid tolerant mutants, was prepared and stored at −80 °C. The glycerol stock was used to inoculate a new pre-culture, and the above-described enrichment procedure was repeated for additional three rounds.
Re-transformation of strain CEN.PK113-13D haa1Δ with plasmids isolated from single cell colonies obtained after the enrichment procedure
After the fourth round of enrichment, an aliquot of the cell culture was streaked on solid synthetic medium to obtain single cell colonies. Plasmid DNA was extracted from several of these colonies according to the method described by Singh and Weil [33] with some modifications. In particular, cells originating from a single cell colony were used to inoculate 50 mL of synthetic medium, and subsequently cultivated overnight in an orbital shaker. The cells were then pelleted by centrifugation at 1811g for 5 min, and resuspended in 1 mL of P1 buffer from the miniprep kit. The cell suspension was transferred to a microcentrifuge tube, and approximately 1 g of acid-washed glass beads (diameter of 425–600 µm) was added. The cells were subsequently lysed by vortexing the tube vigorously for 5 min. After the glass beads had settled, 250 µL of the cell lysate were transferred to a new microcentrifuge tube, and further treated according to the guidelines given in the kit’s manual. The plasmid DNA was afterwards eluted from the miniprep column with 100 µL of water, and used to transform strain CEN.PK113-13D haa1Δ.
Targeted introduction of single nucleotide mutations into the genome
Introduction of the single nucleotide mutations C404T and G1447T in the native HAA1 gene of strain CEN.PK113-7D was achieved in two steps using markers for selection and subsequent counterselection. In the first step, a part of the HAA1 gene (containing the locus with the mutation to be introduced) was replaced by a cassette containing the kanMX gene and a galactose-inducible growth inhibitory sequence (referred to as GALp-GIN11M86). The GALp-GIN11M86 sequence was amplified from plasmid pGG119 [34] by PCR using primers HAA1_(mutation)_GIN11_fw and HAA1_GIN11_rv (Table 2), while the kanMX gene was amplified from plasmid pUG6 using primers HAA1_kanMX_fw and HAA1_(mutation)_kanMX_rv (Table 2). Primers HAA1_GIN11_rv and HAA1_kanMX_fw were designed to result in a complementary sequence between the two generated PCR products downstream of the GALp-GIN11M86 sequence and upstream of the kanMX gene, so that both cassettes could be assembled by homologous recombination upon co-transformation. Primers HAA1_(mutation)_GIN11_fw and HAA1_(mutation)_kanMX_rv contained at their 5′ terminal ends 40-bp sequences complementary to regions upstream and downstream of the HAA1 sequence to be exchanged in the genome. Both PCRs were performed using Phusion High-Fidelity DNA Polymerase according to the manufacturer’s guidelines. After purification of the PCR products, strain CEN.PK113-7D was co-transformed with equimolar amounts of each product, and transformants were selected on YD medium containing 100 mg L−1 G418. The correct integration of the cassette was verified by PCR using primers HAA1_control_fw and GIN11_rv (Table 2).
In the second step, the kanMX/GALp-GIN11M86 cassette in strain CEN.PK113-7D was removed by transformation of the strain with a PCR product containing the HAA1 allele with the respective mutation, flanked by sequences homologous to regions upstream and downstream of the chromosomal position of the kanMX/GALp-GIN11M86 cassette. The PCR was performed with Phusion High-Fidelity DNA Polymerase using primers HAA1_(mutation)_fw and HAA1_(mutation)_rv (Table 2) and plasmid pRS416-HAA1 C404T_G1447T as a template. After transformation, cells were plated on solid synthetic medium containing 2% galactose as the sole carbon source for induction of the growth inhibitory sequence GIN11M86. Correct integration of the single nucleotide mutations into the genome was verified by PCR and sequencing.
Overexpression of HAA1 in strain CEN.PK113-7D
Overexpression of the HAA1 gene in strain CEN.PK113-7D was achieved by integration of an additional CEN.PK113-7D wild-type HAA1 allele (including native promoter and terminator) at the YGLCτ3 site on chromosome VII (integration site 8 according to Flagfeldt et al. [35]). The method used was similar to the one described for the introduction of single nucleotide mutations into the genome; only the primers used were different. For the first step, the GALp-GIN11M86 sequence was amplified using primers HAA1_OE_GIN11_fw and HAA1_GIN11_rv (Table 2), while the kanMX gene was amplified using primers HAA1_kanMX_fw and HAA1_OE_kanMX_rv (Table 2). For the second step, the HAA1 allele was amplified from genomic DNA of strain CEN.PK113-7D using primers HAA1_OE_fw and HAA1_OE_rv (Table 2). Correct integration of the gene was verified by PCR using primers HAA1_OE_control_fw and HAA1_OE_control_rv (Table 2).
Quantitative analysis of acetic acid tolerance in liquid medium using the Growth Profiler 1152, and on solid medium
For pre-culture, 3 mL of synthetic medium were inoculated with cells originating from a single cell colony on plate. The cells were cultivated overnight in an orbital shaker at 200 rpm and 30 °C. The pre-culture was used to inoculate 3 mL of fresh synthetic medium to an OD600 of 0.2. This culture (referred to as the intermediate culture) was cultivated under the same conditions as the pre-culture until mid-exponential phase was reached (i.e. OD600 between 1.0 and 1.5). For acetic acid tolerance assays in liquid medium, an appropriate amount of cells from the intermediate culture to obtain an OD600 of 0.2 in 5 mL was collected by centrifugation (800g for 5 min), and subsequently resuspended in 5 mL of synthetic medium either with or without acetic acid (pH 4.5). An aliquot of 750 µL from the latter culture was transferred immediately into a well of a white Krystal 24-well clear bottom microplate (Porvair Sciences, Leatherhead, UK). Growth was recorded using the Growth Profiler 1152 (Enzyscreen, Haarlem, The Netherlands) as previously described [5].
For acetic acid tolerance assays on solid medium, an appropriate amount of cells from the intermediate culture to obtain an OD600 of 0.2 in 1 mL was collected by centrifugation (800g for 5 min), and subsequently resuspended in 1 mL of synthetic medium without acetic acid and without glucose. This cell suspension was then serially diluted in the same medium to obtain dilutions of 10−1 to 10−4. An aliquot of 250 µL of each dilution was spread on solid synthetic medium either with or without acetic acid (pH 4.5). Plates were then incubated in a static incubator at 30 °C. The incubation time was 2 days for the medium without acetic acid, and 4 days for the medium with acetic acid. Dilutions resulting in colony forming units (CFU) in the range of 50–150 per plate were included for counting.
Determination of the transcription profiles of HAA1 and Haa1-regulated genes in cells exposed to acetic acid
Real-time RT-PCR was performed to determine the mRNA levels from HAA1 and Haa1-target genes TPO2, TPO3, YGP1 and YRO2 in cells of strain CEN.PK113-7D expressing either wild-type or mutant Haa1 proteins immediately before and at different time points after exposure to acetic acid. The FPS1 gene was included as a negative control since its regulation is Haa1-independent, and the ACT1 gene was included as an internal control. Cells of each strain were used to inoculate 30 mL of synthetic medium, and subsequently cultivated overnight in an orbital shaker at 200 rpm and 30 °C. This pre-culture was used to inoculate 110 mL of fresh synthetic medium, after which the culture was cultivated until an OD600 of 1.0–1.5 was reached. Cells from an adequate volume of culture were then collected by filtration and resuspended in 200 mL of fresh synthetic medium in order to obtain an OD600 of 0.5. After 20 min of incubation in an orbital shaker at 200 rpm and 30 °C, cells of 20 mL of the culture were collected by centrifugation (5000g for 3 min) at 4 °C, after which the cell pellet was frozen immediately in liquid nitrogen and stored at −80 °C. In parallel, acetic acid was added to the remaining culture to a final concentration of 50 mM, and cells of 20 mL of the culture were collected after 30, 60 and 120 min using the same protocol as described for the non-stressed cells. Total RNA was extracted from the frozen cell pellets by the hot phenol method [36], and treated with DNAseI (Invitrogen, Carlsbad, USA) according to the manufacture’s guidelines. In total 1 µg of the treated RNA was used in the reverse transcription step using Taqman® reverse transcription reagents (Applied Biosystems, Branchburg, USA), and 62.5 ng of the synthesized cDNA were used in the PCR amplification step using Power SYBR® Master Mix reagents (Applied Biosystems, Warrington, UK). Primers used for the amplification of the selected cDNAs were designed using the Primer Express Software (Applied Biosystems, Foster City, USA) and are listed in Table 2. The mRNA level of ACT1 was used for normalization of the mRNA levels of all other genes. For each of the latter genes, the mRNA levels shown are relative to the level registered in Haa1 wild-type cells cultivated in the absence of acetic acid, which was set as 1.
Subcellular localization of Haa1 in non-stressed and acetic acid stressed cells
Fusion of the green fluorescent protein (GFP) to the C-terminus of Haa1 was achieved by insertion of the GFP(S65T)-ADH1terminator-natMX6 cassette directly upstream of the stop codon of the HAA1 sequence. The cassette was PCR-amplified from plasmid pFA6a-GFP(S65T)-natMX6 [37] using primers HAA1_GFP-NAT_fw and HAA1_GFP-NAT_rv (Table 2) for insertion of the cassette downstream of the wild-type HAA1 or mutant HAA1 C404T allele, or primers HAA1_STOP483_GFP-NAT_fw and HAA1_STOP483_GFP-NAT_rv (Table 2) for insertion of the cassette downstream of the mutant HAA1 G1447T allele, which contains a premature stop codon. The cassettes were purified from the reaction mixtures, and subsequently used for transformation of the respective wild-type and mutant CEN.PK113-7D strains. Transformants were selected on solid YPD medium containing 100 µg mL−1 nourseothricin, after which the correct integration of the GFP(S65T)-ADH1terminator-natMX6 cassette was verified by sequencing. Notably, in the CEN.PK113-7D strain overexpressing HAA1, only one if the two alleles were fused with the GFP(S65T)-ADH1terminator-natMX6 cassette.
Cells expressing Haa1-GFP(S65T) fusions were cultivated in the same way as cells used for the determination of the transcription profiles of HAA1 and Haa1-regulated genes. For nuclear staining of the cells, 5 µL of 4′,6-diamidino-2-phenylindole (DAPI, 1 mg mL−1) was added to 1 mL of cell suspension. The cells were subsequently incubated for 2 min at room temperature, collected by centrifugation (5000g for 30 s), resuspended in about 50 µL of supernatant, and visualized using fluorescence microscopy. Fluorescence images were captured with a cooled CCD camera (Cool SNAPFX, Roper Scientific Photometrics, Tucson, USA) coupled to a Zeiss Axioplan microscope (Carl Zeiss MicroImaging GmbH, Oberkochen, Germany) using excitation and emission filters of 365/12 and 397 nm to detect DAPI fluorescence signal, and 450–490 and 515 nm to detect GFP signal. ImageJ software was used to overlay the images.
Results
Identification of mutant HAA1 alleles that improve acetic acid tolerance
The first part of this study aimed at generating a plasmid-based library of mutant HAA1 alleles, and isolating alleles able to improve the acetic acid tolerance of S. cerevisiae strain CEN.PK113-13D haa1Δ in comparison to the wild-type HAA1 allele (HAA1 WT). Briefly, mutant alleles of the HAA1 coding sequence were created by error-prone PCR, after which the PCR products were placed between the native HAA1 promoter and terminator in the backbone of the low-copy (CEN/ARS) pRS416 plasmid by recombination-based cloning in the strain CEN.PK113-13D haa1Δ. The transformation resulted in more than 80,000 clones. Sequencing of plasmids isolated from 15 randomly selected clones revealed a mutation frequency between 2 and 10 non-synonymous mutations within the HAA1 coding sequence. In order to select clones with improved acetic acid tolerance, two parallel enrichments of the library were conducted by serial batch cultivations of the cells in the presence of a relatively high concentration of acetic acid (200 mM at pH 4.5). In between each round of enrichment, the cells were brought back to non-stress conditions as described in the “Methods” section. As a control, the strain CEN.PK113-13D haa1Δ expressing HAA1 WT from the same plasmid backbone was included in the enrichment experiment. After four rounds of enrichment, all cultures showed a clear improvement in acetic acid tolerance, however, the improvement was more pronounced for the library cultures than for the control culture (Fig. 2a). Aliquots from the two enriched library cultures were then streaked for single cell colonies, after which plasmid DNA was isolated from 9 clones from each library culture and introduced into a fresh CEN.PK113-13D haa1Δ background. Out of the 18 resulting strains, 11 showed a growth performance comparable to the enriched library cultures (data not shown), implying that the improved acetic acid tolerance of strain CEN.PK113-13D haa1Δ was truly caused by the expression of the mutant HAA1 alleles. In fact, the originally isolated clones could have obtained mutations that have naturally arisen in the genomic DNA during the enrichment procedure. Sequencing of the 11 plasmids that improved acetic acid tolerance revealed five different HAA1 alleles (Fig. 2b, c).
Fig. 2.
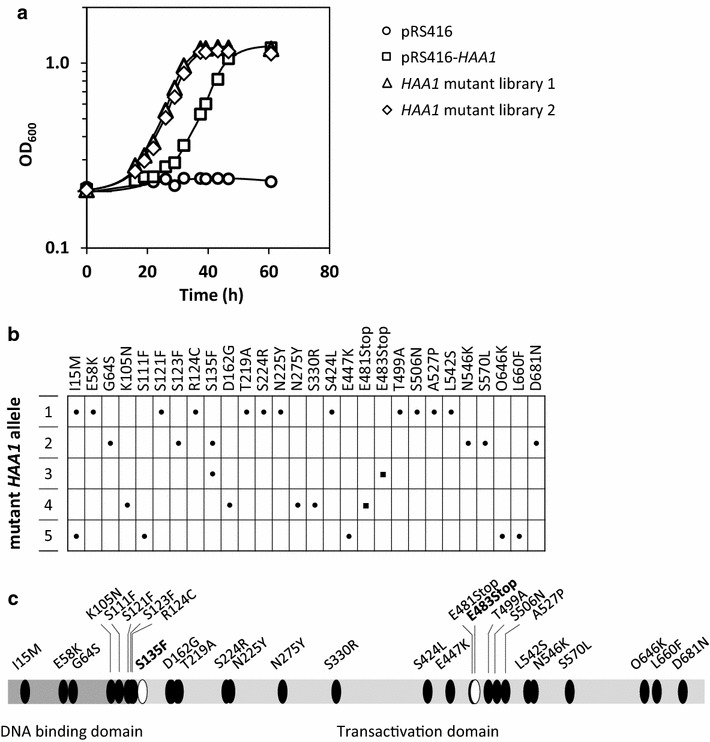
Selection of mutant HAA1 alleles with improved tolerance to acetic acid. a Growth performance of two parallel cultures of an HAA1 mutant library during the fourth round of enrichment in synthetic medium containing 200 mM acetic acid (pH 4.5) is shown. First, a CEN/ARS plasmid-based HAA1 mutant library was generated by means of error-prone PCR and recombinatorial cloning in the S. cerevisiae strain CEN.PK113-13D haa1Δ. Two aliquots of the library were then cultivated under non-stress conditions until exponential growth phase, after which the cells were transferred to synthetic medium containing 200 mM acetic acid (pH 4.5). This procedure of alternating non-stress and acetic acid stress conditions was repeated four times. For comparison, cells of an isogenic strain containing the wild-type HAA1 allele cloned in the same plasmid backbone (pRS416-HAA1) were subjected to the same enrichment procedure, and the growth performance in acetic acid containing medium during the fourth round of enrichment is shown as well. The strain CEN.PK113-13D haa1Δ containing the empty plasmid (pRS416) was always included as a control. b Modifications in protein sequence deduced from five non-redundant mutant HAA1 alleles isolated from the enriched library cultures that significantly improved acetic acid tolerance. c Relative position of the identified mutations in the Haa1 DNA binding and transactivation domains
Detailed characterization of the strain CEN.PK113-7D Haa1S135F_Truncated and dissection of each mutation’s contribution to the phenotype
Out of the five mutant HAA1 alleles shown in Fig. 2b, the allele with the lowest number of mutations was selected for further study. This allele contained a cytosine to thymine mutation at position 404 (resulting in a serine to phenylalanine exchange at position 135 in the protein), and a guanine to thymine mutation at position 1447 (resulting in a truncation of the protein from position 483 onwards).
In order to rule out any potential side effects of the episomal expression of HAA1 as well as of the auxotrophy of the used strain, the HAA1 C404T_G1447T allele was used to replace the native HAA1 coding sequence in the genome of the prototrophic strain CEN.PK113-7D (hereafter referred to as strain CEN.PK113-7D Haa1S135F_Truncated). The allele swapping was conducted in a seamless way, meaning that no markers or foreign sequences were left behind. For comparative analyses, a derivative of strain CEN.PK113-7D with a second copy of the native HAA1 gene was also constructed (hereafter referred to as strain CEN.PK.113-7D Haa1OE). The acetic acid tolerance of strains CEN.PK113-7D Haa1S135F_Truncated and Haa1OE as compared to strain CEN.PK113-7D Haa1WT was first evaluated by quantifying the effect of acetic acid on the maximum specific growth rate and duration of the latency phase. As shown in Fig. 3a, b, strains CEN.PK113-7D Haa1S135F_Truncated and Haa1OE did not show a significantly improved growth rate but do show a shorter latency phase in the presence of 160 mM acetic acid (pH 4.5) as compared to strain CEN.PK113-7D Haa1WT. Our data also shows that the expression of HAA1 S135F_Truncated led to a slightly (reproducible but not significantly) higher acetic acid tolerance than the overexpression of HAA1. In addition, the fraction of cells that resume proliferation in the presence of acetic acid was quantified for each strain by counting colony forming units on solid acetic acid containing medium. This method has been previously demonstrated to properly represent the fraction of cells able to resume proliferation in liquid acetic acid containing medium [5]. Consistent with the theory and in congruence with the duration of the latency phases, both strains CEN.PK113-7D Haa1S135F_Truncated and CEN.PK113-7D Haa1OE showed a significantly higher fraction of proliferating cells in the presence of 160 mM acetic acid as compared to strain CEN.PK113-7D Haa1WT (Fig. 3c).
Fig. 3.
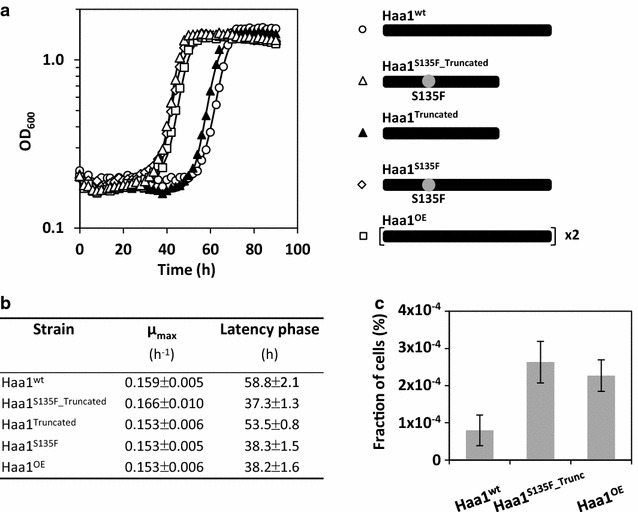
Dissection of the phenotypic contributions of each individual mutation in the identified HAA1 mutant allele HAA1 C404T_G1447T (protein Haa1S135F_Truncated). The effects of the modified proteins were characterized after allele swapping in strain CEN.PK113-7D, respectively. a Growth curves, b maximum specific growth rates and duration of latency phases, and c fraction of cells that resume proliferation are shown after shifting exponentially growing cells from medium without acetic acid to medium with 160 mM acetic acid at pH 4.5 are shown. The isogenic wild type CEN.PK113-7D (Haa1WT) and the strain containing a second copy of the endogenous HAA1 expression cassette (Haa1OE) were also included in these experiments. Mean values and standard deviations of at least three biological replicates are shown
After showing the effect of the expression of the HAA1 C404T_G1447T allele on the acetic acid tolerance of strain CEN.PK113-7D, the effect of the two individual point mutations was studied in more detail. For this purpose, the two mutations were separately introduced into the genome of strain CEN.PK113-7D, generating strains CEN.PK113-7D Haa1S135F and CEN.PK113-7D Haa1Truncated. Our data shows that both mutations have an additive effect on the acetic acid tolerance of strain CEN.PK113-7D (Fig. 3a, b); even though the amino acid exchange at position 135 exerts a by far more severe impact than the Haa1 protein truncation. In fact, the latency phase of strain CEN.PK113-7D Haa1S135F was only slightly reduced in the presence of 160 mM acetic acid as compared to strain CEN.PK113-7D Haa1S135F_Truncated (Fig. 3a, b).
We next sought to understand the mechanisms underlying the differences in acetic acid tolerance between the Haa1 wild-type, mutant and overproduction strains. At high acetic acid concentrations such as those used in the first part of the current work, molecular studies at the population level are hampered since only a few cells in the population resume proliferation while the remaining cells arrest growth [5, 6]. In order to rule out the effect of cell-to-cell heterogeneity, we significantly reduced the stress level to 50 mM acetic acid at pH 4.5. At this condition, all cells of strain CEN.PK113-7D have been demonstrated to resume proliferation [6], whereas differences between the strains with regard to the duration of the latency phase are still apparent (although to a lesser extent) (Fig. 5).
Fig. 5.
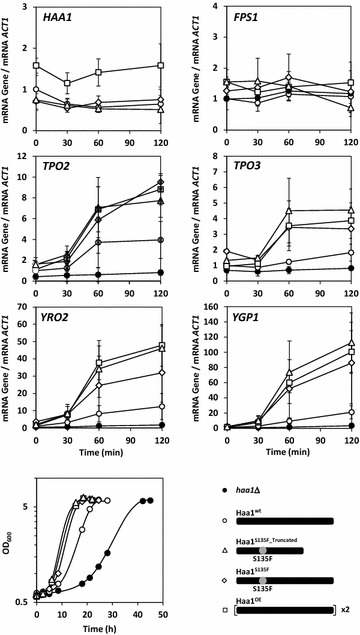
Effect of the different Haa1 modifications on the time courses of transcription levels of the HAA1 gene, four genes belonging to the Haa1-regulon (TPO2, TPO3, YRO2 and YGP1), and a reference gene (FPS1) whose expression has been shown to be not affected by acetic acid. Cells were cultivated in synthetic medium without acetic acid until exponential phase (time point 0) and then transferred to synthetic medium containing 50 mM acetic acid (pH 4.5). Immediately before and 30, 60 and 120 min after exposure to acetic acid, aliquots of the cell suspension were taken to determine the respective mRNA levels by real-time RT-PCR. Values represent the average of at least three independent experiments, and ACT1 mRNA level was used as an endogenous control. The mRNA level measured in cells of the wild-type CEN.PK113-7D strain (Haa1WT) cultivated in the absence of acetic acid was set as 1 and used to normalize the mRNA levels determined in the other strains and conditions. The corresponding growth curves recorded by OD600 measurement are also shown
Both Haa1 wild-type and mutant proteins translocate from the cytosol to the nucleus in response to acetic acid-induced stress
In a previous study it was shown that Haa1 translocates from the cytosol to the nucleus upon exposure to lactic acid [14]. In the current study it was examined whether similar Haa1 translocation also occurs upon exposure to acetic acid, and whether this translocation is affected by the different mutations. A GFP-tag was therefore inserted downstream of the wild-type and mutant HAA1 coding sequences within the genome of the Haa1 wild-type, mutant and overproduction strains, respectively. Afterwards, the localization of Haa1 was studied in cells of all strains cultivated in the absence and presence of 50 mM acetic acid at pH 4.5. In all tested strains, Haa1 was translocated from the cytosol to the nucleus upon acetic acid exposure, and this translocation was already apparent after 5 min (Fig. 4).
Fig. 4.
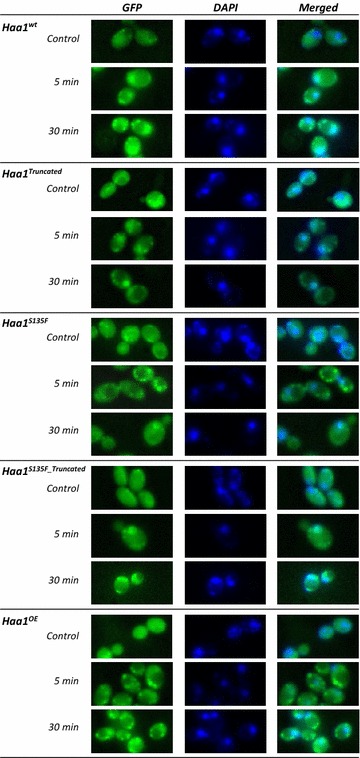
Subcellular localization of Haa1 before and after exposure of the cells to acetic acid. Results are shown for the wild-type CEN.PK113-7D strain (Haa1WT) and the Haa1S135F_Truncated, Haa1Truncated, Haa1S135F and Haa1OE CEN.PK113-7D strains. Haa1 localization was determined in cells immediately before and 5 and 30 min after the transfer to medium containing 50 mM acetic acid (pH 4.5). Cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) to label nuclear DNA. Visualization was performed using fluorescence microscopy to detect DAPI and GFP fluorescence signals. ImageJ software was used to overlay the images
Expression of the mutant HAA1 alleles increases transcript levels from selected Haa1-target genes
While overexpression of HAA1 was expected to result in an increase of HAA1 transcript level, the question arose as to whether expression of the mutant HAA1 alleles would also modify HAA1 mRNA abundance, for example, by affecting the stability of the corresponding mRNAs. For this purpose, HAA1 transcript levels were determined in Haa1 wild-type, mutant and overproduction strains before and at different time points up to 2 h after exposure of the cells to 50 mM acetic acid (pH 4.5). Our data shows similar HAA1 transcript levels in the Haa1 wild-type and mutant strains, while an about 1.5-fold increase was shown in the Haa1 overproduction strain (Fig. 5). No detectable fluctuation in HAA1 transcript levels was found in any of the Haa1 mutant strains as compared to the Haa1 wild-type strain during a period of up to 2 h after exposure of the cells to acetic acid.
In order to investigate whether the expression of the mutant HAA1 alleles as well as the overexpression of HAA1 result in an increased transcription of acetic acid-responsive genes of the Haa1-regulon, the transcript levels from four of these genes (TPO2, TPO3, YRO2, and YGP1) [7, 19] were determined during the first 2 h following sudden exposure to acetic acid (Fig. 5). FPS1 was used as a negative control in the experiment, given that the gene is a determinant of S. cerevisiae susceptibility to acetic acid [38] and that its expression has been shown to be independent of Haa1 [15]. The latter is supported by our data, which shows that FPS1 transcript levels are maintained constant during acetic acid exposure and no significant differences are detected among the strains (Fig. 5). With regard to the genes TPO2, TPO3, YRO2, and YGP1, transcript levels were clearly increased in strain CEN.PK113-7D Haa1OE, correlating with the increased HAA1 mRNA abundance in this strain; however, a similar increase was also observed in strains CEN.PK113-7D Haa1S135F_Truncated and Haa1S135F (Fig. 5). It is also noteworthy that no detectable strain-specific differences in the low basal transcription level of the respective target genes before acetic acid addition were found, and that no significant transcriptional activation of the four selected Haa1-target genes was registered in the haa1 deletion mutant (Fig. 5). Remarkably, the level of transcriptional activation from all selected Haa1-target genes by Haa1 wild-type and mutant proteins correlates with the level of acetic acid tolerance exhibited by the corresponding strains (Fig. 5).
Discussion
Previous studies have shown that the acetic acid tolerance of S. cerevisiae strains S288c and Ethanol Red can be improved by exchanging the native HAA1 promoter for the strong and constitutive TDH3 promoter [15, 16, 39]. This overexpression of HAA1 was shown to result in an increased transcript level of Haa1-target genes TPO2, TPO3, YRO2 and YGP1 even in the absence of acetic acid [15], implying that the higher Haa1 protein level resulted in a constitutive activation of the Haa1-regulon. It is assumed that the latter better arms the cells once they are exposed to the acid. In the current study, overexpression of HAA1 was achieved by introducing an additional copy of the HAA1 coding sequence under the control of the endogenous HAA1 promoter and terminator. This engineered strain showed an about 1.5-fold increase in HAA1 transcript level in unstressed cells, which is in clear contrast to the Haa1 overproduction strain constructed by Inaba et al. [16] that showed a sevenfold increase. The strain constructed in the current study did also not show an increase in the expression level of tested Haa1-target genes in the absence of acetic acid which is in contrast to what has been observed in the study of Tanaka et al. [15]. Although it remains to be clarified whether an even higher expression of HAA1 in the CEN.PK background could further increase its acetic acid tolerance, we assume that a higher expression rather represents a burden to the cells. In fact, the studies of Tanaka et al. [15] (2.5-fold increase) and Inaba et al. [16] (sevenfold increase) recognized a reduction in growth under non-stress conditions, which would be a disadvantage for industrial applications and suggests a serious limitation of cell endurance against acetic acid. Notably, the Haa1 overproduction strain constructed in the current study did not show any growth deficit in the absence of acetic acid (data not shown). It was also interesting to see that the Haa1 overproduction strain constructed in the current study showed a higher fraction of cells resuming proliferation in the presence of acetic acid as compared to the wild-type strain. To the best of our knowledge, this has been the first time that a positive impact of an engineered Haa1 variant or of an increased cellular Haa1 abundance on this parameter has been measured. In order to understand this phenomenon at the molecular level, respective studies at the single-cell level are envisaged.
The isolation of mutant HAA1 alleles that significantly improve acetic acid tolerance demonstrates the success of engineering a regulon-specific transcription factor to improve a complex phenotype that is controlled by this regulon. In fact, the strains CEN.PK113-7D Haa1S135F and CEN.PK113-7D Haa1S135F_Truncated showed an acetic acid tolerance that was similar or even slightly higher than the strain CEN.PK113-7D Haa1OE. Interestingly, in contrast to the Haa1 overproduction strain, the Haa1 mutant strains showed an improved acetic acid tolerance that was not concomitant with an increased HAA1 transcript level. Hence, the question arises why the different genetic modifications and underlying molecular mechanisms observed in the Haa1 mutant and overproduction strains can result in the same tolerance phenotype. In principle it is possible that a higher stability of the mutant Haa1 proteins, in particular of the truncated variant, may underlie their increased activity. We tried to test this hypothesis by quantitative immunodetection of Haa1 proteins using the constructed Haa1-GFP fusion proteins and antibodies against GFP (data not shown). However, the naturally low cellular levels of the transcription factor did not allow an unambiguous quantification of the different protein variants.
In addition to an altered protein stability, it is also conceivable that the mutant Haa1 proteins may exhibit different structures that improve their ability to recruit the transcription machinery to the target genes of the Haa1-regulon. According to the PhosphoGRID database, the critical point mutation identified in our study (S135F) substitutes a serine residue located within a putative kinase target site [40]. It is likely that this mutation influences the phosphorylation level and consequently biological activity of the Haa1 protein, considering that the position of this residue is relatively close to the DNA-binding domain.
It is also noteworthy that the loss of one-third of the entire Haa1 sequence (the last 212 C-terminal residues) leads to a small, although significant increase of the protein biological activity. Remarkably, a recent study identified acetic-acid tolerant mutant HAA1 alleles that exhibit multiple mutations in the transactivation domain, mainly in the end of the C-terminal region (mutant 2: F440Y, P518S, I591V, H605Y, S622F, S639F, S673L; and mutant 40: D508Y, N510K, A527V, N554Stop) [41], while another study identified the single mutation S506N as contributing to the difference in acetic acid tolerance between a strain with unusually high tolerance and an industrial reference strain [42]. With the exception of the F440Y mutation, all identified mutations are located in the sequence that was lost in the Haa1Truncated protein identified in the current study. Although little is known about the structure of the Haa1 transactivation domain, our results suggest that the C-terminal region of the Haa1 sequence may harbor domains involved in the negative regulation of the protein’s activity. Nevertheless, full understanding of the mechanisms underlying the different biological activity of the mutant HAA1 alleles identified will require additional work, including work on the structural characterization of the protein-DNA complexes.
Conclusion
In the current study, mutant HAA1 alleles have been identified that improve the acetic acid tolerance of the strain CEN.PK113-7D, representing a prototrophic version of the industrially-relevant S. cerevisiae CEN.PK family. Initial investigations with regard to the biochemical mechanisms underlying the improved phenotype suggests that the alleles encode proteins that are more active compared to the wild-type protein. The mutant HAA1 alleles could be used for the construction of robust strains for biotechnological processes using lignocellulosic hydrolysates as feedstock, particularly since the reverse engineered strains did not show any impairment with regard to growth under non-stress conditions. The alleles may also inspire further studies to better understand the underlying molecular basis for the increased activity of the respective proteins in inducing the Haa1-regulon.
Authors’ contributions
SS, SFH, ISC and EN conceived and designed the experiments. SS, SFH, RS and PWH carried out the experimental work. SS, SFH, ISC and EN wrote and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Martina Carrillo for technical assistance.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was developed in the framework of the Era-Net Industrial Biotechnology, research project INTACT-“Integral Engineering of Acetic Acid Tolerance in Yeast” funded in Portugal by FCT (Fundação para a Ciência e a Tecnologia) (ERA-IB/0002/2010) and in Germany by the German Federal Ministry of Education and Research (Project No. 0315933). ISC acknowledges funding received through FCT/MEC (PIDDAC) (Project PTDC/BBB-BEP/0385/2014) and by iBB-Institute for Bioengineering and Biosciences from Programa Operacional Regional de Lisboa 2020 (N. 007317) and from FCT (UID/BIO/04565/2013). SFH is the recipient of a Ph.D. fellowship (SFRH/BD/78058/2011) funded by FCT.
Footnotes
Steve Swinnen and Sílvia F. Henriques contributed equally to this work
Contributor Information
Steve Swinnen, Email: steve.swinnen@globalyeast.com.
Sílvia F. Henriques, Email: silvia.henriques@tecnico.ulisboa.pt
Ranjan Shrestha, Email: ranjansth@gmail.com.
Ping-Wei Ho, Email: p.ho@jacobs-university.de.
Isabel Sá-Correia, Phone: +351-218417682, Email: isacorreia@tecnico.ulisboa.pt.
Elke Nevoigt, Phone: +49-421-2003541, Email: e.nevoigt@jacobs-university.de.
References
- 1.Borodina I, Nielsen J. Advances in metabolic engineering of yeast Saccharomyces cerevisiae for production of chemicals. Biotechnol J. 2014;9:609–620. doi: 10.1002/biot.201300445. [DOI] [PubMed] [Google Scholar]
- 2.Jonsson LJ, Alriksson B, Nilvebrant NO. Bioconversion of lignocellulose: inhibitors and detoxification. Biotechnol Biofuels. 2013;6:16. doi: 10.1186/1754-6834-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmqvist E, Hahn-Hagerdal B. Fermentation of lignocellulosic hydrolysates. II: inhibitors and mechanisms of inhibition. Bioresour Technol. 2000;74:25–33. doi: 10.1016/S0960-8524(99)00161-3. [DOI] [Google Scholar]
- 4.Mira NP, Palma M, Guerreiro JF, Sa-Correia I. Genome-wide identification of Saccharomyces cerevisiae genes required for tolerance to acetic acid. Microb Cell Fact. 2010;9:79. doi: 10.1186/1475-2859-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swinnen S, Fernandez-Nino M, Gonzalez-Ramos D, van Maris AJ, Nevoigt E. The fraction of cells that resume growth after acetic acid addition is a strain-dependent parameter of acetic acid tolerance in Saccharomyces cerevisiae. FEMS Yeast Res. 2014;14:642–653. doi: 10.1111/1567-1364.12151. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-Nino M, Marquina M, Swinnen S, Rodriguez-Porrata B, Nevoigt E, Arino J. The cytosolic pH of individual Saccharomyces cerevisiae cells is a key factor in acetic acid tolerance. Appl Environ Microbiol. 2015;81:7813–7821. doi: 10.1128/AEM.02313-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mira NP, Becker JD, Sa-Correia I. Genomic expression program involving the Haa1p-regulon in Saccharomyces cerevisiae response to acetic acid. OMICS. 2010;14:587–601. doi: 10.1089/omi.2010.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawahata M, Masaki K, Fujii T, Iefuji H. Yeast genes involved in response to lactic acid and acetic acid: acidic conditions caused by the organic acids in Saccharomyces cerevisiae cultures induce expression of intracellular metal metabolism genes regulated by Aft1p. FEMS Yeast Res. 2006;6:924–936. doi: 10.1111/j.1567-1364.2006.00089.x. [DOI] [PubMed] [Google Scholar]
- 9.Li BZ, Yuan YJ. Transcriptome shifts in response to furfural and acetic acid in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2010;86:1915–1924. doi: 10.1007/s00253-010-2518-2. [DOI] [PubMed] [Google Scholar]
- 10.Abbott DA, Knijnenburg TA, de Poorter LM, Reinders MJ, Pronk JT, van Maris AJ. Generic and specific transcriptional responses to different weak organic acids in anaerobic chemostat cultures of Saccharomyces cerevisiae. FEMS Yeast Res. 2007;7:819–833. doi: 10.1111/j.1567-1364.2007.00242.x. [DOI] [PubMed] [Google Scholar]
- 11.Bajwa PK, Ho CY, Chan CK, Martin VJJ, Trevors JT, Lee H. Transcriptional profiling of Saccharomyces cerevisiae T2 cells upon exposure to hardwood spent sulphite liquor: comparison to acetic acid, furfural and hydroxymethylfurfural. Antonie Van Leeuwenhoek Int J General Mol Microbiol. 2013;103:1281–1295. doi: 10.1007/s10482-013-9909-1. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y, Nasution O, Choi E, Choi IG, Kim W, Choi W. Transcriptome analysis of acetic-acid-treated yeast cells identifies a large set of genes whose overexpression or deletion enhances acetic acid tolerance. Appl Microbiol Biotechnol. 2015;99:6391–6403. doi: 10.1007/s00253-015-6706-y. [DOI] [PubMed] [Google Scholar]
- 13.Hasunuma T, Sakamoto T, Kondo A. Inverse metabolic engineering based on transient acclimation of yeast improves acid-containing xylose fermentation and tolerance to formic and acetic acids. Appl Microbiol Biotechnol. 2016;100:1027–1038. doi: 10.1007/s00253-015-7094-z. [DOI] [PubMed] [Google Scholar]
- 14.Sugiyama M, Akase SP, Nakanishi R, Horie H, Kaneko Y, Harashima S. Nuclear localization of Haa1, which is linked to its phosphorylation status, mediates lactic acid tolerance in Saccharomyces cerevisiae. Appl Environ Microbiol. 2014;80:3488–3495. doi: 10.1128/AEM.04241-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka K, Ishii Y, Ogawa J, Shima J. Enhancement of acetic acid tolerance in Saccharomyces cerevisiae by overexpression of the HAA1 gene, encoding a transcriptional activator. Appl Environ Microbiol. 2012;78:8161–8163. doi: 10.1128/AEM.02356-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inaba T, Watanabe D, Yoshiyama Y, Tanaka K, Ogawa J, Takagi H, Shimoi H, Shima J. An organic acid-tolerant HAA1-overexpression mutant of an industrial bioethanol strain of Saccharomyces cerevisiae and its application to the production of bioethanol from sugarcane molasses. AMB Express. 2013;3:74. doi: 10.1186/2191-0855-3-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakihama Y, Hasunuma T, Kondo A. Improved ethanol production from xylose in the presence of acetic acid by the overexpression of the HAA1 gene in Saccharomyces cerevisiae. J Biosci Bioeng. 2015;119:297–302. doi: 10.1016/j.jbiosc.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Fernandes AR, Mira NP, Vargas RC, Canelhas I, Sa-Correia I. Saccharomyces cerevisiae adaptation to weak acids involves the transcription factor Haa1p and Haa1p-regulated genes. Biochem Biophys Res Commun. 2005;337:95–103. doi: 10.1016/j.bbrc.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Mira NP, Henriques SF, Keller G, Teixeira MC, Matos RG, Arraiano CM, Winge DR, Sa-Correia I. Identification of a DNA-binding site for the transcription factor Haa1, required for Saccharomyces cerevisiae response to acetic acid stress. Nucleic Acids Res. 2011;39:6896–6907. doi: 10.1093/nar/gkr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de-Carvalho J, Rodrigues RMM, Tome B, Henriques SF, Mira NP, Sa-Correia I, Ferreira GNM. Conformational and mechanical changes of DNA upon transcription factor binding detected by a QCM and transmission line model. Analyst. 2014;139:1847–1855. doi: 10.1039/C3AN01682J. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigues RM, de-Carvalho J, Henriques SF, Mira NP, Sa-Correia I, Ferreira GN. Transmission line model analysis of transcription factors binding to oligoduplexes—differentiation of the effect of single nucleotide modifications. Analyst. 2014;139:3871–3874. doi: 10.1039/C4AN00709C. [DOI] [PubMed] [Google Scholar]
- 22.Tomitori H, Kashiwagi K, Asakawa T, Kakinuma Y, Michael AJ, Igarashi K. Multiple polyamine transport systems on the vacuolar membrane in yeast. Biochem J. 2001;353:681–688. doi: 10.1042/bj3530681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Destruelle M, Holzer H, Klionsky DJ. Identification and characterization of a novel yeast gene: the YGP1 gene product is a highly glycosylated secreted protein that is synthesized in response to nutrient limitation. Mol Cell Biol. 1994;14:2740–2754. doi: 10.1128/MCB.14.4.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takabatake A, Kawazoe N, Izawa S. Plasma membrane proteins Yro2 and Mrh1 are required for acetic acid tolerance in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2015;99:2805–2814. doi: 10.1007/s00253-014-6278-2. [DOI] [PubMed] [Google Scholar]
- 25.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 26.Alper H, Moxley J, Nevoigt E, Fink GR, Stephanopoulos G. Engineering yeast transcription machinery for improved ethanol tolerance and production. Science. 2006;314:1565–1568. doi: 10.1126/science.1131969. [DOI] [PubMed] [Google Scholar]
- 27.Verduyn C, Postma E, Scheffers WA, Vandijken JP. Effect of benzoic-acid on metabolic fluxes in yeasts—a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–517. doi: 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- 28.Gietz RD, Schiestl RH, Willems AR, Woods RA. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995;11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Russell DW. Molecular cloning: a laboratory manual, vol 1. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 30.Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- 31.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 32.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh MV, Weil PA. A method for plasmid purification directly from yeast. Anal Biochem. 2002;307:13–17. doi: 10.1016/S0003-2697(02)00018-0. [DOI] [PubMed] [Google Scholar]
- 34.Akada R, Hirosawa I, Kawahata M, Hoshida H, Nishizawa Y. Sets of integrating plasmids and gene disruption cassettes containing improved counter-selection markers designed for repeated use in budding yeast. Yeast. 2002;19:393–402. doi: 10.1002/yea.841. [DOI] [PubMed] [Google Scholar]
- 35.Flagfeldt DB, Siewers V, Huang L, Nielsen J. Characterization of chromosomal integration sites for heterologous gene expression in Saccharomyces cerevisiae. Yeast. 2009;26:545–551. doi: 10.1002/yea.1705. [DOI] [PubMed] [Google Scholar]
- 36.Kohrer K, Domdey H. Preparation of high molecular weight RNA. Methods Enzymol. 1991;194:398–405. doi: 10.1016/0076-6879(91)94030-G. [DOI] [PubMed] [Google Scholar]
- 37.Van Driessche B, Tafforeau L, Hentges P, Carr AM, Vandenhaute J. Additional vectors for PCR-based gene tagging in Saccharomyces cerevisiae and Schizosaccharomyces pombe using nourseothricin resistance. Yeast. 2005;22:1061–1068. doi: 10.1002/yea.1293. [DOI] [PubMed] [Google Scholar]
- 38.Mollapour M, Piper PW. Hog1 mitogen-activated protein kinase phosphorylation targets the yeast Fps1 aquaglyceroporin for endocytosis, thereby rendering cells resistant to acetic acid. Mol Cell Biol. 2007;27:6446–6456. doi: 10.1128/MCB.02205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakihama Y, Hasunuma T, Kondo A. Improved ethanol production from xylose in the presence of acetic acid by the overexpression of the HAA1 gene in Saccharomyces cerevisiae. J Biosci Bioeng. 2014;119:297–302. [DOI] [PubMed]
- 40.Sadowski I, Breitkreutz BJ, Stark C, Su TC, Dahabieh M, Raithatha S, Bernhard W, Oughtred R, Dolinski K, Barreto K, Tyers M. The PhosphoGRID Saccharomyces cerevisiae protein phosphorylation site database: version 2.0 update. Database (Oxford) 2013;2013:026. doi: 10.1093/database/bat026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zahn K, Jacobson S. Acetate resistance in yeast based on introduction of a mutant Haa1 allele. PCT/US2013/051500 edition. US; 2014.
- 42.Meijnen JP, Randazzo P, Foulquie-Moreno MR, van den Brink J, Vandecruys P, Stojiljkovic M, Dumortier F, Zalar P, Boekhout T, Gunde-Cimerman N, et al. Polygenic analysis and targeted improvement of the complex trait of high acetic acid tolerance in the yeast Saccharomyces cerevisiae. Biotechnol Biofuels. 2016;9:5. doi: 10.1186/s13068-015-0421-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 44.van Dijken JP, Bauer J, Brambilla L, Duboc P, Francois JM, Gancedo C, Giuseppin ML, Heijnen JJ, Hoare M, Lange HC, et al. An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb Technol. 2000;26:706–714. doi: 10.1016/S0141-0229(00)00162-9. [DOI] [PubMed] [Google Scholar]