

Abstract
Background
Cardiovascular diseases are the leading cause of death in many countries and myocardial ischemia-reperfusion (I/R) injury is the cause of many serious heart diseases. Recent reports suggested that endoplasmic reticulum (ER) stress is associated with the progress of ischemia/reperfusion (I/R) injury. In a previous study, we illustrated that 4-phenylbutyric acid (4-PBA) reduces I/R-induced cell death in vitro through inhibiting the ER stress-initiated cell apoptosis. In the present study we investigated whether 4-PBA improves heart function in isolated rat hearts subjected to I/R and elucidated the potential mechanisms involved in 4-PBA-induced cardioprotective effects.
Material/Methods
The isolated rat hearts were subjected to global ischemia and reperfusion in the absence or presence of 4-PBA. Hemodynamic parameters (LVSP, LVEDP, ±dP/dtmax, and HR) were monitored and histopathological examination was applied. The biomarkers related to oxidative stress were detected by LDH, ROS, MDA, CK, SOD, and GSH-Px kits. A TUNEL apoptosis assay kit was used to detect apoptosis. The expression levels of ER stress and apoptosis proteins were evaluated by Western blotting.
Results
We found that 4-PBA (5 mM, 10 mM) pretreatment significantly attenuated cardiac dysfunction and depressed oxidative stress induced by I/R. Moreover, I/R activated the ER stress proteins Grp78 and PERK, which are all decreased by 4-PBA. 4-PBA pretreatment also inhibited the expression of CHOP, Caspase-12, and Bax, reduced the phosphorylation of JNK, and enhanced the expression of anti-apoptotic protein Bcl-2.
Conclusions
We elucidated the significant protective effects of 4-PBA against I/R injuries by inhibition of ER stress, oxidative stress, and their associated apoptosis.
MeSH Keywords: Apoptosis, Endoplasmic Reticulum Stress, Reperfusion Injury, Unfolded Protein Response
Background
Cardiovascular diseases are the leading cause of death in many countries and myocardial ischemia is a primary cause of morbidity and mortality among them [1]. During ischemia, the supply of nutrients and oxygen to heart tissue is impeded, which will eventually lead to heart failure and can be fatal if untreated. Once blood flow is restored to the ischemic zone, ischemic cells undergo further injury due to toxicity from the burst of ensuing reactive oxygen species, which may trigger apoptosis [2,3]. Myocardium cell apoptosis has been reported to be an essential form of cell death in ischemia/reperfusion (I/R) injury and is characterized by nuclear condensation, shrinkage of the cell membrane, and formation of apoptotic bodies [4–6].
Evidence has shown that endoplasmic reticulum (ER) stress and ER stress-initiated apoptotic signaling pathways are involved in the development of myocardium I/R injury [7]. Various stimuli, such as ischemia, hypoxia, free radical exposure, elevated protein synthesis, and gene mutations, can perturb ER homeostasis and cause the pathological accumulation of unfolded/misfolded proteins in the ER [8,9]. The unfolded protein response (UPR) is triggered in cells when ER transmembrane protein sensors (PERK, IRE1, and ATF6) detect the accumulation of unfolded proteins [10]. However, if the stress is prolonged or overwhelming, the pro-survival effects of UPR switch to pro-apoptotic signaling, which is mostly mediated by transcriptional induction of CHOP or by activation of the JNK/c-JUN and/or Caspase-12-dependent pathways [11].
4-PBA (Figure 1A) is a chemical chaperone with high in vivo safety [12]. Its physiochemical properties enable it to stabilize peptide structures and to improve the luminal folding capacity and traffic of aberrant proteins [13,14]. In the previous study, we found that 4-PBA reduces I/R-induced cardiomyocytes apoptosis in vitro through inhibiting the specific ER stress-associated cell apoptosis pathways [15]. However, whether 4-PBA provides cardioprotection against I/R injury in rat hearts, or whether its protective effects are connected with the inhibition of oxidative stress and ER stress-associated pathways, remains unclear. In the current study, we aimed to determine whether 4-PBA improves heart function in isolated rat hearts subjected to I/R and to elucidate the potential mechanisms involved in 4-PBA-induced cardioprotective effects. Our results showed that pretreatment of 4-PBA alleviated I/R-induced cardiac dysfunction, oxidative stress, and ER stress responses, suggesting that 4-PBA can be a novel and useful therapeutic agent for myocardium I/R injury and possibly for heart failure.
Figure 1.
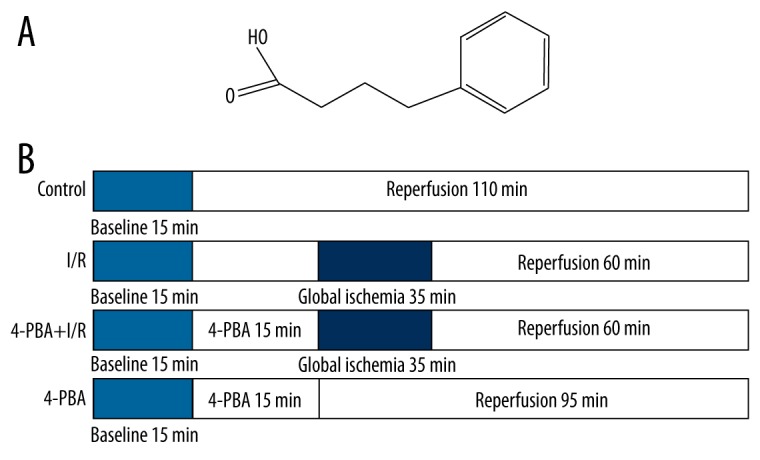
The chemical structure of 4-phenylbutyric acid and experimental protocol. (A) The chemical structure of 4-phenylbutyric acid. (B) All hearts were equilibrated for 15 min after being mounted on the Langendorff apparatus. Hearts in the control group were constantly perfused with K-H buffer for another 110 min. Hearts in I/R group were perfused with K-H buffer for another 15 min, then subjected to 35 min of globe myocardial ischemia and 60 min of reperfusion. Hearts in the 4-PBA (5 mM, 10 mM) +I/R group were perfused with 4-PBA (5 mM, 10 mM) for 15 min before the ischemia and reperfusion process. Hearts in the 4-PBA (10mM) group were administered 4-PBA (10 mM) for 15 min and then perfused with K-H buffer for 95 min. There were 10 rats in each group.
Material and Methods
Ethics statements
All of the experimental procedures were carried out according to the guidelines of the Experimental Laboratory Animal Committee of the Chinese Academy of Medical Sciences and the principles and guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Reagents
4-phenylbutiric acid (4-PBA) and dimethyl sulfoxide (DMSO) were purchased from Sigma Chemical Co. (St. Louis, MO). The kits for determining lactate dehydrogenase (LDH), total reactive oxygen species (ROS), malondialdehyde (MDA) contents, total creatine kinase (CK), superoxide dismutase (SOD) activity, and glutathione peroxidase (GSH-Px) were obtained from Jiancheng Bioengineering Institute (Nanjing, China). In Situ Cell Death Detection Kits (Fluorescein) were purchased from Roche (Basel, Switzerland). Primary antibodies against Grp78, p-PERK, PERK, CHOP, Caspase-12, JNK, p-JNK, Bax, Bcl-2, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Experimental Protocol
Male Sprague-Dawley rats weighing 200–220 g were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The Langendorff operation was performed as shown in Figure 1B. Briefly, rats were anesthetized with urethane (20%), and their hearts were rapidly removed and mounted to a Langendorff perfusion apparatus. The hearts were perfused with oxygenated Krebs-Henseleit (KH) buffer (95% O2, 5% CO2) at a constant temperature of 37°C at a cycle length of 200 ms (300 bpm). LV pressure was measured using a water-filled wrap balloon connected to a pressure transducer and the signals were recorded using PowerLab and analyzed using Chart V 7.3.3. (AD Instruments). Rats were randomly assigned to 5 groups with 10 rats in each group: (1) control group; (2) I/R group; (3) (4) 4-PBA (5 mM, 10 mM) +I/R groups; and (5) 4-PBA (10 mM) group. All hearts were equilibrated with KH buffer for 15 min before the application of experimental protocols. For those undergoing ischemia/reperfusion, the hearts were subjected to 35 min of ischemia and 60 min of reperfusion. The following hemodynamic parameters were monitored during this process: left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), maximum rate of contraction (+dP/dtmax), maximum rate of relaxation (−dP/dtmax), heart rates (HR), and LVDP (Left Ventricular Developed Pressure) ×HR.
Tissue processing and analysis
Frozen tissues of the isolated hearts were quickly homogenized in cooled phosphate buffer (pH 7.40), and the homogenates were centrifuged at 5000× g for 10 min at 4°C. The supernatants were then used to measure the content of LDH, ROS, MDA, and activities of SOD, CK, and GSH-Px using commercial kits (JianCheng Bioengineering Institute, Nanjing, China).
Heart histopathological examination
The isolated rat hearts were fixed with 4% paraformaldehyde for more than 48 h. Afterwards, the left ventricles of the hearts were trimmed and embedded in paraffin blocks, sectioned, stained with hematoxylin and eosin (HE), and examined under a light microscope (CKX41, Tokyo, Japan) by a pathologist blinded to the groups under study.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining
The apoptotic cells were determined by terminal deoxynucleotidyl transferase – mediated dUTP nick-end labeling (TUNEL) assay using the In Situ Cell Death Detection Kit (Roche, Basel, Switzerland) according to the manufacturer’s protocol. Briefly, the paraffin-embedded tissue sections were deparaffinized, rehydrated, and fixed in 4% paraformaldehyde solution. After being dewaxed, rehydrated, and treated with 20 mg/ml proteinase K for 10 min, the tissue sections were rinsed with PBS 3 times. Then the tissue was permeabilized with 0.2% Triton X-100 for 5 min. Tissue slices were labeled with fluorescein TUNEL reagent mixture for 1 h at 37°C according to the manufacturer’s suggested protocol. After that, slides were examined by fluorescence microscopy and the number of TUNEL-positive (apoptotic) nuclei was counted in 5 randomly chosen fields on each section. DAPI (10 mg/ml) was used to stain nuclei.
Western blotting analysis
Total soluble protein was extracted from the left ventricle of the hearts with extraction buffer supplemented with 1 mM PMSF. Immunoblotting analysis was performed by incubating the membrane overnight with corresponding primary antibodies (Grp78, p-PERK, PERK, CHOP, Caspase-12, JNK, p-JNK, Bax, Bcl-2, and β-actin). Afterwards, the membrane was incubated with secondary antibody conjugated with horseradish peroxidase at a 1:1000 dilution. β-actin was used as an internal standard. At least 3 separate experiments were performed with different lysates to confirm changes in the protein levels.
Statistical analysis
All experiments were repeated at least 3 times. The results are presented as mean ±S.E.M. or mean ±S.D. The differences between groups were analyzed using one-way analysis of variance (ANOVA) and the post hoc Tukey test to identify specific differences between groups. The level of statistical significance was set at p<0.05.
Results
4-PBA ameliorated I/R-induced heart dysfunction in isolated rat hearts
To determine the therapeutic implications of 4-PBA on I/R injuries, we employed an ex vivo Langendorff model. As illustrated in Figure 1B, after a 15-min stabilization period and the drug processing, adult rat hearts were subjected to 35 min of global ischemia followed by 60 min of reperfusion. The LVSP (left ventricular systolic pressure), LVEDP (left ventricular end-diastolic pressure), +dP/dtmax (maximum rate of contraction), −dP/dtmax (maximum rate of relaxation), HR (heart rates), and LVDP (Left Ventricular Developed Pressure) ×HR were recorded at the end of the experiments to evaluate the heart function. The results presented in Table 1 show that I/R treatment significantly decreased the levels of heart LVSP, −dP/dtmax, +dP/dtmax, heart rate, and LVDP×HR, and increased the level of LVEDP compared with the control group (all p<0.05). However, 4-PBA (5 mM, 10 mM) treatment significantly reduced this I/R-induced effect on the heart. Especially in the 4-PBA high concentration group (10 mM), the levels of heart LVSP, −dP/dtmax, +dP/dtmax, heart rate, and LVDP×HR were increased to about 25.7%, 104%, 130.6%, 48.1%, and 222% of that in I/R group, respectively, and LVEDP was deceased from 4.4-fold to 2.3-fold of the control compared with I/R group (all p<0.05). Moreover, histopathological examination results represented in Figure 2A confirmed the results. The left ventricles of I/R-impaired hearts were seriously damaged, with widespread edema, necrosis, inflammatory cells infiltration, and separation of cardiac muscle fibers. However, pretreatment with 4-PBA at a dose of 5 or 10 mM significantly alleviated the I/R-induced myocardial injury. Taken together, these results suggest that 4-PBA ameliorated I/R-induced heart dysfunction in Langendorff-perfused isolated rat hearts.
Table 1.
Cardiac function of normal and I/R-impaired rat hearts treated with or without 4-PBA in perfusion solution (mean ±SE, n=10).
| LVSP | LVEDP | +dP/dtmax | −dP/dtmax | HR | LVDP×HR | |
|---|---|---|---|---|---|---|
| Control | 82.77±17.2 | 8.03±2.9 | 2078±402 | 1901±298 | 267.7±20.3 | 20007.9±354.0 |
| I/R | 64.73±8.8# | 35.7±8.2## | 824.8±158## | 629.6±97## | 123.5±22.7## | 3585.2±273.1### |
| 4-PBA (5 mM) +I/R | 74.68±4.3* | 24.5±7.9* | 1569±299** | 1217±211* | 171.4±19.3* | 8600.9±173.5** |
| 4-PBA (10 mM) +I/R | 81.34±7.8** | 18.3±7.5* | 1683±352** | 1452±271** | 182.9±28.3* | 11530.0±306.2*** |
| 4-PBA | 79.57±5.2 | 9.2±3.0 | 1993±315 | 1868±217 | 250.4±11.3 | 17620.6±67.8 |
LVSP – left ventricular systolic pressure; LVEDP – left ventricular end-diastolic pressure; +dP/dtmax – maximum rate of contraction; −dP/dtmax – maximum rate of relaxation; HR – heart rate; LVDP – left ventricular developed pressure.
p<0.05 versus untreated control group;
p<0.01 versus untreated control group;
p<0.001 versus untreated control group;
p<0.05 versus I/R-impaired group;
p<0.01 versus I/R-impaired group,
p<0.01 versus I/R- impaired group.
Figure 2.

Histopathological examination and detection of intracellular antioxidant enzyme activities. (A) Histopathological examination showed the cardioprotective effect of 4-PBA on I/R-impaired hearts (n=6). (B) Intracellular oxidation-reduction condition and antioxidant enzyme activities of isolated hearts were examined by measuring LDH, ROS, CK, MDA, SOD, and GSH-Px (n=6). #4p<0.05 versus untreated control group; ## p<0.01 versus untreated control group; ### p<0.001 versus untreated control group; * p<0.05 versus I/R-impaired group; ** p<0.01 versus I/R-impaired group; *** p<0.001 versus I/R-impaired group.
4-PBA suppressed I/R-induced oxidative stress in isolated rat hearts
In the process of ischemia and reperfusion, oxygen-derived free radicals are thought to play an important role in the genesis of tissue injury [16]. The effects of I/R treatment on antioxidant enzyme activities were further evaluated in Langendorff rat hearts. Results presented in Figure 2B indicate that I/R caused significant decreases in SOD and GSH-Px activities (p<0.01 or p<0.001) and increases in LDH, ROS, MDA, and CK production (p<0.01 or p<0.05). However, these changes were effectively improved by 4-PBA pretreatment in a dose-dependent manner, indicating that 4-PBA protects myocardium from injuries induced by oxidative stress.
4-PBA suppressed I/R-induced myocardial apoptosis
Apoptotic cells in the paraffin sections were identified by TUNEL (In Situ Cell Death Detection Kit, Roche). There were significant increases in TUNEL-positive cells, indicating cell apoptosis and cell death of I/R-treated hearts as compared to controls. Treatment with 4-PBA (5 mM, 10 mM) significantly decreased the number of TUNEL-positive cells in the left ventricle region of the heart (Figure 3).
Figure 3.

4-PBA attenuated I/R-induced myocardial apoptosis in rat hearts. (A) TUNEL staining of myocardium from the control group, I/R-treated group, 4-PBA group, and 4-PBA (5 mM, 10 mM) plus I/R group (C). Original magnification ×100. (B) TUNEL-positive cells were counted in more than 300 myocytes in 3 random fields and expressed as percentage of total nuclei. Data are mean ± SEM (n=3). ### p<0.001 versus untreated control group; ** p<0.01 versus I/R- impaired group; *** p<0.001 versus I/R-impaired group.
4-PBA suppressed I/R-induced overexpression of ER stress-associated apoptosis pathway proteins, thus alleviating ER stress and providing cardioprotection
The potential cardioprotective effects of 4-PBA against I/R-induced heart injury were further explored by immunoblotting analysis. Total soluble protein of isolated rat hearts was extracted and subjected to the following experiments. The ER protein chaperone BiP, also known as Grp78, plays a major role in the progress of ER stress and is highly expressed in many diseases [17,18]. According to the results in Figure 4A–4C, the expression level of Grp78 was markedly increased in the I/R group by 1.5-fold as compared with the control group (p<0.01), and the phosphorylation of PERK was increased to 2.8-fold of that in the control group (p<0.001), suggesting that I/R initiated the ER stress response. The induction of CHOP by the PERK-dependent pathway plays a convergent role in the UPR and has been identified as one of the most important mediators of ER stress-induced apoptosis protein [19]. In addition, the phosphorylation of JNK and the downstream Bcl-2 family proteins activation are widely involved in ER stress-induced apoptosis [20]. As shown in Figure 4A, 4D–4G, I/R significantly increased the relative protein levels of CHOP to 1.9-fold of that in the control group, and Caspase-12 to 3.5-fold, Bax to 8.8-fold, p-JNK to 3.2-fold of that in the control group, respectively. However, this I/R-induced processing was significantly suppressed by 4-PBA. Compared to the I/R group, 4-PBA treatment significantly increased the expression of anti-apoptotic protein Bcl-2, decreased pro-apoptotic proteins CHOP, Caspase-12, Bax expression, and inhibited the phosphorylation of JNK (Figure 4, p<0.05, p<0.01 or p<0.001). However, the 4-PBA treatment alone had no significant effects on the expression levels of the protein mentioned above (p>0.05). Collectively, these results indicate that ER stress is involved in the process of I/R-induced myocardium injury, and 4-PBA can inhibit the PERK-related ER stress pathway and its downstream apoptosis pathways, thus providing cardioprotective effects in isolated rat hearts.
Figure 4.

Involvement of ER stress and associated apoptosis signaling pathways in the cardioprotective effects of 4-PBA. (A) The expression levels of Grp78, p-PERK, PERK, CHOP, Bax, Bcl-2, p-JNK, and JNK were detected by immunoblotting assay. The normalized relative levels of Grp78 (B), p- PERK (C), CHOP (D), Caspase-12 (E), Bax (F), and p-JNK (G) were determined by densitometry. The results are represented as means ±SD from 3 independent experiments (n=3). # p<0.05 versus untreated control group; ## p<0.01 versus untreated control group; ### p<0.001 versus untreated control group; * p<0.05 versus I/R-impaired group; ** p<0.01versus I/R-impaired group; *** p<0.001 versus I/R-impaired group.
Discussion
Ischemic heart diseases are severe health problem worldwide, and ischemia-reperfusion injury plays a major role in their high morbidity and mortality [21]. Our previous study investigated whether 4-PBA reduces I/R-induced cardiomyocyte apoptosis in vitro [15]. In the present experiments, we demonstrated that 4-PBA pretreatment alleviates the deterioration of cardiac contractile function, prevents oxidative stress, and decreases myocardium apoptosis in I/R-impaired isolated rat hearts. Our results indicate that 4-PBA ameliorates the I/R- induced myocardial dysfunction and cell apoptosis, and delays the onset of ER stress by inhibiting the overexpression of Grp78 and phosphorylation of PERK. The inhibition of oxidative stress was also involved in the cardioprotective effects of 4-PBA.
Cardiovascular diseases are the leading cause of death in many countries. In order to prevent myocardium from further damage, the best therapeutic strategy for myocardial infarction is to reestablish the blood flow [22,23]. Nevertheless, ischemia/reperfusion (I/R) injury is inevitable, and causes a gradual decline of cardiac function [24]. Therefore, the exploration of new therapeutic agents that reduce I/R injury is very important. In a previous study, we comprehensively illustrated the inhibitory effects of 4-PBA on cardiomyocytes death and apoptosis induced by I/R in vitro [15]. We showed that 4-PBA could effectively inhibit I/R injury-mediated apoptosis pathways by modulating the expression of ER stress moderator proteins (Grp78, ATF6, and PERK) and their associated apoptosis pathways, including CHOP, Caspase-12, JNK, and the Bcl-2 family proteins (Bax and Bcl-2). In this regard, the central aim of this study was to explore whether 4-PBA improves heart function in isolated rat hearts subjected to I/R and elucidated the potential mechanisms involved in 4-PBA-induced cardioprotection. To address this question, we established a myocardial I/R model in isolated rat hearts. We observed that I/R injury irreversibly decreased the levels of heart LVSP, −dP/dtmax, +dP/dtmax, heart rate, and LVDP× HR, while increasing the level of LVEDP (Tab. 1, p<0.05 or p<0.01). However, 4-PBA pretreatment largely ameliorated this I/R-induced heart dysfunction. Also, histopathological examination confirmed that 4-PBA pretreatment inhibited the I/R-initiated widespread myocardial edema, necrosis, inflammatory cells infiltration, and cardiac muscle fiber separation (Figure 2A).
Cell death during I/R is a widely accepted reason for myocardium I/R injury. Numerous studies in humans and animals have indicated the association between the progression of endoplasmic reticulum (ER) stress and apoptotic loss of cardiomyocytes [25,26]. Treatment with 4-PBA (5 mM, 10 mM) significantly decreased the number of TUNEL-positive cells (Figure 3), indicating the inhibition of apoptosis by 4-PBA in the process of I/R injury. ER stress is the result of the accumulation of unfolded or misfolded proteins in the ER lumen. Misfolding of proteins can be caused by endogenous factors such as genetic mutations, as well as exogenous factors such as I/R-initiated abnormal oxidative status and disrupted calcium homeostasis [27,28]. Glucose-regulated protein 78 (Grp78) is an important ER molecular chaperone regulating protein folding, facilitating, translocation, and secretion in the ER [29]. Under homeostatic conditions, Grp78 is bound to 3 ER transmembrane proteins that act as signal transducers, activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and protein kinase-like ER kinase (PERK) [30]. Increased numbers of unfolded proteins within the ER prompt Grp78 to “undock” from IRE1, PERK, and ATF6. We found that I/R significantly increased the relative protein levels of Grp78 and p-PERK (Figure 3, p<0.05, p<0.01, and p<0.001), indicating the activation of ER stress. In addition, I/R activated the downstream apoptosis proteins CHOP, Caspase-12, p-JNK, and Bax, and decreased the level of Bcl-2, thus initiating the associated apoptosis (Figure 3, p<0.05, p<0.01, and p<0.001). CHOP is a critical pro-apoptotic transcription factor during ER-initiated apoptosis; it can mediate transcriptional induction of BIM, a pro-apoptotic BH3-only protein, and inhibit Bcl-2 at the same time [31,32]. Caspase-12 and phosphorylated JNK are also involved in this process [21,33,34]. However, this I/R-induced processing is significantly suppressed by 4-PBA. Compared to the I/R group, 4-PBA treatment significantly increased the expression level of Bcl-2, and decreased protein expression of Grp78, p-PERK, and apoptosis proteins CHOP, Caspase-12, p-JNK, and Bax (Figure 3, p<0.05, p<0.01, and p<0.001), suggesting that 4-PBA can prevent I/R-initiated ER stress and its associated apoptosis.
The relationship between reactive oxygen species (ROS) and myocardium I/R injury is dynamic in the body [35]. However, in some pathological conditions, for example I/R injury, the generation of ROS is a signal generated by misfolded proteins in the ER that causes UPR activation and cell death. However, how protein misfolding and oxidative stress affect each other has not yet been explored in plant systems [35,37]. Endogenous antioxidants, including SOD and GSH-Px, are depleted and ROS are accumulated. In the present study, I/R injury was associated with increased oxidative stress, as evidenced by increases in myocardial LDH, ROS, CK, and MDA, and depletion of myocardial endogenous antioxidants such as SOD and GSH-Px (Figure 2, p<0.05, p<0.01, and p<0.001). On the other hand, the ROS produced by I/R can be scavenged directly by 4-PBA, thereby preventing lipid peroxidation and helping to maintain membrane integrity. This is further supported by the decreased levels of LDH, ROS, CK, and MDA in the 4-PBA-treated group (Figure 2, p<0.05, p<0.01, and p<0.001).
ER stress-related accumulation of unfolded/misfolded proteins and the subsequent apoptosis are important pathways for the progression of many diseases [38,39]. Myocardium ischemia-reperfusion injury causes cardiovascular disorders by the disturbance of the UPR and leads to apoptotic cell death [40]. 4-PBA, a molecule currently used to clinically treat urea cycle disorders, has received attention as a small chemical chaperone that modulates UPR activation [41,42]. In the present study we elucidated the significant protective effects of 4-PBA against I/R injuries in isolated rat hearts and illustrated its suppression of ER stress and associated apoptosis. The effect of 4-PBA on alleviation of ER stress may be associated with the inhibition of oxidative stress. This study provides new insights into the progression of ER stress in I/R, and also provides a novel drug candidate for preventing myocardium I/R injury in the future. However, the deeper mechanism involved in the cardioprotective effect of 4-PBA requires further study.
Conclusions
The results of this study revealed that a chemical chaperone 4-phenylbutyric acid has potent cardioprotective activity against heart ischemia-reperfusion injuries. We demonstrate that 4-PBA ameliorates the I/R-induced myocardial dysfunction and delays the onset of ER stress by inhibiting the overexpression of Grp78 and phosphorylation of PERK and that the inhibition of oxidative stress is involved in the cardioprotection effects of 4-PBA.
Footnotes
Conflict of interest statement
The authors report that they have no conflicts of interest.
Source of support: Departmental sources
References
- 1.Logue SE, Gustafsson AB, Samali A, et al. Ischemia/reperfusion injury at the intersection with cell death. J Mol Cell Cardiol. 2005;38:21–33. doi: 10.1016/j.yjmcc.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 2.Bagur R, Tanguy S, Foriel S, et al. The impact of cardiac ischemia/reperfusion on the mitochondria-cytoskeleton interactions. Biochim Biophys Acta. 2016;1862:1159–71. doi: 10.1016/j.bbadis.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Bliksoen M, Baysa A, Eide L, et al. Mitochondrial DNA damage and repair during ischemia-reperfusion injury of the heart. J Mol Cell Cardiol. 2015;78:9–22. doi: 10.1016/j.yjmcc.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Yuan Y, Guo Q, Ye Z, et al. Ischemic postconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PI3K-Akt pathway. Brain Res. 2011;1367:85–93. doi: 10.1016/j.brainres.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 5.Ong SB, Samangouei P, Kalkhoran SB, et al. The mitochondrial permeability transition pore and its role in myocardial ischemia-reperfusion injury. J Mol Cell Cardiol. 2015;78:23–34. doi: 10.1016/j.yjmcc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Scarabelli TM, Gottlieb RA. Functional and clinical repercussions of myocyte apoptosis in the multifaceted damage by ischemia/reperfusion injury: Old and new concepts after 10 years of contributions. Cell Death Differ. 2004;11(Suppl 2):S144–52. doi: 10.1038/sj.cdd.4401544. [DOI] [PubMed] [Google Scholar]
- 7.Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–10. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 8.Sozen E, Karademir B, Ozer NK. Basic mechanisms in endoplasmic reticulum stress and relation to cardiovascular diseases. Free Radic Biol Med. 2015;78C:30–41. doi: 10.1016/j.freeradbiomed.2014.09.031. [DOI] [PubMed] [Google Scholar]
- 9.Powell KS, Latterich M. The making and breaking of the endoplasmic reticulum. Traffic. 2000;1:689–94. doi: 10.1034/j.1600-0854.2000.010901.x. [DOI] [PubMed] [Google Scholar]
- 10.Nishitoh H. Life and death under the ER stress condition. Journal of Oral Biosciences. 2004;46:259–69. [Google Scholar]
- 11.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–70. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolb PS, Ayaub EA, Zhou W, et al. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol. 2015;61:45–52. doi: 10.1016/j.biocel.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Li P, Zhang L, Zhang M, et al. Uric acid enhances PKC-dependent eNOS phosphorylation and mediates cellular ER stress: A mechanism for uric acid-induced endothelial dysfunction. Int J Mol Med. 2016;37:989–97. doi: 10.3892/ijmm.2016.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy D, Kumar V, James J, et al. Evidence that chemical chaperone 4-phenylbutyric acid binds to human serum albumin at fatty acid binding sites. PLoS One. 2015;10(7):e0133012. doi: 10.1371/journal.pone.0133012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jian L, Lu Y, Lu S, Lu C. Chemical chaperone 4-phenylbutyric acid protects H9c2 cardiomyocytes from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis. Mol Med Rep. 2016;13:4386–92. doi: 10.3892/mmr.2016.5063. [DOI] [PubMed] [Google Scholar]
- 16.Hanada S, Harada M, Kumemura H, et al. Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol. 2007;47:93–102. doi: 10.1016/j.jhep.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 17.Yan XH, Guo XY, Jiao FY, et al. Activation of large-conductance Ca(2+)-activated K(+) channels inhibits glutamate-induced oxidative stress through attenuating ER stress and mitochondrial dysfunction. Neurochem Int. 2015;90:28–35. doi: 10.1016/j.neuint.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Chan JY, Biden TJ, Laybutt DR. Cross-talk between the unfolded protein response and nuclear factor-kappaB signalling pathways regulates cytokine-mediated beta cell death in MIN6 cells and isolated mouse islets. Diabetologia. 2012;55:2999–3009. doi: 10.1007/s00125-012-2657-3. [DOI] [PubMed] [Google Scholar]
- 19.Allagnat F, Fukaya M, Nogueira TC, et al. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells. Cell Death Differ. 2012;19:1836–46. doi: 10.1038/cdd.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bravo R, Gutierrez T, Paredes F, et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int J Biochem Cell Biol. 2012;44:16–20. doi: 10.1016/j.biocel.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duehrkop C, Rieben R. Ischemia/reperfusion injury: Effect of simultaneous inhibition of plasma cascade systems versus specific complement inhibition. Biochem Pharmacol. 2014;88:12–22. doi: 10.1016/j.bcp.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Ilic MD, Ilic S. [Prevalence and prognostic significance of silent myocardial ischemia in patients after myocardial infarction]. Vojnosanit Pregl. 2007;64:519–23. doi: 10.2298/vsp0708519d. [in Serbian] [DOI] [PubMed] [Google Scholar]
- 23.Chien CY, Chien CT, Wang SS. Progressive thermopreconditioning attenuates rat cardiac ischemia/reperfusion injury by mitochondria-mediated antioxidant and antiapoptotic mechanisms. J Thorac Cardiovasc Surg. 2014;148:705–13. doi: 10.1016/j.jtcvs.2013.12.065. [DOI] [PubMed] [Google Scholar]
- 24.Wallis EJ, Ramsay LE. Primary and secondary prevention of coronary heart disease. Medicine. 2002;30:59–64. [Google Scholar]
- 25.Qi X, Vallentin A, Churchill E, et al. deltaPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart. J Mol Cell Cardiol. 2007;43:420–28. doi: 10.1016/j.yjmcc.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duan SR, Wang JX, Wang J, et al. Ischemia induces endoplasmic reticulum stress and cell apoptosis in human brain. Neurosci Lett. 2010;475:132–35. doi: 10.1016/j.neulet.2010.03.058. [DOI] [PubMed] [Google Scholar]
- 27.McLaughlin M, Vandenbroeck K. The endoplasmic reticulum protein folding factory and its chaperones: New targets for drug discovery? Br J Pharmacol. 2011;162:328–45. doi: 10.1111/j.1476-5381.2010.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Wang L, Kern TS, et al. Inhibition of poly(ADP-ribose) polymerase inhibits ischemia/reperfusion induced neurodegeneration in retina via suppression of endoplasmic reticulum stress. Biochem Biophys Res Commun. 2012;423:276–81. doi: 10.1016/j.bbrc.2012.05.109. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Lv M, Jia J, et al. Expression of the 78 kD glucose-regulated protein is induced by endoplasmic reticulum stress in the development of hepatopulmonary syndrome. Gene. 2014;537:115–19. doi: 10.1016/j.gene.2013.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo T, Kim JK, Chen B, et al. Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis. Chem Biol Interact. 2015;225:90–98. doi: 10.1016/j.cbi.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao J, Zhang C, Wang P, et al. C/EBP homologous protein (CHOP) contributes to hepatocyte death via the promotion of ERO1alpha signalling in acute liver failure. Biochem J. 2015;466:369–78. doi: 10.1042/BJ20140412. [DOI] [PubMed] [Google Scholar]
- 32.Yin XL, Zhang W, Yang Y, et al. Increasing expression of (CCAAT enhancer binding protein) homologous protein induced by endoplasmic reticulum stress in myocardium after cardiac arrest and resuscitation in rat. Resuscitation. 2012;83:378–85. doi: 10.1016/j.resuscitation.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 33.Testai L, Martelli A, Marino A, et al. The activation of mitochondrial BK potassium channels contributes to the protective effects of naringenin against myocardial ischemia/reperfusion injury. Biochem Pharmacol. 2013;85:1634–43. doi: 10.1016/j.bcp.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa A, Sullivan KD, Xue D. Caspase-activated phosphoinositide binding by CNT-1 promotes apoptosis by inhibiting the AKT pathway. Nat Struct Mol Biol. 2014;21:1082–90. doi: 10.1038/nsmb.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozgur R, Turkan I, Uzilday B, et al. Endoplasmic reticulum stress triggers ROS signalling, changes the redox state, and regulates the antioxidant defence of Arabidopsis thaliana. J Exp Bot. 2014;65:1377–90. doi: 10.1093/jxb/eru034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pisarenko O, Shulzhenko V, Studneva I, et al. Structural apelin analogues: Mitochondrial ROS inhibition and cardiometabolic protection in myocardial ischaemia reperfusion injury. Br J Pharmacol. 2015;172:2933–45. doi: 10.1111/bph.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng XS, Jia JJ, Ma LF. Gensenoside Rb1 protects rat PC12 cells from oxidative stress-induced endoplasmic reticulum stress: The involvement of thioredoxin-1. Mol Cell Biochem. 2015;410:239–46. doi: 10.1007/s11010-015-2557-1. [DOI] [PubMed] [Google Scholar]
- 38.Zha X, Yue Y, Dong N, et al. Endoplasmic reticulum stress aggravates viral myocarditis by raising inflammation through the IRE1-associated NF-kappaB pathway. Can J Cardiol. 2015;31:1032–40. doi: 10.1016/j.cjca.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Liu G, Liu N, Xu Y, et al. Endoplasmic reticulum stress-mediated inflammatory signaling pathways within the osteolytic periosteum and interface membrane in particle-induced osteolysis. Cell Tissue Res. 2016;363:427–47. doi: 10.1007/s00441-015-2205-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo XF, Yang XJ. Endoplasmic reticulum stress response in spontaneously hypertensive rats is affected by myocardial ischemia-reperfusion injury. Exp Ther Med. 2015;9:319–26. doi: 10.3892/etm.2014.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuang X, Hu W, Yan M, et al. Phenylbutyric acid suppresses protein accumulation-mediated ER stress in retrovirus-infected astrocytes and delays onset of paralysis in infected mice. Neurochem Int. 2010;57:738–48. doi: 10.1016/j.neuint.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srinivasan K, Sharma SS. Sodium phenylbutyrate ameliorates focal cerebral ischemic/reperfusion injury associated with comorbid type 2 diabetes by reducing endoplasmic reticulum stress and DNA fragmentation. Behav Brain Res. 2011;225:110–16. doi: 10.1016/j.bbr.2011.07.004. [DOI] [PubMed] [Google Scholar]