

Abstract
Background:
The molecular genetic research showed the association between X-linked hearing loss and mutations in POU3F4. This research aimed to identify a POU3F4 mutation in a nonsyndromic X-linked recessive hearing loss family.
Methods:
A series of clinical evaluations including medical history, otologic examinations, family history, audiologic testing, and a high-resolution computed tomography scan were performed for each patient. Bidirectional sequencing was carried out for all polymerase chain reaction products of the samples. Moreover, 834 controls with normal hearing were also tested.
Results:
The pedigree showed X-linkage recessive inheritance pattern, and pathogenic mutation (c.499C>T) was identified in the proband and his family member, which led to a premature termination prior to the entire POU domains. This mutation co-segregated with hearing loss in this family. No mutation of POU3F4 gene was found in 834 controls.
Conclusions:
A nonsense mutation is identified in a family displaying the pedigree consistent with X-linked recessive pattern in POU3F4 gene. In addition, we may provide molecular diagnosis and genetic counseling for this family.
Keywords: c.499C>T, Nonsyndromic Hearing Loss, POU3F4, X-linked
Introduction
Hearing loss is one of the common sensory disorders in humans, affecting 1 in 1000 newborns. More than one half of it may attribute to genetic factors. Among them, it showed that hearing loss is transmitted with a heredity form concurrent with autosomal recessive (75%), dominant (20%), X-linked (3%), and others (2%). So far, five loci (DFNX1, DFNX2, DFNX3, DFNX4, and DFNX6) and four genes (PRPS1, POU3F4, SMPX, and COL4A6) have been found for X-linked hearing impairment.[1] The clinical features of DFNX3 comprise profound mixed hearing loss and vestibular problems in males.[2]
The molecular genetic research showed the association between X-linked hearing loss and mutations in POU3F4.[3] This gene belongs to a superfamily of POU domain transcription factors (transcription factor 4 of the POU domain, Class III), which encodes transcription factor with bipartite DNA-binding domains, a 75-amino acid POU-specific domain that is linked by 17 amino acids to a 63-amino acid homeobox domain. Both subdomains contain helix-turn-helix motifs that directly associate with the two components of bipartite DNA-binding sites. Animal models also showed that the expression of Brn-4 (POU3F4 synonyms) was highly associated with remodeling of the otic capsule.[4]
There are 52 variants reported in the Human Gene Mutation Database, including intragenic mutations, complete or partial deletions, duplications, inversions, and other chromosomal deletions.[5] To identify the mutation in the POU3F4 gene, in this study, we performed the analysis in a Chinese nonsyndromic X-linked hereditary hearing loss family.
Methods
Clinical evaluation
The proband, a 10-year-old boy, presented to the otolaryngology clinic with genetic severe deafness and speech delay. His family history was obtained by his mother. He was subjected to detailed clinical evaluations including medical history, family history, otologic examinations, audiologic testing (pure-tone audiometry, tympanometry, acoustic reflex, auditory brainstem response, and 40 Hz auditory events-related potential), and high-resolution axial computed tomography (CT) scan of temporal bones. This research was approved by the collectors’ Institutional Review Board of the Ethics Committee at Chinese People's Liberation Army General Hospital.
Molecular genetic analysis
After getting informed consent from all participants and parents of persons <18 years, genomic DNA was extracted from peripheral blood leukocytes of the proband (V1) and other members of family (IV1, IV2, IV3, IV4, IV5, IV6, III1) using standard procedures. Moreover, 834 controls with normal hearing were also tested. The coding region of POU3F4 was amplified by polymerase chain reaction (PCR) with two pairs of primers. The first pair primers were a forward primer (5′-TAACCCGTGCTAGCGTCTTT-3′) and a reverse primer (5′-GAACCTGCAGATGGTGGTCT-3′) and the second pair primers were a forward primer (5′-CAACCTCTGATGAGTTGGAACA-3′) and a reverse primer (5′-AAAGGAAGAGATGGAAGGGAAG-3′). The PCR procedure was as follows: 95°C for 5 min, followed by 32 cycles of denaturation at 94°C for 45 s; annealing at 56°C for 30 s, extension at 72°C for 30 s, and a final elongation at 72°C for 7 min. All the PCR products were purified and subjected to sequencing in both directions using a DNA sequencer (ABI 3730).
Results
Clinical investigations
The five-generation pedigree presented with an X-linkage recessive inheritance pattern of hearing loss. Otomicroscopical examination showed intact tympanic membranes and no middle ear effusion in proband (V1). Moreover, pure tone audiometry indicated bilateral severe to profound deafness, but bone hearing thresholds were normal at 250 Hz. Further, high-resolution axial CT scan revealed a dilatation of the lateral end of the internal auditory meatus and a deficit or absence in the basal turn of the cochlea [Figure 1].
Figure 1.

The clinical features and genetic testing results of the proband and the family member. (a) Pedigree of the family. The incidence pattern of the pedigree shows X-linked hereditary; (b) The imaging tests of the proband. The high-resolution axial computed tomography reveals a dilatation of lateral end of the internal auditory meatus and a deficit or absence in the basal turn of the cochlea; (c) Audiograms of the proband. The pure tone audiometry shows belated severe to profound hearing loss, but bone hearing thresholds was normal (10 dB) at 250 Hz; (d) Sequencing chromatograms of POU3F4. The molecular testing result of proband and so family members.
Identification of the mutation in POU3F4 gene
Variant analysis of the POU3F4 was performed for the radiologic results. Sequence screening of all the family members showed a nonsense mutation at nucleotide position c.499C>T in the proband (V1) and his uncle with hearing loss phenotype (IV3). The proband's mother (IV1), one aunt (IV2), and grandmother (III1) carried the c.499C>T mutation without hearing loss phenotype. The proband's other two aunts (IV4, IV5) and another uncle (IV6) showed normal hearing without any mutation. No mutation of POU3F4 gene was found in 834 controls. The location of POU3F4 gene on the X chromosome and the variations found on this gene were shown in Figure 2.
Figure 2.
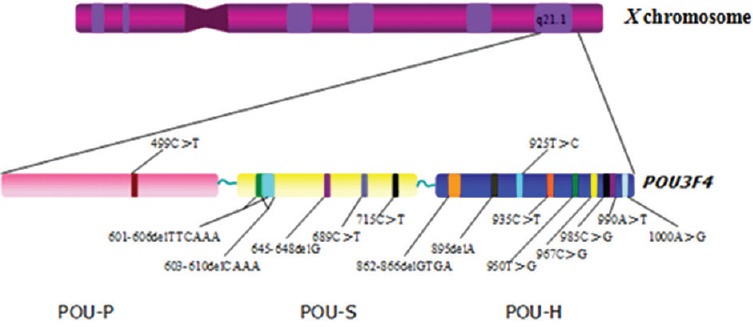
The location of POU3F4 gene on the X chromosome and the variations found on this gene.
Discussion
In recent years, studies have performed on POU3F4 gene mutations in different countries. Until now, mutations in this gene have been identified containing missense mutations, in-frame deletions, and truncating mutations [Table 1].
Table 1.
The genotype distribution of POU3F4 gene mutations
| Year | Nucleotide | Protein | Authors | Reference |
|---|---|---|---|---|
| 1988 | c.715C>T | p.S228L | Brunner et al. | [6] |
| 1995 | c.604A>T | p.K202X | De Kok et al. | [3] |
| 1995 | c.607_610del4 | – | De Kok et al. | [3] |
| 1995 | c.648del1 | – | De Kok et al. | [3] |
| 1995 | c.862_865del4 | – | Bitner-Glindzicz et al. | [7] |
| 1995 | c.896del1 | – | De Kok et al. | [3] |
| 1995 | c.935C>T | p.A312V | Bitner-Glindzicz et al. | [7] |
| 1995 | c.950T>G | p.L317W | De Kok et al. | [3] |
| 1995 | c.1000A>G | p.K334E | De Kok et al. | [3] |
| 1997 | c.689C>T | p.T230I | Friedman et al. | [8] |
| 1997 | c.985C>G | p.R329G | Friedman et al. | [8] |
| 1997 | c.990A>T | p.R330S | De Kok et al. | [9] |
| 1998 | c.601_606del6 | – | Hagiwara et al. | [10] |
| 2000 | c.200G>A | p.W67X | Cremers et al. | [11] |
| 2000 | c.907C>T | p.P303S | Cremers et al. | [11] |
| 2000 | c.967C>G | p.R323G | Cremers et al. | [11] |
| 2000 | c.983A>C | p.N328T | Cremers et al. | [11] |
| 2005 | c.683C>T | p.S228L | Vore et al. | [12] |
| 2006 | c.925T>C | p.S309P | Wang et al. | [13] |
| 2009 | c.293C>A | p.S98X | Marlin et al. | [14] |
| 2009 | c.346del1 | – | Lee et al. | [15] |
| 2009 | c.383del1 | – | Lee et al. | [16] |
| 2009 | c.623T>A | p.L208X | Lee et al. | [16] |
| 2009 | c.923T>A | p.I308N | Marlin et al. | [14] |
| 2009 | c.927_929del3 | – | Lee et al. | [15] |
| 2009 | c.986G>C | p.R329P | Lee et al. | [15] |
| 2010 | c.499C>T | p.R167X | Stankovic et al. | [17] |
| 2010 | c.647G>A | p.G216E | Li et al. | [18] |
| 2011 | c.341G>A | p.W114 | Waryah et al. | [19] |
| 2011 | c.406C>T | p.Q136 | Waryah et al. | [19] |
| 2011 | c.973T>A | p.W325R | Schild et al. | [20] |
| 2013 | c.235C>T | p.Q79X | Parzefall et al. | [21] |
| 2013 | c.632C>T | p.T211M | Choi et al. | [22] |
| 2013 | c.686A>G | p.Q229R | Choi et al. | [22] |
| 2013 | c.853_854delAT | – | Parzefall et al. | [21] |
| 2013 | c.950dupT | – | Choi et al. | [22] |
| 2013 | c.1060delA | – | Choi et al. | [22] |
| 2013 | c.1084T>C | p.X362R | Choi et al. | [22] |
This is the first study to identify a nonsense mutation c.499C>T in POU3F4 gene in Asians. So far, the most identified mutations of POU3F4 gene are located in nucleotide poison-encoding POU domain.[4,5,9,18] The c.499C>T mutation changes an arginine (CGA) to a stop code (ATG), which leads to a premature termination prior to the entire POU domains in amino acid level.[17] This mutation co-segregates with hearing loss in this family. Male members carried c.499C>T mutation. However, females with this mutation are normal hearing. Therefore, we consider that this mutation may be the cause of disease etiology of the pedigree. By molecular testing, we provided definitive diagnosis and genetic counseling for this family and further enriched pathogenic mutation spectrum of POU3F4 gene.
Pedigree analysis is an important approach to search the underlying cause of hearing loss. However, more patients with hearing loss were sporadic and there is a lack of characteristic phenotype in clinical practice. Hence, using all the available phenotypic clues to direct molecular testing would be cost-effective. The clinical features of CT images of the temporal bones are now well recognized through reported cases.[11,21,23,24] Besides the characteristics of CT images, audiological phenotype of DFNX3 is defined as profound deafness with or without a conductive element.[7,14,19,25] The conductive component in the audiogram could also be concerned about because of an atypical communication between an internal auditory canal and the inner ear leading to a pathologic third window which deteriorates air-conducted thresholds and increases bone-conducted thresholds. The supposed improvement in bone conduction sensitivity could be concealed by a true sensorineural deafness following the disorder.[15,26]
The proband had a profound deafness but displayed normal bone conductive hearing at 250 Hz. Based on other authors’ records and our experience, it was revealed that patients with this type of audiogram of low-frequency air–bone gap increased suspicion for inner malformation. Subsequent axial CT images of the temporal bones showed the defects of the bony labyrinth in proband such as a symmetric bulbous dilatation of the internal auditory canals in both ears and partial separation of the cochlea with the fundus of internal auditory canals, which was consistent with DFNX3. It was the above clinical information that guided us to reveal the causative mutation of this family. Last but not the least, DFNX3 has already been diagnosed in molecular level, so POU3F4 gene should be screened in the outpatient persons who have the clinical phenotypes of DFNX3.[27,28,29] Our study also has some limitations. On one hand, more pedigree and sporadic people should be recruited. On the other hand, new and rapid technique should be adopted to detect the patients.
In conclusion, a nonsense mutation is identified in a Chinese family displaying the pedigree consistent with X-linked recessive pattern in POU3F4 gene. Furthermore, we may provide molecular diagnosis and genetic counseling for this family.
Financial support and sponsorship
This work was supported by the grants from the National Key Basic Research Program of China (No.2014CB943001), the National Natural Science Foundation of China (No. 81120108009 and No. 81530032), and the China Postdoctoral Science Foundation (No.2015M572690).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Li-Min Chen
References
- 1.Hereditary Hearing Loss Homepage. [Last accessed on 2016 Apr 01]. (http://hereditaryhearingloss.org)
- 2.Nance WE, Setleff R, McLeod A, Sweeney A, Cooper C, McConnell F. X-linked mixed deafness with congenital fixation of the stapedial footplate and perilymphatic gusher. Birth Defects Orig Artic Ser. 1971;07:64–9. [PubMed] [Google Scholar]
- 3.de Kok YJ, van der Maarel SM, Bitner-Glindzicz M, Huber I, Monaco AP, Malcolm S, et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995;267:685–8. doi: 10.1126/science.7839145. doi: 10.1126/science.7839145. [DOI] [PubMed] [Google Scholar]
- 4.Phippard D, Heydemann A, Lechner M, Lu L, Lee D, Kyin T, et al. Changes in the subcellular localization of the Brn4 gene product precede mesenchymal remodeling of the otic capsule. Hear Res. 1998;120:77–85. doi: 10.1016/s0378-5955(98)00059-8. doi: 10.1016/S0378-5955(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 5.The Human Gene Mutation Database. [Last accessed on 2016 Apr 01]. Available from: http://www.hgmd.org .
- 6.Brunner HG, van Bennekom A, Lambermon EM, Oei TL, Cremers WR, Wieringa B, et al. The gene for X-linked progressive mixed deafness with perilymphatic gusher during stapes surgery (DFN3) is linked to PGK. Hum Genet. 1988;80:337–40. doi: 10.1007/BF00273647. doi: 10.1007/BF00273647. [DOI] [PubMed] [Google Scholar]
- 7.Bitner-Glindzicz M, Turnpenny P, Höglund P, Kääriäinen H, Sankila EM, van der Maarel SM, et al. Further mutations in brain 4 (POU3F4) clarify the phenotype in the X-linked deafness, DFN3. Hum Mol Genet. 1995;4:1467–9. doi: 10.1093/hmg/4.8.1467. doi: 10.1093/hmg/4.8.1467. [DOI] [PubMed] [Google Scholar]
- 8.Friedman RA, Bykhovskaya Y, Tu G, Talbot JM, Wilson DF, Parnes LS, et al. Molecular analysis of the POU3F4 gene in patients with clinical and radiographic evidence of X-linked mixed deafness with perilymphatic gusher. Ann Otol Rhinol Laryngol. 1997;106:320–5. doi: 10.1177/000348949710600411. doi: 10.1177/000348949710600411. [DOI] [PubMed] [Google Scholar]
- 9.de Kok YJ, Cremers CW, Ropers HH, Cremers FP. The molecular basis of X-linked deafness type 3 (DFN3) in two sporadic cases: Identification of a somatic mosaicism for a POU3F4 missense mutation. Hum Mutat. 1997;10:207–11. doi: 10.1002/(SICI)1098-1004(1997)10:3<207::AID-HUMU5>3.0.CO;2-F. doi: 10.1002/(SICI)1098-1004(1997)10:3<207::AID-HUMU5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 10.Hagiwara H, Tamagawa Y, Kitamura K, Kodera K. A new mutation in the POU3F4 gene in a Japanese family with X-linked mixed deafness (DFN3) Laryngoscope. 1998;108:1544–7. doi: 10.1097/00005537-199810000-00022. doi: 10.1097/00005537-199810000-00022. [DOI] [PubMed] [Google Scholar]
- 11.Cremers FP, Cremers CW, Ropers HH. The ins and outs of X-linked deafness type 3. Adv Otorhinolaryngol. 2000;56:184–95. doi: 10.1159/000059101. doi: 10.1159/000059101. [DOI] [PubMed] [Google Scholar]
- 12.Vore AP, Chang EH, Hoppe JE, Butler MG, Forrester S, Schneider MC, et al. Deletion of and novel missense mutation in POU3F4 in 2 families segregating X-linked nonsyndromic deafness. Arch Otolaryngol Head Neck Surg. 2005;131:1057–63. doi: 10.1001/archotol.131.12.1057. doi: 10.1001/archotol.131.12.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang QJ, Li QZ, Rao SQ, Zhao YL, Yuan H, Yang WY, et al. Anovel mutation of POU3F4 causes congenital profound sensorineural hearing loss in a large Chinese family. Laryngoscope. 2006;116:944–50. doi: 10.1097/01.MLG.0000215285.53045.24. doi: 10.1097/01.MLG.0000215285.53045.24. [DOI] [PubMed] [Google Scholar]
- 14.Marlin S, Moizard MP, David A, Chaissang N, Raynaud M, Jonard L, et al. Phenotype and genotype in females with POU3F4 mutations. Clin Genet. 2009;76:558–63. doi: 10.1111/j.1399-0004.2009.01215.x. doi: 10.1111/j.1399-0004.2009.01215.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee HK, Song MH, Kang M, Lee JT, Kong KA, Choi SJ, et al. Clinical and molecular characterizations of novel POU3F4 mutations reveal that DFN3 is due to null function of POU3F4 protein. Physiol Genomics. 2009;39:195–201. doi: 10.1152/physiolgenomics.00100.2009. doi: 10.1152/physiolgenomics.00100.2009. [DOI] [PubMed] [Google Scholar]
- 16.Lee HK, Lee SH, Lee KY, Lim EJ, Choi SY, Park RK, et al. Novel POU3F4 mutations and clinical features of DFN3 patients with cochlear implants. Clin Genet. 2009;75:572–5. doi: 10.1111/j.1399-0004.2009.01181.x. doi: 10.1111/j.1399-0004.2009.01181.x. [DOI] [PubMed] [Google Scholar]
- 17.Stankovic KM, Hennessey AM, Herrmann B, Mankarious LA. Cochlear implantation in children with congenital X-linked deafness due to novel mutations in POU3F4 gene. Ann Otol Rhinol Laryngol. 2010;119:815–22. doi: 10.1177/000348941011901205. doi: 10.1177/000348941011901205. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Cheng J, Lu Y, Lu Y, Chen A, Sun Y, et al. Identification of a novel mutation in POU3F4 for prenatal diagnosis in a Chinese family with X-linked nonsyndromic hearing loss. J Genet Genomics. 2010;37:787–93. doi: 10.1016/S1673-8527(09)60096-5. doi: 10.1016/S1673-8527(09)60096-5. [DOI] [PubMed] [Google Scholar]
- 19.Waryah AM, Ahmed ZM, Bhinder MA, Choo DI, Sisk RA, Shahzad M, et al. Molecular and clinical studies of X-linked deafness among Pakistani families. J Hum Genet. 2011;56:534–40. doi: 10.1038/jhg.2011.55. doi: 10.1038/jhg.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schild C, Prera E, Lüblinghoff N, Arndt S, Aschendorff A, Birkenhäger R. Novel mutation in the homeobox domain of transcription factor POU3F4 associated with profound sensorineural hearing loss. Otol Neurotol. 2011;32:690–4. doi: 10.1097/MAO.0b013e318210b749. doi: 10.1097/MAO.0b013e318210b749. [DOI] [PubMed] [Google Scholar]
- 21.Parzefall T, Shivatzki S, Lenz DR, Rathkolb B, Ushakov K, Karfunkel D, et al. Cytoplasmic mislocalization of POU3F4 due to novel mutations leads to deafness in humans and mice. Hum Mutat. 2013;34:1102–10. doi: 10.1002/humu.22339. doi: 10.1002/humu.22339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi BY, Kim DH, Chung T, Chang M, Kim EH, Kim AR, et al. Destabilization and mislocalization of POU3F4 by C-terminal frameshift truncation and extension mutation. Hum Mutat. 2013;34:309–16. doi: 10.1002/humu.22232. doi: 10.1002/humu.22232. [DOI] [PubMed] [Google Scholar]
- 23.Phelps PD, Reardon W, Pembrey M, Bellman S, Luxom L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology. 1991;33:326–30. doi: 10.1007/BF00587816. [DOI] [PubMed] [Google Scholar]
- 24.Talbot JM, Wilson DF. Computed tomographic diagnosis of X-linked congenital mixed deafness, fixation of the stapedial footplate, and perilymphatic gusher. Am J Otol. 1994;15:177–82. [PubMed] [Google Scholar]
- 25.Snik AF, Hombergen GC, Mylanus EA, Cremers CW. Air-bone gap in patients with X-linked stapes gusher syndrome. Am J Otol. 1995;16:241–6. [PubMed] [Google Scholar]
- 26.Merchant SN, Rosowski JJ. Conductive hearing loss caused by third-window lesions of the inner ear. Otol Neurotol. 2008;29:282–9. doi: 10.1097/mao.0b013e318161ab24. doi: 10.1097/mao.0b013e318161ab24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi JW, Min B, Kim A, Koo JW, Kim CS, Park WY, et al. De novo large genomic deletions involving POU3F4 in incomplete partition type III inner ear anomaly in East Asian populations and implications for genetic counseling. Otol Neurotol. 2015;36:184–90. doi: 10.1097/MAO.0000000000000343. doi: 10.1097/MAO.0000000000000343. [DOI] [PubMed] [Google Scholar]
- 28.Bademci G, Lasisi A, Yariz KO, Montenegro P, Menendez I, Vinueza R, et al. Novel domain-specific POU3F4 mutations are associated with X-linked deafness: Examples from different populations. BMC Med Genet. 2015;16:9. doi: 10.1186/s12881-015-0149-2. doi: 10.1186/s12881-015-0149-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong WX, Gong RZ, Zhao B. HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int J Pediatr Otorhinolaryngol. 2014;78:1756–62. doi: 10.1016/j.ijporl.2014.08.013. doi: 10.1016/j.ijporl.2014.08.013. [DOI] [PubMed] [Google Scholar]