

Abstract
Research on extracting biomedical relations has received growing attention recently, with numerous biological and clinical applications including those in pharmacogenomics, clinical trial screening and adverse drug reaction detection. The ability to accurately capture both semantic and syntactic structures in text expressing these relations becomes increasingly critical to enable deep understanding of scientific papers and clinical narratives. Shared task challenges have been organized by both bioinformatics and clinical informatics communities to assess and advance the state-of-the-art research. Significant progress has been made in algorithm development and resource construction. In particular, graph-based approaches bridge semantics and syntax, often achieving the best performance in shared tasks. However, a number of problems at the frontiers of biomedical relation extraction continue to pose interesting challenges and present opportunities for great improvement and fruitful research. In this article, we place biomedical relation extraction against the backdrop of its versatile applications, present a gentle introduction to its general pipeline and shared resources, review the current state-of-the-art in methodology advancement, discuss limitations and point out several promising future directions.
Keywords: biomedical relation extraction, natural language processing, graph mining, machine learning, scientific literature, clinical narratives
Introduction
Relation extraction from text documents is an important task in knowledge representation and inference to create or augment structured knowledge bases and in turn support question answering and decision making. The task generally involves annotating unstructured text with named entities and identifying the relations between these annotated entities. State-of-the-art named entity recognizers can automatically annotate text with high accuracy [1, 2], but relation extraction is not as straightforward. General domain relation extraction is an active research area for decades [3]. In the biomedical and clinical domain, extracting relations from scientific publications and clinical narratives has been gaining traction over the past decade, and is the focus of this review.
To illustrate the importance of biomedical relation extraction, consider that in lymphoma pathology reports, immunophenotypic features are expressed as relations among medical concepts. For example, in ‘[large atypical cells] are positive for [CD30] and negative for [CD15]', ‘large atypical cells', ‘CD30' and ‘CD15' are medical concepts; ‘CD30' and ‘CD15' are cell surface antigens. A bag-of-words or bag-of-concepts representation would fail to capture whether ‘large atypical cells' are positive or negative for ‘CD30' or ‘CD15'. In this and many other similar cases, the biomedical concepts need to be linked through syntax and/or semantics to be informative and to resolve ambiguities by putting the concepts into context.
In this article, we define a relation as a tuple r(c1,c2,…, cn), n ≥ 2, where ci’s are concepts (named entities) and the ci’s are semantically and/or syntactically linked to form relation r, as expressed in text. Thus, a single named entity is generally not regarded as a relation, neither is an assertion. In other words, a relation involves at least two concepts. If n is two (three), we call the relation a binary (ternary) relation, and for general n an n-ary relation. Some researchers use the term relation to focus on triples that represent binary relations [e.g. positive-expression (large atypical cells, CD30), negative-expression (large atypical cells, CD15)]. Others also consider composite relations, e.g. and[positive-expression (large atypical cells, CD30), negative-expression (large atypical cells, CD15)].
We also use the term relation to include what are often referred to as events, e.g. the ternary relation treated_by (patient, Imatinib regimen, 5 months) as expressed in ‘[the patient] was put on [Imatinib regimen] for [5 months]' can also be parsed as an event, where the event trigger is ‘put', theme is ‘Imatinib regimen' and target argument is ‘patient'. Nested events may occur when one event takes other events as arguments; Figure 1 shows another more complex example, which can be interpreted as a nested event with solid and dashed boxes indicating two argument events. In computational linguistics, events are often defined as grammatical objects that combine lexical elements, logical semantics, and syntax [4]. Figure 1 shows that the notions of binary relations, n-ary relations, events and nested events are closely related. As will be evidenced in the section on state-of-the-art methods, the natural language processing (NLP) techniques for extracting relations and events are often similar in principle. Thus, we include both relation and event extraction in our review, and we use both ‘relation' and ‘event', with the choice made to be consistent with the literature being referenced.
Figure 1.

Relations from an example sentence, using graph representation, where nodes are named entities and edges indicate the relations between two nodes (or multiple named entities connected by multiple edges can be considered as one relation). Named entities considered are in bold in the sentence. The dashed box denotes a binary relation, i.e. with two named entities. The solid box denotes a relation with multiple named entities, which alternatively can be viewed as a collection of three binary relations. These relations (in solid box and dashed box) can also be regarded as events, and the entire graph can be interpreted as a nested event (where the solid box and the dashed box are nodes and are connected by ‘evaluate' as indicated by the heavy stroke edge). Because both relations and graphs may have directed and undirected representations, we show edges as undirected to be more general.
The representation of relations has been a subject of knowledge representation research for decades [5], with various alternatives. One representation uses composed simple logical forms. For example, Resource Description Framework (RDF) or Web Ontology Language encodes complex relations by multiple triples, where the elements of these triples can themselves be other composed forms. Thus, binary relations such as positive-expression (large atypical cells, CD30) have the following subject-predicate-object triple representation: large atypical cells-positively express-CD30. A more powerful alternative is the sentential logic (or propositional logic) representation [5], in which relations are propositions or composed propositions using logical connectives (e.g. ‘and' for conjunction, ‘or' for disjunction). Compared with RDF triples, propositional logic has additional constructs such as connectives and inference rules, thus is more expressive. A third alternative is the graph-based representation in which nodes are named entities and edges indicate relationships, or multiple named entities connected by multiple edges can be regarded as one relation, as shown in Figure 1.
This review focuses on NLP methods using graph-based representations and algorithms to extract biomedical relations from unstructured text. Regarding alternative representations, the sentential logic representation can be noted with graph-based representation [6]. Biomedical relations (including events) can be universally represented as graphs by converting biomedical concepts to nodes and syntactic/semantic links to edges. Other propositional representations may require specific interpretation of the graphs. For instance, representing the negation of a proposition may require the introduction of nested graphs, and to give special semantics to a relation labeled NOT. Furthermore, although composition leads to complexity (e.g. n-ary relations or nested relations), by adopting a graph-based representation, we can focus on common syntactic and semantic graphical patterns that provide good ways to capture relations. In fact, as will become clear later in this review, almost all state-of-the-art methods for extracting relations are graph-based. This article assumes that the reader has a basic knowledge of biomedical NLP; see [7–12] for introductions and surveys on latest applications.
The reader should also be aware of a body of research on curated structured knowledge bases, with manual annotations of biomedical relations by experts. Some of these knowledge bases are biologically focused, such as KEGG [13], STRING [14], InterPro [15] and InterDom [16]. Others are more clinically focused, such as PharmGKB [17], VARIMED [18] and ClinVar [19]. However, the expert sourcing methods often scale poorly with the exponentially growing body of biomedical free text. Thus, automated methods present a promising direction for discovering relations to augment existing knowledge bases; this motivated many methods discussed in this article.
Application of biomedical relation extraction
Extracting biomedical relations has numerous applications that vary from advancing basic sciences to improving clinical practices, as shown in Figure 2. These applications include but are not limited to biomolecular information extraction, clinical trial screening, pharmacogenomics, diagnosis categorization, as well as discovery of adverse drug reactions (ADRs) and drug–drug interactions (DDIs).
Figure 2.
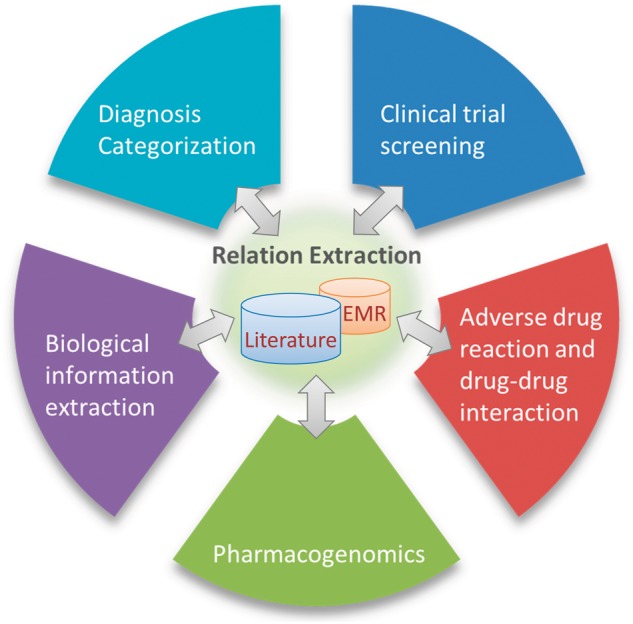
Applications of biomedical relation extraction. The bidirectional arrows indicate that on the one hand, automated methods for relation extraction can help biological and clinical investigations; on the other hand, these applications can in turn provide shared resources (e.g. corpora and knowledge base).
Biomolecular information extraction
To keep up with the exponential growth of the literature, automated methods have been applied to mining protein–protein interactions (PPIs) [20, 21], gene–phenotype associations [22, 23], gene ontology [24] and pathway information [25], which we collectively call biomolecular information extraction. Such relation mining has shown its value in the prioritization of cancerous genes for further validation from a large number of candidates [26]. Many of these approaches apply NLP methods to extract known disease–gene relations from the literature, which are then used to predict novel disease–gene relations [27–31].
Clinical trial screening
Archived clinical and research data have been made available by governmental agencies and corporations, such as ClinicalTrials.gov [32]. Clinical trials are in large part characterized by eligibility criteria, some of which can be captured via relations (e.g. no [diagnosis] for [rheumatoid arthritis] for at least [6 months]). Electronic screening can improve efficiency in clinical trial recruitment, and intelligent query over trials can support clinical knowledge curation [33]. Recently, NLP support has proved useful in automatically detecting named entities in eligibility criteria [34, 35], and further in extracting relations between named entities to characterize eligibility criteria [36–38].
Pharmacogenomics
Pharmacogenomics aims to understand how different patients respond to drugs by studying relations between drug response phenotypes and patient genetic variations. Much of this knowledge can be mined from scientific literature and curated in databases to enable discovering new relationships. One such database is the Pharmacogenetics Research Network and Knowledge Base (PharmGKB [39]). Initial efforts to populate PharmGKB included a mixture of expert annotation and rule-based approaches. Recent approaches have extended to using semantic and syntactic analysis as well as statistical machine learning tools to mine targeted pharmacogenomics relations from scientific literature and clinical records [40–42].
Diagnosis categorization
Diagnosis categorization enables automated billing and patient cohort selection for secondary research. Developed systems can automatically code and classify diagnoses from Electronic Medical Records (EMRs) [43–47]. Recent work demonstrated the success of extracting semantic relations and using these relations as additional features in diagnosis categorization, some through better grouping features using curated relations [48], others through unsupervised learning to extract more expressive representation of relations between medical concepts [49, 50].
ADR and DDI
ADR refers to unexpected injuries caused by taking a medication. DDI happens when a drug affects the activity of another drug simultaneously administered. ADR is an important cause of morbidity and mortality [51], and DDIs may cause reduced drug efficacy or drug overdose. Detecting potential ADRs and DDIs can guide the process of drug development. An increasing number of methods have leveraged the scientific literature and clinical records using NLP. These systems often explore the relations between drugs, genes and pathways, and discover ADRs [52–54] and DDIs [55, 56] stated in text. A large amount of research in recent years also explored user-generated content in social networks to detect ADRs, see [57] for a recent review.
General pipeline for biomedical relation extraction
In Figure 3, we first present a general pipeline, summarized from the reviewed approaches, as a cookbook to follow either in part or as a whole for extracting biomedical relations. The reader can refer the components discussed in the state-of-the-art methods to this cookbook to gain comprehensive understanding. For completeness, we assume documents as input and extracted relations as output. The pipeline starts with section recognition, which distinguishes text under different section headings (e.g. ‘Chief Complaints' or ‘Past Medical History'). Sentence breaking then automatically decides where sentences in a paragraph begin and end. Typographical analysis investigates features such as capitalization and usage of alphanumeric characters. Stemming reduces the inflected words to the root form (e.g. performed to perform). POS tagging assigns a part-of-speech tag for each word in the sentence (e.g. VBN for ‘performed' in the sentence in Figure 1). Parsing is the process of assigning a syntactic structure to a sentence (e.g. the constituency or dependency structure obtained by Stanford Parser). The results from typographical analysis, stemming, POS tagging and parsing can provide features for recognizing anaphora (coreference resolution) and typed concepts (concept recognition). Coreference resolution and concept resolution can also improve parsing accuracy. Together with parsing, they are essential in generating the graph representation for a sentence and labeling semantic roles of concepts in the graph representation (Semantic Role Labeling). The graph representation is the foundation for graph mining, and along with upstream steps including direct regular expression feature extraction, leads to the generation of semantically and syntactically enriched features. These features then support either rule-based, feature-space-based or kernel-based relation extraction system. Many biomedical relation extraction systems rely on external knowledge sources [e.g. Unified Medical Language System (UMLS) [58]].
Figure 3.

General workflow of biomedical relation extraction. See Section General Pipeline for Biomedical Relation Extractionfor a description of each step. The shaded cloud denotes that the external resources (terminology, ontology and knowledge bases) can be used by some or all of the above steps.
The pipeline can also be used as a foundation for downstream applications such as logical inference with extracted relations. The pipeline covers steps for breaking the documents to sentences, understanding the semantic and syntactic structures of sentences and constructing a multitude of features for relation extraction. We refer the reader to the description of each step in the caption of Figure 3. We emphasize the role of graph mining in the pipeline as a central concept. The mined graphs provide a converging point for methods that combine local features (e.g. tokens and part-of-speech tags), a diverging point where more integrated features (e.g. relations as features) are constructed, and a bridge to connect the syntax and semantics.
Introduction to graph representations and graph algorithms for biomedical relation extraction
Graph representation for narrative sentences naturally breaks down to the choice of information represented by nodes and edges. Common node choices include tokens (e.g. biopsy), named entities (e.g. bone marrow biopsy), semantically labeled named entity (e.g. bone marrow biopsy-Diagnostic Procedure, where Diagnostic Procedure is a UMLS semantic type) and relations themselves within nested relations. Those choices are in the order of encoding increasingly enriched and complex semantic information. Common edge choices include syntactic dependency (e.g. dobj for direct object, in dependency parsing), syntactic constituency link (e.g. NP-NN, in constituency parsing), event argument (e.g. event theme) and association (e.g. based on co-occurrence or customized statistics) as edge. These choices integrate semantic and syntactic information into the graph representation to different degrees. Graph mining algorithms can be applied to extract relations directly or construct useful features. Frequently used algorithms can be categorized as follows: some identify shortest path (or its variant) between concept pair, which can be performed using standard algorithms such as Dijkstra’s algorithm [59]; some create association graphs then try to apply customized labels to them; some use subgraph matching to compare the similarity between subgraphs based on a score that aggregates node distances and edge distances; some carry out frequent subgraph or subtree pattern mining to directly extract candidate relations; some directly parse graph representation of relations from sentences by integrating the graph structure into the learning objective of the parsers. When reviewing state-of-the-art methods in the next section, we characterize each method along the axis of graph-encoded information, graph algorithms and intended usage, which is summarized in Table 1 for the reader’s convenience.
Table 1.
Summarization and characterization of relation extraction systems
| Methods | Parsersa | Graph algorithm and usageb | Information | CoRef | External resources |
|---|---|---|---|---|---|
| Liu et al. [60, 61], Mackinlay et al. [62], Ravikumar et al. [63] | McCCJ, SD | Exact subgraph matching, approximate subgraph matching to match dependency graphs to event graphs | TN, DE | No | PDB, Uniprot, Biothesaurus |
| Björne et al. [64, 66], Hakala et al. [67] | McCCJ, SD | Identifying shortest path to generate features for event arguments, rule-based graph pruning to exclude invalid event arguments | NEN, REN, EAE | No | Uniprot, SubtiWiki, Wordnet, DrugBank |
| Kilicoglu et al. [68, 69] | McCCJ, SD | Transforming dependency graphs to embedding graphs, and rule-based traversal of embedding graphs to map them to events | NEN, REN, DE | Yes | Compiled dictionaries |
| Hakenberg et al. [70, 71] | BioLG | Using customized query language and post-processing rules to match subgraph patterns to events | TN, DE | Yes | Compiled dictionaries, Uniprot, GO |
| Riedel et al. [72, 73] | McCCJ | Scoring candidate graphs to rank the event arguments | NEN, EAE | No | Compiled dictionaries |
| Van Landeghem et al. [74] | Stanford | Identifying minimal event containing subgraph patterns to construct event extraction rules | TN, DE | No | Compiled dictionaries |
| Kaljurand et al. [75] | Pro3Gres | Enumerating dependency paths between the concept pairs to count frequencies of paths in events and calculate likelihood of event arguments | NEN, DE | Yes1 | IntAct |
| Vlachos et al. [76] | RASP | Enumerating dependency paths between the concept pairs to manually identify paths that are likely to generate events | TN, DE | Yes | No |
| McClosky et al. [77, 78] | McCCJ, SD | Using minimum spanning tree algorithm to parse events directly from sentences | NEN, EAE | No | No |
| Quirk et al. [79] | McCCJ, SD; Enju | Identifying shortest paths between the concept pairs to generate dependency chain as features | TN, DE | No | No |
| Miwa et al. [80, 81] | Enju, GDep | Enumerating dependency paths between the concept pairs to generate dependency chain as features | TN, DE | No | UMLS, Wordnet |
| Coulet et al. [40, 82], Percha et al. [42] | Stanford | Enumerating dependency paths between a named entity and a verb, then merging paths ending in the same verb to form binary relations | TN, DE | No | PharmGKB |
| Hakenberg et al. [83] | Stanford | Enumerating subtrees rooted at the lowest common ancestors of candidate concept pairs to subsequently pick the closest pairs | TN, CE | No | UMLS, SIDER, DrugBank, PharmGKB, GNAT |
| Wang et al. [84] | No | Assigning KL divergence as distance of an edge in a association graph, identifying an entity with shortest weighted distance path from target entity to form putative relations and label the graphs | Nodes and edges from a curated association graph, AE | No | Chem2Bio2RDF |
| Bui et al. [85] | Stanford | Using grammatical rules to traverse the tree structures to extract relations | TN, CE | Yes | HIVDB, RegaDB |
| Katrenko et al. [86] | LGP, Minipar, Charniak | Enumerating the lowest common ancestors of concept pairs in the dependency tree to be used as features | TN, DE | No | No |
| Sætre et al. [87] | Enju, GDep | Enumerating dependency paths between the concept pairs to generate dependency chain as features | NEN, DE | No | UniProt, Entrez Gene, GENA |
| Weng et al. [36] | In-house parser | Frequent subtree pattern mining to construct relations representing clinical research eligibility criteria | SNEN, DE | No | UMLS |
| Thomas et al. [88] | McCCJ, SD | Using Graph kernels (e.g. APG, kBSPS) to compare relation instance similarity | TN, DE | No | No |
| Chowdhury et al [89–91] | Stanford, McCCJ, SD | Using tree kernel MEDT to compare relation instance similarity | TN, DE | No | No |
| Luo et al. [49, 50, 92] | Stanford (augmented by UMLS) | Frequent subgraph mining with redundancy removing to extract complex relations without supervision | SNEN, DE | No | UMLS |
| Roberts et al. [93] | Stanford | Identifying shortest paths between the concept pairs to compute edit distances between relation instances | TN, DE | No | UMLS, Wordnet Wikipedia |
| deBruijn et al. [94] | McCCJ, SD | Identifying minimal trees over concept pairs to generate dependence paths between concept covering minimal trees as features | TN, DE | No | UMLS |
| Xu et al. [95] | Kay | Customized parsing to produce conceptual graph representation | NEN, EAE | No | UMLS |
| Solt et al. [96] | Stanford, McCCJ, Enju | Using graph kernels (e.g. APG, kBSPS) to compare relation instance similarity | TN, DE | No | Compiled dictionaries |
| Xu et al. [97] | Stanford | Extracting dependencies (edges) directly as features to classify relations; using CRF to classify body location relation | TN, DE (PE for CRF) | No | UMLS |
| Pathak et al. [98] | No | Using dictionary augmented CRF to detect relation keywords | TN, PE | No | Compiled dictionaries, UMLS |
Abbreviation used in this table include: CoRef, coreference resolution; CRF, conditional random field; HMM, hidden Markov model; APG, all paths graph kernel [20]; kBSPS, k-band shortest path spectrum kernel [99]; MEDT, mildly extended dependency tree kernel [100]; PDB, Protein Data Bank [101]; UMLS, Unified Medical Language System. The key for the parsers are: Stanford—Stanford Parser, McCCJ—McClosky-Charniak-Johnson Parser, Chart—Kay Chart Parser, Enju—Enju Parser, Bikel—Bikel Parser, SD—Stanford Dependency. The key for the information represented by graphs are: TN—token as node, NEN—named entity as node, SNEN—semantically labeled named entity as node, REN—relation/event as node, AE—association as edge, PE—position adjacency as edge (e.g. connecting adjacent tokens), DE—dependency as edge, CE—constituency as edge, EAE—event argument as edge. When Stanford Parser is used with dependency as edge, Stanford Dependency is assumed by default.
aParsers column is filled for only those systems that use dependency or constituency information from the parses.
bGraph Algorithm and Usage column is filled using the format [graph algorithm] to [usage].
State-of-the-art methods for biomedical relation extraction
Methodology for biomedical relation extraction has received increasing attention. Conventional approaches focus on using co-occurrence statistics as a proxy for relatedness [41, 102–105]. Some clinical NLP systems apply hand-crafted syntactic and semantic rules to extract prespecified semantic relations, such as MedLEE [106] and SemRep [107], but are hard to adapt to new subdomains. Recent research focuses more on syntactic parsing, to develop generalizable methods to extract relations that fully explore the constituency and dependency structures of natural language. In this section, we review the state-of-the-art approaches where graph (including tree) mining techniques are used to derive relations from syntactic or semantic parses. We categorize the methods according to whether their corpora mainly concern scientific publications or clinical narratives, as this content difference often has implications for the methods and resources used to extract relations. On the other hand, community challenges continuously help to promote the development of state-of-the-art methods. Thus, in each category, we organize research work around the challenge they participate or as non-challenge-participating research. We summarize the algorithms and systems in Table 1.
Relation extraction from the scientific literature
Over the past decade, continuous effort has been directed to extracting semantic relations from biomedical literature text, often in the form of shared-task community challenges that aim to assess and advance NLP techniques. Notable community challenges include BioNLP shared tasks on event mining, BioCreative shared tasks on PPI extraction and DDIExtraction challenges on DDI extraction. We observed that an increasing number of teams applied graph-based techniques to characterize the semantic relations in these shared tasks. These techniques are frequently placed among the top-performing echelon. This section reviews the graph-based methodologies developed for these challenges. We consider only the papers accepted into the shared task proceedings as full publications, and focus on the top-performing systems. We summarize the f-measures of the best systems in each shared task as a representative evaluation, and refer the reader to the challenge overviews for detailed and comprehensive evaluations. Perhaps through learning the lessons from these challenges, real-world applications such as the field of pharmacogenomics also saw significant momentum in development of graph-based text-mining methods. We devote the last part of this section to recent advances in pharmacogenomics and demonstrate the transfer and adaptation of graph-based algorithms from methodology-oriented research to application-oriented research in biomedical relation extraction.
BioNLP event extraction shared tasks
Three BioNLP shared tasks (ST) have focused on recognizing biological events (relations) from the literature. These tasks provided the protein mentions as input and asked the participating teams to identify a predefined set of semantic relations. Teams were not required to discover the protein mentions. BioNLP-ST-2009 consisted of three tasks, including core event detection, event argument recognition and negation/speculation detection, all based on the GENIA corpus [108]. BioNLP-ST-2011 expanded the tasks and resources to cover more text types, event types and subject domains [109]. Besides the continued GENIA task (GE), BioNLP-ST-2011 added the following tasks: epigenetics and post-translational modification (EPI), infectious diseases (ID), bacteria biotope (BB) and bacteria interaction (BI). BioNLP-ST-2013 further expanded the application domains with tasks of GE, BB, cancer genetics (CG), pathway curation (PC) and gene regulation ontology (GRO) [110]. Table 2 describes those tasks in more detail.
Table 2.
BioNLP event extraction tasks from the 2009, 2011 and 2013 shared tasks
| Tasks | Task descriptions | Year |
|---|---|---|
| GE | Extracting the biomolecular events related to NFB proteins. | 2009, 2011, 2013 |
| EPI | Extracting epigenetic and post-transcriptional modification events. | 2011 |
| ID | Extracting events describing the biomolecular foundations of infectious diseases. | 2011 |
| BB | Extracting the association between bacteria and their habitats. | 2011, 2013 |
| BI | Extracting the bacterial molecular interactions and transcriptional regulations. | 2011 |
| CG | Extracting cancer-related molecular- and cellular-level foundations, tissue- and organ-level effects and organism-level outcomes. | 2013 |
| PC | Extracting signaling and metabolic pathway-related biomolecular reactions. | 2013 |
| GRO | Extracting regulatory events between genes. | 2013 |
The typical event extraction workflow can be broken into two general steps: trigger detection and argument detection. For example, in ‘[the patient] was put on [Imatinib regimen]', the first step detects the event trigger ‘put', and the second step detects the theme ‘Imatinib regimen' and target argument ‘patient'. Björne et al. [64, 66] converted sentences to a dependency graph (Stanford Dependency [111]) using the McClosky-Charniak-Johnson parser [112, 113] and explored the graphs to construct features for both steps. The McClosky-Charniak-Johnson parser is based on the constituency parser of Charniak and Johnson [112] and retrained with the biomedical domain model of McClosky [113]. Björne et al. generated n-gram features connecting event arguments based on the shortest path of syntactic dependencies (also see [114] for their original use in kernel method) between the arguments. They included as features the types and super-types of trigger nodes from event-type generalization, to address feature sparsity. They also applied semantic post-processing to prune graph edges that violate semantic compatibility as required by the event definition to hold between event arguments. Their system TEES performed best in the 2009 GE (0.52 f-measure), 2011 EPI (0.5333 f-measure), 2013 CG (0.5541 f-measure), 2013 GRO (0.215 f-measure, being the only participating system) and 2013 BB full-event extraction (0.14 f-measure). Hakala et al. [67] built on top of the TEES system and re-ranked its output by enriched graph-based features, including paths connecting nested events and occurrence of gene–protein pairs in general subgraphs mined from external PubMed abstracts and the PubMed Central full-text corpus. The system by Hakala et al. placed first in 2013 GE (0.5097 f-measure), whereas the TEES system placed second (0.5074 f-measure). The strong performance of both systems with heavy utilization of graph-based features, especially the fact that Hakala et al. extended Björne et al. using enriched graph-based features and obtained better performance (first versus second place), suggests the potential benefits of exploring graph-based features.
Miwa et al. [80, 81] built the EventMine system that can extract not only biomedical events but also their negations and uncertainty statements. For event extraction, they used the Enju parser [115] and the GENIA Dependency parser (GDep) [116] to generate path features along with dictionary-based features (e.g. UMLS Specialist lexicon [117] and Wordnet [118]). Their entry in BioNLP-ST-2013 placed first in the PC task. In particular, their path features include not only paths between event arguments but also paths between event argument and non-argument named entities. The latter paths likely account for the strong performance by providing more local context features.
Another vein of work proposed joint event extraction in which triggers and arguments for all events in the same sentence are predicted simultaneously. McClosky et al. [77, 78] integrated event extraction into the overall dependency parsing objective. They applied the McClosky-Charniak-Johnson parser and converted the parses to Stanford Dependency. They converted the annotated event structures in the training data to event dependency graphs that take event arguments (named entities) as nodes and argument slot names as edge labels. They mapped the event dependency graphs to Stanford Dependency graphs and generated graph-based features to train an extended MSTParser [119] for extracting event dependency graphs from test data. Their graph-based features included paths between nodes in the Stanford Dependency graph, as well as subgraphs consisting of parents, children and siblings of the path nodes. They then converted the top-n extracted event dependency graphs back to event structures and re-ranked event structures for the best one, using the same graph-based features from event dependency graphs. Riedel et al. [120] first applied Markov Logic Networks to learn event structures and later switched to graph-based methods [72, 73]. They projected events to labeled graphs, and scored candidate graphs using a function that captures constraints on event triggers and arguments. The scoring function considers token features, dictionary features and dependency path features. Riedel et al. further used a stacking model to combine their system with the system by McClosky et al. [77, 78]. The combined system obtained first place in 2011 GE task (0.56 f-measure) and 2011 ID task (0.556 f-measure).
Most of the remaining BioNLP systems that performed competitively also used graph-based features to various extents. Liu et al. [60] developed an Exact Subgraph Matching (ESM) method, and later a more flexible Approximate Subgraph Matching (ASM) method [61–63]. They processed sentences with the McClosky-Charniak-Johnson parser and transformed the parses to directed dependency graphs. They constructed the graph of an event by computing unions of dependency paths between event arguments. Liu et al. then applied ESM/ASM from sentence graphs to event graphs, using a customized distance metric that accounts for subgraph differences in graph structure, node-covered stemmed words and edge directionality. They extended their method by integrating Protein Data Bank (PDB) [101], Uniprot [121] and Biothesaurus [122] to recognize protein/residue names and also adapted their method to other corpora including BioInfer [123] and Uniprot corpus [124]. This work falls along the lines of graph kernel-based methods. In general, kernel methods weight similarity function explicitly, and features are only used to evaluate similarity function on instance pairs. Thus kernel methods usually cannot directly weight/rank features. Kilicoglu et al. [68, 69] also adopted the McClosky-Charniak-Johnson Parser/Stanford Dependency pipeline. They converted the dependency graphs to embedding graphs, where nodes themselves can be small dependency graphs, to apply post-processing rules to traverse embedding graphs and extract nested events. However, their embedding graphs also lead to argument error propagation and hurt precision.
Besides the popular McClosky-Charniak-Johnson Parser/Stanford Dependency pipeline, some systems experimented with different parsers and/or dependency representations. Hakenberg et al. [70] applied BioLG [125], a Link Grammar Parser [126] extension, to generate parse trees. They stored parse trees in a database and designed a query language to match subgraph patterns, which are manually generated from training data, against parse trees. They pointed out that generalization of event types could further improve their results. Van Landeghem et al. [74] analyzed dependency graphs from the Stanford Parser [127], identified minimal event-containing subgraph patterns from training data and constructed extraction rules based on these patterns. Their post-processing rules handled overlapping triggers of different event types and events based on the same trigger, trading recall for precision.
The other systems generally used the dependency paths connecting the concept pairs as features for event extraction. For example, the dependencies were obtained through applications of different parsers including the Pro3Gres parser [128] (Kaljurand et al. [75]), the RASP parser [129] (Vlachos et al. [76]) or both McClosky-Charniak-Johnson parser and Enju parser (Quirk et al. [79]). However, most of these methods attained inferior performance compared with the best systems in the same tasks. We believe that there are at least two reasons: the McClosky-Charniak-Johnson parser with the self-trained biomedical parsing model is probably the most accurate parser in this domain; the enriched graph-based features and event type generalization as used by the best systems likely produced more useful features for event extraction.
PPI extraction and BioCreative shared tasks
BioCreative shared tasks focused on automatic named entity recognition on genes and proteins in biomedical text and on extraction of the interactions between these entities [130–133]. Among the participants of the PPI task of BioCreative-II [132], most systems used co-occurrence statistics, pattern templates and shallow linguistic features (e.g. context words and part-of-speech tags), with either statistical machine learning or rule-based systems. Some systems observed the need for capturing cross sentence mentions of interacting proteins. For example, Huang et al. [134] developed a profile-based method that creates a vector representation for candidate protein pairs by aggregating features from multiple sentences in the document. The profile features included n-grams, manually constructed templates and relative positions of protein mentions. In BioCreative-II.5, based on the top teams in the PPI task, the organizers pointed out that the BioNLP techniques using deep parsing and dependency tree/graph mining were necessary to achieve significant results [133]. In particular, Hakenberg et al. [71] used a system similar to their BioNLP-ST-2009 entry system [70]. They manually generated subgraph patterns from training data and matched them against parse trees. Their f-measure was 0.30. Sætre et al. [87] applied the Enju parser and the GDep parser and considered the dependency paths between concept pairs as features for relation extraction. They achieved an f-measure of 0.374. The PPI tasks of BioCreative-III consisted of detecting PPI-related articles that provide evidence to specified PPIs, but did not include the actual extraction of PPIs, which is the focus of this review [131]. Several follow-up studies to BioCreative-II.5 evaluated the usage of kernels in PPI extraction [99, 135], based on corpora including AIMed [136], BioInfer [123], HPRD50 [137], IEPA [138] and LLL [139]. They categorized kernels into the following categories: (1) shallow linguistic (SL) kernels [140]; (2) constituent parse tree-based kernels, including subtree (ST) [141], subset tree (SST) [142] and partial tree (PT) [143] kernels that use increasingly generalized forms of subtrees, as well as a spectrum tree (SpT) [144] kernel that uses path structures from constituent parse trees; (3) dependency parse tree-based kernels, including edit distance and cosine similarity kernels that are based on shortest paths [145], k-band shortest path spectrum (kBSPS) [99] that additionally allows k-band extension of shortest paths, all-path graph (APG) kernel [20] that differently weights shortest paths and extension paths in similarity calculation, as well as Kim’s kernels [146] that combine lexical, part-of-speech and syntactic information along with the shortest path structures. The comparative studies and error analyses showed that: (1) dependency tree-based kernels generally outperform constituent tree-based kernels; (2) kernel method performances heavily depend on corpus-specific parameter optimization; (3) APG, kBSPS and SL are top-performing kernels; (4) ensembles based on dissimilar kernels can significantly improve performance; (5) non-kernel-based methods (e.g. rule-based method, BayesNet) can perform on par with or better than all non-top kernel methods. From these observations, it is evident that richer dependency graph/tree structures (e.g. in APG, kBSPS) than shortest paths are important to better performance of graph/tree-based kernels, which is consistent with the analysis of BioNLP participating systems. The limited advantage of the kernel methods over non-kernel methods and the interpretation difficulty associated with kernel methods seem to favor investigating novel feature sets rather than novel kernel functions.
DDI extraction and DDIExtraction shared tasks
The two DDIExtraction challenges (2011 and 2013) aimed at automated extraction of DDI from biomedical text [55, 56]. The organizers of the two challenges recognized the extended delays in updating manually curated DDI databases. They observed that the medical literature and technical reports are the most effective sources for the detection of DDIs but contain an overwhelming amount of data. Thus, DDIExtraction was motivated by the pressing need for accurate automated text-mining approaches. DDIExtraction-2011 focused on classifying whether there is any interaction between candidate drug pairs. DDIExtraction-2013 additionally pursued detailed classification of DDIs into one of the four possible subtypes: advice (advice regarding the concomitant use of two drugs), effect (effect of DDI), mechanism (pharmacodynamics or pharmacokinetic mechanism of DDI) and int (general mention of interaction without further detail). In DDIExtraction-2011, Thomas et al. [88] applied the McClosky-Charniak-Johnson parser/Stanford dependency pipeline. They used voting to combine the following kernels to implicitly capture features for relation extraction: APG [20], kBSPS [99] and SL [140] kernels. Their system achieved the best f-measure of 0.657. Chowdhury et al. [89, 90] applied the Stanford parser to obtain dependency trees and experimented with both feature-based methods and kernel-based ensemble methods for relation extraction. They experimented with SL [140], mildly extended dependency tree [100] (expanding shortest paths to also cover important verbs, modifiers or subjects) and path-encoded tree [147] (based on constituency tree) kernels. By combining feature-based and kernel-based methods, Chowdhury et al. achieved the second best f-measure of 0.6398. In DDIExtraction-2013, Chowdhury et al. [89, 90] used their previous kernel method but switched to the McClosky-Charniak-Johnson parser and converted the parses to Stanford Dependency [91]. They attained an f-measure of 0.80 for general classification and 0.65 for detailed classification and placed first. Thomas et al. [148] followed a two-step approach to first detect general DDIs and then classify detected DDIs into subtypes. For the general DDI task, they used voting to combine kernels including APG [20], subtree (ST) [141], SST [142], SpT [144] and SL [140] kernels. For the subtype classification step, they used TEES directly [66]. Their system performed second best with an f-measure of 0.76 for general classification and 0.609 in detailed classification. It is interesting to see that adoption of systems originally developed for PPI extraction or event extraction has led to top performances in DDIExtraction. This further corroborates that these tasks are closely related, and technical solutions for one are generalizable to others.
Pharmacogenomics
In pharmacogenomics, numerous efforts have centered on the utilization of literature and clinical text to mine interesting relations between genetic mutations and drug response phenotypes. Although it is difficult to compare their performances because the experiments are not on shared corpora, these approaches do illuminate the translational application and adaptation of some state-of-the-art biomedical relation extraction techniques to problems directly asked by clinicians and pharmacologists.
Some systems used path-based approaches. Coulet et al. [40, 82] aimed at extracting binary relations between genes, drugs and phenotypes to build semantic networks for pharmacogenomics. They first converted the Stanford Parser output on sentences (from collected PubMed abstracts) into dependency graphs. They tracked the paths starting from named entities and ending at a verb, and merged paths ending with the same verb to form binary relations. Coulet et al. retained frequent relations and normalized both the collected entities and relation types (verbs). Without requiring prior enumeration of relation types, they created a semantic network knowledge base from 17 million MEDLINE abstracts, providing semantically rich summaries of pharmacogenomics relations. Percha et al. [42] extended this approach to use breadth-first search to yield the shortest path between two named entities in the dependency graph to generate features for relation extraction. They combined the extracted gene–drug relations to infer DDIs for those drugs that interact with the same gene product. Wang et al. [84] used Latent Dirichlet Allocation to create a semantic representation of biomedical named entities and used Kullback-Leibler (KL) divergence to calculate the association distance between pairs of entities in the Chem2Bio2RDF [149] semantic network. They ranked candidate associations between named entity pairs by summing distances along the path connecting the pairs. They demonstrated uses cases on novel knowledge discovery including searching and predicting novel gene–drug associations, traversing the mined semantic network to compare the molecular therapeutic and toxicological profiles of candidate drugs.
Other systems used tree-based approaches. Katrenko et al. [86] studied gene–protein relation and protein–protein relation extraction and included as features the subtrees rooted at the lowest common ancestors of two named entities in the dependency parse trees. Their experiment used several parsers including the Link Grammar Parser [126], Minipar [150] and the Charniak Parser [151]. Compared with individual parser’s results separately, they reported improved performance from adopting ensemble methods (stacking and AdaBoost) and combining multiple parsers’ results [152]. Hakenberg et al. [83] aimed at extracting relations among genes, single-nucleotide polymorphism variants, drugs, ADRs. They relied on co-occurrence for extraction of certain relations (e.g. gene–drug, gene–disease and drug–disease), but augmented co-occurrence with subtrees from the Stanford Parser output for other types of relations. They considered binary relations and used subtrees rooted at the lowest common ancestors of named entity pairs. The mined relations are cross-referenced with knowledge bases including EntrezGene [153], PharmGKB [17] and PubChem [154]. Bui et al. [85] aimed at extracting relations between drugs and virus mutations from the literature to predict HIV drug resistance. They used Stanford Parser to generate constituent parse trees for sentences and developed grammatical rules that traverse the tree structures to extract drug–gene relations. Their system is in research use at five hospitals to preselect novel HIV drug resistance candidates.
Both path-based and tree-based systems in pharmacogenomics tend to focus on precision over recall in their evaluation, differing from the balanced f-measure used in multiple shared tasks. This likely stems from their specific goals of harvesting reliable relations to build and grow pharmacogenomics semantic networks. Too much noise will likely cloud the initial semantic network, while missing relations still have a chance to be later discovered with growing literature. In fact, reported precisions for pharmacogenomics relation extraction systems typically range from 70% to >80%. In addition, these systems often check extracted relations against curated database such as PharmGKB. We believe that these systems can further benefit from adopting parsers trained with biomedical models and using enriched graph-based features, two of the most recent lessons learned in shared tasks.
Relation extraction from clinical narrative text
The medical informatics community has also extensively studied relation extraction in the form of shared tasks and separately motivated research. For example, significant advances in extracting semantic relations from narrative text in EMRs have been documented in the i2b2/VA-2010 challenge (i2b2—Informatics for Integrating Biology to the Bedside, VA—Veterans Association) [1].
i2b2/VA challenge
The challenge had three tasks including concept extraction, assertion classification and relation classification, participated by numerous international teams [1]. Concept extraction can be considered the basic task, as assertions and relations all refer to the extracted concepts. As the challenge allows relation classification to use the ground truth of concepts extraction, the performance metrics for relation classification should be interpreted as an upper bound for the end-to-end relation extraction task (same as the challenges from BioNLP, BioCreative and DDIExtraction). In this section, we review only the relation classification systems, where the target relations are predefined among medical problems, tests and treatments. These relations include ‘treatment improves / worsens / causes / is administered for / is not administered because of medical problem', ‘test reveals / conducted to investigate medical problem' and ‘medical problem indicates medical problem'. As in reviewing the above challenges, we review only those systems that represented sentences as graphs and explored such graphs during the feature-generation step.
Roberts et al. [93] classified the semantic relations using a rather comprehensive set of features: context features (e.g. n-grams, GENIA part-of-speech tags surrounding medical concepts), nested relation features (relations in the text span between candidate pairs of concepts), single concept features (e.g. covered words and concept types), Wikipedia features (e.g. concepts matching Wikipedia titles), concept bi-grams features and similarity features. The latter were computed using edit distance on language constructs including GENIA phrase chunks and Stanford Dependency shortest paths. Their system reached the highest f-measure on relation classification (0.737). deBruijn et al. [94] applied a maximum entropy classifier with down sampling applied to balance the relation distribution. They applied the McClosky-Charniak-Johnson parser/Stanford Dependency pipeline, and included as features the dependency paths between the minimal trees that cover the concept pairs. They used word clusters as features to address the problem of unseen words. Their system reached the second best f-measure of 0.731. Solt et al. [96] then experimented with several parsers including the Stanford Parser, the McClosky-Charniak-Johnson Parser and the Enju Parser. They used the resulting dependency graphs with two graph kernels including the all paths graph (APG) kernel [20] and kBSPS [99], which produced only moderate performance. This likely reflects the difficulty in tuning the graph/tree kernel-based systems, consistent with the observations from the experience in relation/event extraction from the scientific literature.
SemEval 2015 Task 14
The SemEval 2015 Task 14 included disorder identification and disorder slot filling tasks [155]. Disorder identification is essentially named entity detection, and disorder slot filling is similar to BioNLP event extraction tasks but in clinical subdomain. The challenge further divided the slot filling task into two subtasks, one with gold-standard disorder spans (task 2a) and one without (task 2b). Thus, task 2b has stricter evaluation results than task 2a. The attribute slots defined by the challenge include concept unique identifier (CUI), negation (NEG), subject (SUB), uncertainty (UNC), course (COU), severity (SEV), conditional (CND), generic (GEN) and body location (BL). Identifying the CUI is the named entity-detection problem, and identifying negation and uncertainty is the assertion classification problem. Identifying SUB, COU, SEV, CND, GEN and BL are more analogous to binary relation extraction. They are not completely equivalent to binary relation extraction, as the challenge limited the possible values for those slots, adding a layer of abstraction.
The challenge used weighted accuracy to rank the participants. Xu et al. [97] and Pathak et al. [98] consistently ranked as the top two teams in both task 2a (0.886 and 0.880, respectively) and task 2b (0.808 and 0.795, respectively). Xu et al. used Conditional Random Field (CRF) as the classifier for BL slot filling and SVM as the classifier for the other slots. The SVM classifier additionally used dependencies coming into and out of the disorder mentions. Note that these dependencies cannot capture multi-hop syntax dependence, but the authors observed that NEG/UNC/COU/SEV/GEN always have one-hop dependence. On the other hand, CRF (for BL) is itself a graph-based model that treats tokens and hidden states as nodes (integrating semantic and syntactic features including n-grams, context words, dictionaries and section names) and interconnects nodes with transition and emission edges [156]. Pathak et al. divided slot detection into two parts: detecting keywords and relating keywords with disorder mentions. They used dictionary look-up combined with CRF trained on features such as bag-of-words and orthographic features to detect keywords. To relate keyword with disorder mentions, they trained SVM using features similar to Xu et al. plus Part-of-Speech tags. Other teams used explicit graph-mining algorithms [157, 159] and but did not perform as competitively. For example, Hakala et al. [157] tackled task 2a by adapting TEES system to work with SemEval data format and achieved a weighted accuracy of 0.857, placing the third. This is not surprising, as given many slots only involve one-hop dependencies, full-fledged graph-based approach only offers limited benefits. In addition, the controlled vocabulary and controlled format nature of challenge tasks makes themselves suitable for CRF, as limited number of states and state-transitions lead to less sparse and more robust probability estimation.
Separately motivated clinical relation extraction
After the i2b2 challenges, several authors aimed at combining the concept and relation extraction steps into an integral pipeline and/or generalizing to the extraction of complex or even nested relations. Xu et al. [95] developed a rule-based system MedEx to extract medications and specific relations between medications and their associated strengths, routes and frequencies. The MedEx system converts narrative sentences in clinical notes into conceptual graph representations of medication relations. To do so, Xu et al. designed a semantic grammar directly mappable to conceptual graphs and applied the Kay Chart Parser [160] to parse sentences according to this grammar. They also used a regular-expression-based chunker to capture medications missed by the Kay Chart Parser. Weng et al. [36] applied a customized syntactic parser on text specifying clinical eligibility criteria. They mined maximal frequent subtree patterns and manually aggregated and enriched them with the UMLS to form a semantic representation for eligibility criteria, which aims to enable semantically meaningful search queries over ClinicalTrials.gov. Luo et al. [49] augmented the Stanford Parser with UMLS-based concept recognition to accurately generate graph representations for sentences in pathology reports where the graph nodes correspond to medical concepts. Frequent subgraph mining was then used to collect important semantic relations between medical concepts (e.g. which antigens are expressed on neoplastic lymphoid cells), which serve as the basis for classifying lymphoma subtypes. Extending the subgraph-based feature generation into unsupervised learning, Luo et al. [50] further used tensor factorization to group subgraphs. The intuition is that each subgraph corresponds to a test result, and a subgraph group represents a panel of test results, as typically used in diagnostic guidelines. The tensors incorporated three dimensions: patients, common subgraphs and individual words in each report. The words helped better group subgraphs to recover lymphoma subtype diagnostic criteria.
Shared resources for relation extraction
The shared tasks and separately motivated research on biomedical relation extraction have not only advanced the state-of-the-art in methodology, but also created and/or demonstrated the utilization of a repository of shared resources that range from knowledge bases to shared corpora to graph mining toolkits. We categorize and summarize those resources in Table 3, in the hope that it may serve as a starting point for resource navigation for future research efforts. Some of those resources concern general domain (e.g. general terminology/ontology resources Wordnet [118], Verbnet [163]); some concern the biomedical domain comprehensively (e.g. domain-specific terminology/ontology resources Gene Ontology [161], UMLS [58], Medical Subject Heading [162] and Biothesaurus [122]); some target-specific biomedical subdomain (e.g. knowledge bases such as PDB [101], Uniprot [121], SIDER [164], DrugBank [165], HIVDB [166], RegaDB [167], Entrez Gene [153], GENA [18] and IntAct [169]).
Table 3.
Shared resources for biomedical relation extraction
| Utility category | Data sources |
|---|---|
| Terminology/Ontology/Knowledge Base | GO [161], UMLS [58], MeSH [162], Wordnet [118], Verbnet [163], Biothesaurus [122], PDB [101], Uniprot [121], SIDER [164], DrugBank [165], HIVDB [166], RegaDB [167], Entrez Gene [153], GENA [168], IntAct [169], Chem2Bio2RDF [149], PharmGKB [17], PubChem [154], SubtiWiki (http://subtiwiki.uni-goettingen.de/) |
| Graph Miner | Gaston [170], Mofa [171], GSpan [172], FFSM [173], Graph Spider [174] |
| Tree/Graph Kernel | subtree (ST) kernel [141], subset tree (SST) kernel [142], partial tree (PT) kernel [143], spectrum tree (SpT) kernel [144], mildly extended dependency tree (MEDT) kernel [100], all-path graph (APG) kernel [20], k-band shortest path spectrum (kBSPS) kernel [99], path-encoded tree (PET) kernel [147] |
| Dependency Parsers | Enju Parser [115], GDep Parser [116], Stanford Parser [127], McCCJ Parser [112, 113], RASP Parser [129], Bikel Parser [175], BioLG Parser [125], Pro3Gres Parser [128], Kay Chart Parser [160], C&C [176] |
| Shared Corpora | BioNLP-09 event corpus [108], BioNLP-11 event corpus [109], BioNLP-13 event corpus [110], BioCreative-II relation corpus [130], BioCreative-II.5 relation corpus [133], DDIExtraction relation corpora [55, 56], i2b2/VA corpus [1], AIMed [136], BioInfer [124], HPRD50 [137], IEPA [138], LLL [139], Uniprot corpus [124] |
| Existing Repositories | BioNLP-ST-09: http://www.nactem.ac.uk/tsujii/GENIA/SharedTask/ |
| BioNLP-ST-11: http://2011.bionlp-st.org/home/genia-event-extraction-genia | |
| BioNLP-ST-13: http://2013.bionlp-st.org/ | |
| BioCreative-II: http://biocreative.sourceforge.net/biocreative_2.html | |
| BioCreative-II.5: http://www.biocreative.org/news/biocreative-ii5/ | |
| BioCreative-III: http://www.biocreative.org/events/biocreative-iii/ | |
| DDIExtraction-11: http://labda.inf.uc3m.es/DDIExtraction2011/ | |
| DDIExtraction-13: https://www.cs.york.ac.uk/semeval-2013/task9/ | |
| i2b2/VA-10: https://www.i2b2.org/NLP/Relations/ | |
| SemEval 2015 Task 14: http://alt.qcri.org/semeval2015/task14/ | |
| LDC: https://www.ldc.upenn.edu/language-resources |
The resources are organized by their utility categories. Abbreviations used include: Gene Ontology (GO), Unified Medical Language System (UMLS), Medical Subject Heading (MeSH), Human Protein Reference Database (HPRD), Linguistic Data Consortium (LDC). Existing repositories are those that themselves list biomedical NLP resources.
The road ahead
Although notable progress has taken place in applying graph-based algorithms to improve the extraction of biomedical relations, barriers still exist to developing practical relation extraction methods that are both generalizable and sufficiently accurate. Below we discuss a few such barriers and potential directions to overcome them.
Not all parsers and dependency encodings are synergistic
It has been pointed out repeatedly that the choice of the parser and dependency encodings may play an important role in a relation extraction system’s performance. Buyko et al. [177] performed comparative analysis on the impact of graph encoding based on different parsers (Charniak-Johnson [112], McClosky-Charniak-Johnson, Bikel [175], GDep, MST [119], MALT [178]) and dependency representations (Stanford Dependency and CoNLL dependency) and found that the CoNLL dependency representation performs better in combination with four parsers than the Stanford Dependency representation; and McClosky-Charniak-Johnson parser frequently places as the best performing parser. Miwa et al. [179] compared five syntactic parsers for BioNLP-ST-2009. They concluded that although performances from using individual parsers (GDep, C&C [176], McClosky-Charniak-Johnson, Bikel, Enju) do not differ much, using an ensemble of parsers and different dependency representations (Stanford Dependency, CoNLL, Predicate Argument Structure) can improve the event extraction results. As Stanford Dependency is the most widely used dependency encoding, they also compared the performance of using different Stanford Dependency variants and found that basic dependency performs best if keeping types of dependency edges. On the other hand, if ignoring types of dependency edges, they found that the collapsed dependency variant performs best, which corroborates the finding by Luo et al. [49]. In [49], the task is extracting relations as features to classify lymphoma subtypes instead of classifying relations themselves as in supervised relation classification in the BioNLP-ST event extraction tasks. Thus, recall is favored in the feature learning step, where ignoring types of dependencies helps to improve the coverage of subgraph patterns. The lessons learned in [49, 177, 179] seem to corroborate individual reports from top participants from challenges and are consistent with the popularity of parser-dependency choice from non-challenge applications in sections Pharmacogenomics and Separately motivated clinical relation extraction. In particular, we expect a good combination is to choose from either the McClosky-Charniak-Johnson parser or Stanford parser augmented with medical lexicon and choose from either CoNLL dependency or collapsed Stanford dependency.
Integrating coreference resolution
Coreference occurs frequently in biomedical literature and clinical narrative text, arising from the use of pronouns, anaphora and varied entities for the same concepts. Care must be exercised to transfer the correct relation along the coreference chain. However, many of the reviewed approaches for relation and event extraction did not have a built-in coreference resolution component. Miwa et al. [180] specifically studied the impact of using a coreference resolution system and showed improved event extraction performance. In particular, they developed a rule-based coreference resolution system that consists of detecting rules for mention, antecedent and co-referential link. They used the coreference information to modify syntactic parse results so that antecedent and mention share dependencies. Features were also extended between mentions and antecedents. Recognizing the importance of coreference features, several systems [181–184] subsequently implemented coreference resolution across sentences. They showed that to facilitate extracting specific types of relations, heuristic/rule-based coreference resolution tends to outperform domain-adapted statistical machine learning systems. Part of their lessons concerns the lack of gold-standard coreference annotation in relation extraction corpora; thus, we expect that paired relation and coreference annotations can improve coreference resolution and ultimately relation extraction. Moreover, integrating coreference resolution into the learning objective of relation extraction may lead to more coherent optimization and better end-to-end performance.
General relation and event extraction and domain adaptation
The state-of-the-art relation and event extraction systems from shared tasks are mostly built around domain-specific definitions of relations and events, many of which are in fact binary (e.g. BioCreative PPI challenge [133], DDIExtraction challenge [55, 56] and i2b2/VA challenge [1]). However, there is a gap between the technical advances and the demands from many real-world tasks, including building pharmacogenomics semantic networks [40], extracting clinical trial eligibility criteria [36] and representing test results for automating diagnosis categorization [49, 50]. In those tasks, general relation and event discovery is necessary, where the number of nodes is flexible and even the relation/event structure may not be entirely prespecified. Systems exploring the automation of annotation scheme learning [66] or unsupervised relation extraction [49, 50] attempted to enable generalization but there is still large room for improving accuracy. Another challenge brought by domain-specific relation/event definition concerns the training data. The problem of limited training data often plagues the development of NLP systems, among which the relation extraction systems are no exceptions. To make better use of existing annotated corpora, it is necessary to perform domain adaptation from external training corpora (source) to the target corpora. Merely adapting top systems to the new format and training on the new corpora does not always lead to top performers in the new domains, e.g. the adapted TEES system [157] in SemEval 2015 Task 14. Miwa et al. [180] proposed adding source instances followed by instance reweighting when source and target match on events to be extracted. When source and target corpora have a partial match on events, they proposed to train each event extraction module separately on the source corpora and used its output as additional features for the corresponding modules on the target corpora. Miwa et al. [185] further improved methods of combining corpora by integrating heuristics to filter spurious negative examples. The heuristics aim to correct errors where instances with a ‘None' annotation in one corpus owing to a different focus are all treated as negative instances in the combined corpus. Applying this method on learning from seven event-annotated corpora, they showed improved performance on two tasks in BioNLP-ST-2011. The successes from cascaded training [180] and example filtering heuristics [185] illuminate some promising directions in corpora adaptation. We expect parallel efforts in system generalization and corpora adaptation can complement each other toward effective domain adaptation.
Redundancy in subgraph patterns
Detection of useful subgraph patterns often depends on identifying ones with high frequency. A subgraph occurs once in a corpus whenever it is part of a larger graph in that corpus. Frequent subgraph mining identifies those subgraphs that occur in a corpus more than a given threshold number of times (see [186] for a survey on frequent subgraph mining and see [170–174] for various software). Lessons learned in multiple studies [49, 64, 65, 184] showed that redundancy among collected subgraph patterns creates problems when using subgraphs as features. Many smaller subgraphs are subisomorphic to (part of) other larger frequent subgraphs. Many of these larger subgraphs have the same frequency as their subisomorphic smaller subgraphs. This arises when a larger subgraph is frequent, and therefore all its subgraphs automatically become frequent as well. Furthermore, if the smaller subgraph gs is so unique that it is not subisomorphic to any other larger subgraph than gl, then this pair gs, gl shares identical frequency. Based on such observations, Luo et al. [49] only kept the larger subgraphs in such pairs. Note that it is cost prohibitive to perform a full pairwise check because the subisomorphism comparison between each subgraph pair is NP-complete [170], and a pairwise approach would ask for around a billion comparisons for a collection of several tens of thousands of subgraphs. Luo et al. presented an efficient algorithm using hierarchical hash partitioning that reduces the number of subgraph pairs to compare by several orders of magnitude. The key idea is that one only needs to compare subgraphs whose sizes differ by one, and one can further partition the subgraphs so that only those within the same partition need to be compared. The frequent subgraph redundancy is a systemic problem and is also closely related to the problem of nested terms in automatic term extraction, and a variety of de-duplicating scores (e.g. c-value [187]) proposed for the latter problem could be adapted to address the former problem. Other graph mining approaches may also be applicable, depending on the task. For example, algorithms may be developed to collect subgraph patterns that explore the ‘novelty' of the subgraphs, such as using p-significance to assess how unusual it is to see the subgraphs in the current corpus [188].
Integrating with NER
Most shared task participants were not evaluated based on their relation extraction from scratch. Rather, their systems were evaluated given the gold standard of named entity annotations, which is even true for challenges that include a NER task, such as the i2b2/VA shared tasks. Thus, their evaluation results are likely an upper bound of the end-to-end system performance, the tuning of which is in fact a non-trivial task. Kabiljo et al. [189] evaluated several methods for end-to-end relation extraction including a keyword-based method, a co-occurrence-based method and a method using dependency-graph-based patterns. They noted that in general a significant performance drop will occur when using named entities tagged by NER system such as BANNER [190] instead of using the gold standard. In addition, it is useful but challenging to filter out un-related named entity pairs, for which no relations have been explicitly stated in the text [92]. Such filtering may adopt a hybrid approach that relies on both automatically checking semantic type compatibility and manually sifting through the remaining tuples. However, as the number of non-related tuples often dominates that of related tuples, better automated filtering is necessary and remains an open question. Latest challenges such as SemEval 2015 Task 14 [155] added the end-to-end evaluation on joint detection/inference of entities and relations for prespecified relations, and it is reasonable to expect such evaluation will become more popular and will boost the development of end-to-end systems.
Relation extraction currently concentrates a large amount of efforts in biomedical information extraction. In the future, we anticipate a migration toward more unsupervised relation extraction that are increasingly adaptable across biomedical subdomains. The integration of relation extraction with named entity detection will produce end-to-end systems that can further automate the discovery and curation of novel biomedical knowledge. With advances in deriving better graph representations with more accurate parsers and appropriate dependency choices, in enhancing coreference resolution at document level, and in more efficiently sifting through informative subgraph patterns, the extraction of biomedical relations will continue to improve owing to an increase in the quality of data, and sustained community efforts. Given the rapid progress in the past years, we expect more exciting and promising developments of biomedical relation extraction, which continuously shape the emerging landscape and provide opportunities for researchers to contribute.
Key Points
• The state-of-the-art approaches on extracting biomedical relations from scientific literature and clinical narratives now heavily rely on graph-based algorithms to bridge semantic and syntactic information.
• State-of-the-art methods for biomedical relation extraction present multiple innovations from perspectives including strategies for parsing and dependency generation, graph exploration, as well as heuristics to address feature sparsity. They also create and maintain shared resources and tools for continuing research.
• Important applications of biomedical relation extraction include those in pharmacogenomics, clinical trial screening and adverse drug reaction detection, both advancing basic science and improving clinical practice.
• Challenges remain in biomedical relation extraction, including synergy between parsers and dependency encodings, integrating coreference resolution and named entity recognition, redundant subgraph patterns and domain adaptation. Insights from analyzing these challenges shed light on future directions to battle them.
Acknowledgement
The work was supported in part by Grant Number U54LM008748 from the National Library of Medicine, NIH 154HG007963 from the National Human Genome Research Institute and by the Scullen Center for Cancer Data Analysis.
Yuan Luo is an Assistant Professor at Northwestern University, Department of Preventive Medicine. He works in the area of natural language processing, time series analysis and computational genomics, with a focus on medical and clinical applications.
Özlem Uzuner is an Associate Professor at State University of New York at Albany, Department of Information Studies. Her research interests include information retrieval, medical language processing, information extraction, data anonymization and text summarization.
Peter Szolovits is a Professor at Massachusetts Institute of Technology, Department of Electrical Engineering and Computer Science. His research centers on the application of Artificial Intelligence (AI) methods to problems of medical decision making, natural language processing to extract meaningful data from clinical narratives to support translational medicine and the design of information systems for health care institutions and patients.
Footnotes
1 Relative clause anaphora
References
- 1. Uzuner Ö, South BR, Shen S. et al. 2010 i2b2/VA challenge on concepts, assertions, and relations in clinical text. J Am Med Inform Assoc 2011;18:552–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nadeau D, Sekine S. A survey of named entity recognition and classification. Lingvisticae Investigationes 2007;30:3–26. [Google Scholar]
- 3. Grishman R, Sundheim B. Message understanding conference-6: a brief history. COLING 1996;1:466–71. [Google Scholar]
- 4. Tenny C, Pustejovsky J. A history of events in linguistic theory. Events Grammatical Objects 2000;32:3–37. [Google Scholar]
- 5. Brachman R, Levesque H. Knowledge Representation and Reasoning, San Francisco, CA: Elsevier, 2004. [Google Scholar]
- 6. Sowa JF. Knowledge Representation: Logical, Philosophical, and Computational Foundations, 1999; Brooks/Cole Independence, KY. [Google Scholar]
- 7. Nadkarni PM, Ohno-Machado L, Chapman WW. Natural language processing: an introduction. J Am Med Inform Assoc 2011;18:544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ananiadou S, Mcnaught J. Text Mining for Biology And Biomedicine. Norwood, MA: Artech House, Inc, 2005. [Google Scholar]
- 9. Cohen KB, Demner-Fushman D. Biomedical Natural Language Processing. Philadelphia PA: John Benjamins Publishing Company, 2014. [Google Scholar]
- 10. Cohen AM, Hersh WR. A survey of current work in biomedical text mining. Brief Bioinform 2005;6:57–71. [DOI] [PubMed] [Google Scholar]
- 11. Zweigenbaum P, Demner-Fushman D, Yu H. et al. Frontiers of biomedical text mining: current progress. Brief Bioinform 2007;8:358–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzalez GH, Tahsin T, Goodale BC. et al. Recent advances and emerging applications in text and data mining for biomedical discovery. Brief Bioinform 2015, doi: 10.1093/bib/bbv087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanehisa M, Goto S, Sato Y. et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012;40;D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franceschini A, Szklarczyk D, Frankild S. et al. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013;41:D808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hunter S, Jones P, Mitchell A. et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 2012;40:D306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ng SK, Zhang Z, Tan SH. et al. InterDom: a database of putative interacting protein domains for validating predicted protein interactions and complexes. Nucleic Acids Res 2003:31:251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hewett M, Oliver DE, Rubin DL. et al. PharmGKB: the pharmacogenetics knowledge base. Nucleic Acids Res 2002;30:163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel CJ, Chen R, Butte AJ. Data-driven integration of epidemiological and toxicological data to select candidate interacting genes and environmental factors in association with disease. Bioinformatics 2012;28:i121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landrum MJ, Lee JM, Riley GR. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Airola A, Pyysalo S, Björne J. et al. All-paths graph kernel for protein-protein interaction extraction with evaluation of cross-corpus learning. BMC Bioinformatics 2008;9:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miwa M, Sætre R, Miyao Y. et al. A rich feature vector for protein-protein interaction extraction from multiple corpora. In: Proceedings of the 2009 Conference on Empirical Methods in Natural Language Processing, Volume 1. Singapore: Association for Computational Linguistics, 2009, pp. 121–30. [Google Scholar]
- 22. Chun HW, Tsuruoka Y, Kim JD. et al. Extraction of gene-disease relations from Medline using domain dictionaries and machine learning. In: Pacific Symposium on Biocomputing, Big Island, Hawaii, 2006, pp. 4–15. [PubMed] [Google Scholar]
- 23. Özgür A, Vu T, Erkan G. et al. Identifying gene-disease associations using centrality on a literature mined gene-interaction network. Bioinformatics 2008;24:i277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Camon E, Magrane M, Barrell D. et al. The Gene Ontology annotation (GOA) database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res 2004;32:D262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bader GD, Cary MP, Sander C. Pathguide: a pathway resource list. Nucleic Acids Res 20006;34:D504–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo Y, Riedlinger G, Szolovits P. Text mining in Cancer gene and pathway prioritization. Cancer Inform 2014;13:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen J, Bardes EE, Aronow BJ. et al. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37:W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J, Xu H, Aronow BJ. et al. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics 2007;8:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Driel MA, Bruggeman J, Vriend G. et al. A text-mining analysis of the human phenome. Eur J Hum Genet 2006;14:535–42. [DOI] [PubMed] [Google Scholar]
- 30. Pers TH, Dworzyski P, Thomas CE. et al. MetaRanker 2.0: a web server for prioritization of genetic variation data. Nucleic Acids Res 2013;41:W104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Raychaudhuri S, Plenge RM, Rossin EJ. et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet 2009;5:e1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. US National Library of Medicine. ClinicalTrial.gov. https://clinicaltrial.gov/.
- 33. Thadani SR, Weng C, Bigger JT. et al. Electronic screening improves efficiency in clinical trial recruitment. J Am Med Inform Assoc 2009;16:869–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miotto R, Weng C. Unsupervised mining of frequent tags for clinical eligibility text indexing. J Biomed Inform 2013;46:1145–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. deBruijn B, Carini S, Kiritchenko S. et al. Automated information extraction of key trial design elements from clinical trial publications. In: AMIA Annual Symposium Proceedings. Washington DC: American Medical Informatics Association, 2008, p. 141. [PMC free article] [PubMed] [Google Scholar]
- 36. Weng C, Wu X, Luo Z. et al. EliXR: an approach to eligibility criteria extraction and representation. J Am Med Inform Assoc 2011;18:i116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tu SW, Peleg M, Carini S. et al. A practical method for transforming free-text eligibility criteria into computable criteria. J Biomed Inform 2011;44:239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boland MR, Tu SW, Carini S. et al. EliXR-TIME: a temporal knowledge representation for clinical research eligibility criteria. AMIA Summits Transl Sci Proc 2012; 2012:71. [PMC free article] [PubMed] [Google Scholar]
- 39. Klein T, Chang J, Cho M. et al. Integrating genotype and phenotype information: an overview of the PharmGKB project. Pharmacogenomics J 2001;1:167–70. [DOI] [PubMed] [Google Scholar]
- 40. Coulet A, Shah NH, Garten Y. et al. Using text to build semantic networks for pharmacogenomics. J Biomed Inform 2010;43:1009–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Garten Y, Altman RB. Pharmspresso: a text mining tool for extraction of pharmacogenomic concepts and relationships from full text. BMC Bioinformatics 2009;10:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Percha B, Garten Y, Altman RB. et al. Discovery and explanation of drug-drug interactions via text mining. Pac Symp Biocomput, Big Island, Hawaii, 2012:410–21. [PMC free article] [PubMed] [Google Scholar]
- 43. Pakhomov SV, Buntrock JD, Chute CG. Automating the assignment of diagnosis codes to patient encounters using example-based and machine learning techniques. J Am Med Inform Assoc 2006;13:516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilcox AB, Hripcsak G. The role of domain knowledge in automating medical text report classification. J Am Med Informs Assoc 2003;10:330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aronow DB, Fangfang F, Croft WB. Ad hoc classification of radiology reports. J Am Med Inform Assoc 1999;6:393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aronsky D, Haug PJ. Automatic identification of patients eligible for a pneumonia guideline. In: Proceedings of the AMIA Symposium. Los Angeles, CA: American Medical Informatics Association, 2000, p. 12. [PMC free article] [PubMed] [Google Scholar]
- 47. Fiszman M, Chapman WW, Aronsky D. et al. Automatic detection of acute bacterial pneumonia from chest X-ray reports. J Am Med Inform Assoc 2000;7:593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu S, Liao KP, Shaw SY. et al. Toward high-throughput phenotyping: unbiased automated feature extraction and selection from knowledge sources. J Am Med Inform Assoc 2015;22:993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luo Y, Sohani A, Hochberg E. et al. Automatic Lymphoma classification with sentence subgraph mining from pathology reports. J Am Med Inform Assoc 2014;21:824–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luo Y, Xin Y, Hochberg E. et al. Subgraph Augmented Non-Negative Tensor Factorization (SANTF) for modeling clinical text. J Am Med Inform Assoc 2015;22:1009–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Onder G, Pedone C, Landi F. et al. Adverse drug reactions as cause of hospital admissions: results from the Italian Group of Pharmacoepidemiology in the Elderly (GIFA). J Am Geriatrs Soc 2002;50:1962–8. [DOI] [PubMed] [Google Scholar]
- 52. Zheng H, Wang H, Xu H. et al. Linking biochemical pathways and networks to adverse drug reactions. IEEE Trans NanoBioscience 2014;13:131–7. [DOI] [PubMed] [Google Scholar]
- 53. Liu M, Wu Y, Chen Y. et al. Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J Am Med Inform Assoc 2012;19:e28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Harpaz R, Vilar S, DuMouchel W. et al. Combing signals from spontaneous reports and electronic health records for detection of adverse drug reactions. J Am Med Inform Assoc 2013;20:413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Segura-Bedmar I, Martinez P, Sánchez-Cisneros D. The 1st DDIExtraction-2011 challenge task: extraction of drug-drug interactions from biomedical texts. In: Proceedings of the 1st Challenge Task on Drug-Drug Interaction Extraction, huelva spain, vol. 761, 2011, pp. 1–9. [Google Scholar]
- 56. Segura-Bedmar I, Martinez P, Herrero-Zazo M. Semeval-2013 task 9: extraction of drug-drug interactions from biomedical texts (ddiextraction 201 3) In: Proceedings of Semeval, Atlanta, GA, 2013, pp. 341–50. [Google Scholar]
- 57. Sarker A, Ginn R, Nikfarjam A. Utilizing social media data for pharmacovigilance: a review. J Biomed Inform 2015;54:202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lindberg DA, Humphreys BL, McCray AT. et al. The unified medical language system. Methods Inf Med 1993;32:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dijkstra EW. A note on two problems in connexion with graphs. Numerische Mathematik 1959;1:269–71. [Google Scholar]
- 60. Liu H, Komandur R, Verspoor K. From graphs to events: a subgraph matching approach for information extraction from biomedical text. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 164–72. [Google Scholar]
- 61. Liu H, Hunter L, Kešelj V. et al. Approximate subgraph matching-based literature mining for biomedical events and relations. PloS one 2013;8:e60954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. MacKinlay A, Martinez D, Yepes AJ. et al. Extracting biomedical events and modifications using subgraph matching with noisy training data. In: Proceedings of the BioNLP Shared Task 2013 Workshop. Sofia, Bulgaria: Association for Computational Linguistics, 2013, pp. 35–44. [Google Scholar]
- 63. Ravikumar K, Liu H, Cohn JD. et al. Literature mining of protein-residue associations with graph rules learned through distant supervision. J. Biomedical Semantics 2012;3:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Björne J, Heimonen J, Ginter F. et al. Extracting complex biological events with rich graph-based feature sets. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Singapore: Association for Computational Linguistics, 2009, pp. 10–18. [Google Scholar]
- 65. Björne J, Salakoski T. Generalizing biomedical event extraction. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 183–91. [Google Scholar]
- 66. Björne J, Salakoski T. TEES 2.1: automated annotation scheme learning in the BioNLP 2013 shared task. In: Proceedings of the BioNLP Shared Task 2013 Workshop, Sofia, Bulgaria, 2013, pp. 16–25. [Google Scholar]
- 67. Hakala K, Van Landeghem S, Salakoski T. et al. EVEX in ST’13: Application of a large-scale text mining resource to event extraction and network construction. In: Proceedings of the BioNLP Shared Task 2013 Workshop, Sofia, Bulgaria, 2013, pp. 26–34. [Google Scholar]
- 68. Kilicoglu H, Bergler S. Adapting a general semantic interpretation approach to biological event extraction. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 173–82. [Google Scholar]
- 69. Kilicoglu H, Bergler S. Syntactic dependency based heuristics for biological event extraction. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Association for Computational Linguistics, 2009, pp. 119–27. [Google Scholar]
- 70. Hakenberg J, Solt I, Tikk D. et al. Molecular event extraction from link grammar parse trees. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Association for Computational Linguistics, 2009, pp. 86–94. [Google Scholar]
- 71. Hakenberg J, Leaman R, Ha Vo N. et al. Efficient extraction of protein-protein interactions from full-text articles. IEEE/ACM Trans Comput Biol Bioinform 2010;7:481–94. [DOI] [PubMed] [Google Scholar]
- 72. Riedel S, McCallum A. Robust biomedical event extraction with dual decomposition and minimal domain adaptation. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 46–50. [Google Scholar]
- 73. Riedel S, McClosky D, Surdeanu M. et al. Model combination for event extraction in BioNLP 2011. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Association for Computational Linguistics, 2011, pp. 51–5. [Google Scholar]
- 74. Van Landeghem S, Saeys Y, De Baets B. et al. Analyzing text in search of bio-molecular events: a high-precision machine learning framework. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Association for Computational Linguistics, 2009, pp. 128–36. [Google Scholar]
- 75. Kaljurand K, Schneider G, Rinaldi F. UZurich in the BioNLP 2009 shared task. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Singapore: Association for Computational Linguistics, 2009, pp. 28–36. [Google Scholar]
- 76. Vlachos A, Buttery P, Séaghdha DO. et al. Biomedical event extraction without training data. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Singapore: Association for Computational Linguistics, 2009, pp. 37–40. [Google Scholar]
- 77. McClosky D, Surdeanu M, Manning CD. Event extraction as dependency parsing. In: Proceedings of the 49th Annual Meeting of the Association for Computational Linguistics: Human Language Technologies-Volume 1. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 1626–35. [Google Scholar]
- 78. McClosky D, Surdeanu M, Manning CD. Event extraction as dependency parsing in BioNLP 2011. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 41–5. [Google Scholar]
- 79. Quirk C, Choudhury P, Gamon M. et al. Msr-nlp entry in bionlp shared task 2011. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 155–63. [Google Scholar]
- 80. Miwa M, Thompson P, McNaught J. et al. Extracting semantically enriched events from biomedical literature. BMC Bioinformatics 2012;13:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Miwa M, Ananiadou S. NaCTeM EventMine for BioNLP 2013 CG and PC tasks. In: Proceedings of BioNLP Shared Task 2013 Workshop, Sofia, Bulgaria, 2013, pp. 94–8. [Google Scholar]
- 82. Coulet A, Garten Y, Dumontier M. et al. Integration and publication of heterogeneous text-mined relationships on the Semantic Web. J Biomed Semantics 2011;2:S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hakenberg J, Voronov D, Nguyên VH. et al. A SNPshot of PubMed to associate genetic variants with drugs, diseases, and adverse reactions. J Biomed Inform 2012;45:842–50. [DOI] [PubMed] [Google Scholar]
- 84. Wang H, Ding Y, Tang J. et al. Finding complex biological relationships in recent PubMed articles using Bio-LDA. PLoS One 2011;6:e17243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bui QC, Nualláin BÓ, Boucher CA. et al. Extracting causal relations on HIV drug resistance from literature. BMC Bioinformatics 2010;11:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Katrenko S, Adriaans P. Learning relations from biomedical corpora using dependency trees. In: Knowledge Discovery and Emergent Complexity in Bioinformatics. Berlin Heidelberg: Springer, 2007, pp. 61–80. [Google Scholar]
- 87. Sætre R, Yoshida K, Miwa M. et al. Extracting protein interactions from text with the unified AkaneRE event extraction system. IEEE/ACM Trans Comput Biol Bioinform 2010;7:442–53. [DOI] [PubMed] [Google Scholar]
- 88. Thomas P, Neves M, Solt I. et al. Relation extraction for drug-drug interactions using ensemble learning. In: Proceedings of DDIExtraction-2011 Challenge Task, Huelva Spain, 2011. [Google Scholar]
- 89. Chowdhury MFM, Lavelli A. Drug-drug interaction extraction using composite kernels. In: Proceedings of DDIExtraction-2011 Challenge Task, Huelva Spain, 2011, pp. 27–33. [Google Scholar]
- 90. Chowdhury MFM, Abacha AB, Lavelli A. et al. Two different machine learning techniques for drug-drug interaction extraction. In: Challenge Task on Drug-Drug Interaction Extraction, Huelva Spain, 2011, pp. 19–26. [Google Scholar]
- 91. Chowdhury MFM, Lavelli A. FBK-irst: a multi-phase kernel based approach for drug-drug interaction detection and classification that exploits linguistic information. In: Proceedings of SemEval, Atlanta GA, 2013, pp. 351–5. [Google Scholar]
- 92. Luo Y, Uzuner O. Semi-supervised learning to identify UMLS semantic relations. AMIA Jt Summits Transl Sci Proc 2014;2014:67–75. [PMC free article] [PubMed] [Google Scholar]
- 93. Roberts K, Rink B, Harabagiu S. Extraction of medical concepts, assertions, and relations from discharge summaries for the fourth i2b2/VA shared task. In: Proceedings of the 2010 i2b2/VA Workshop on Challenges in Natural Language Processing for Clinical Data, Boston, MA: i2b2, 2010. [Google Scholar]
- 94. deBruijn B, Cherry C, Kiritchenko S. et al. Machine-learned solutions for three stages of clinical information extraction: the state of the art at i2b2 2010. J Am Med Inform Assoc 2011;18:557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xu H, Stenner SP, Doan S. et al. MedEx: a medication information extraction system for clinical narratives. J Am Med Inform Assoc 2010;17:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Solt I, Szidarovszky F, Tikk D. Concept, assertion and relation extraction at the 2010 i2b2 relation extraction challenge using parsing information and dictionaries. In: Proceedings of i2b2/VA Shared-Task. Washington, DC, 2010. [Google Scholar]
- 97. Xu J, Zhang Y, Wang J. et al. UTH-CCB: the participation of the SemEval 2015 challenge – Task 14. In: Proceedings of the 9th International Workshop on Semantic Evaluation (SemEval 2015). Denver, CO: Association for Computational Linguistics, 2015, pp. 311–14. [Google Scholar]
- 98. Pathak P, Patel P, Panchal V. et al. ezDI: a Supervised NLP System for clinical narrative analysis. Proceedings of the 9th International Workshop on Semantic Evaluation (SemEval 2015). Denver, CO: Association for Computational Linguistics, 2015, pp. 412–16. [Google Scholar]
- 99. Tikk D, Thomas P, Palaga P. et al. A comprehensive benchmark of kernel methods to extract protein–protein interactions from literature. PLoS Comput Biol 2010;6:e1000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chowdhury FM, Lavelli A, Moschitti A. A study on dependency tree kernels for automatic extraction of protein-protein interaction; In: Proceedings of BioNLP 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 124–33. [Google Scholar]
- 101. Berman HM, Westbrook J, Feng Z. et al. The protein data bank. Nucleic Acids Res 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li J, Zhu X, Chen JY. Building disease-specific drug-protein connectivity maps from molecular interaction networks and PubMed abstracts. PLoS Comput Biol 2009;5:e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Blaschke C, Andrade MA, Ouzounis CA. Automatic extraction of biological information from scientific text: protein-protein interactions. In: Ismb, Heidelberg, Germany, 1999, pp. 60–7. [PubMed] [Google Scholar]
- 104. Rosario B, Hearst MA. Classifying semantic relations in bioscience texts. In: Proceedings of the 42nd Annual Meeting on Association for Computational Linguistics. Barcelona, Spain: Association for Computational Linguistics, 2004, p. 430. [Google Scholar]
- 105. Rosario B, Hearst MA. Multi-way relation classification: application to protein-protein interactions. In: Proceedings of the Conference on Human Language Technology and Empirical Methods in Natural Language Processing. Vancouver, Canada: Association for Computational Linguistics, 2005, pp. 732–9. [Google Scholar]
- 106. Hristovski D, Friedman C, Rindflesch TC. et al. Exploiting semantic relations for literature-based discovery. In: AMIA Annual Symposium Proceedings. Washington DC: American Medical Informatics Association, 2006, pp. 349–53. [PMC free article] [PubMed] [Google Scholar]
- 107. Rindflesch TC, Fiszman M. The interaction of domain knowledge and linguistic structure in natural language processing: interpreting hypernymic propositions in biomedical text. J Biomed Inform 2003;36:462–77. [DOI] [PubMed] [Google Scholar]
- 108. Kim JD, Ohta T, Pyysalo S. et al. Overview of BioNLP’09 shared task on event extraction. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Singapore: Association for Computational Linguistics, 2009, pp. 1–9. [Google Scholar]
- 109. Kim JD, Wang Y, Takagi T. et al. Overview of genia event task in bionlp shared task 2011. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 7–15. [Google Scholar]
- 110. Nédellec C, Bossy R, Kim JD. et al. Overview of BioNLP shared task 2013. In: Proceedings of the BioNLP Shared Task 2013 Workshop, Sofia Bulgaria, 2013, pp. 1–7. [Google Scholar]
- 111. De Marneffe MC, MacCartney B, Manning CD. Generating typed dependency parses from phrase structure parses. In: Proceedings of LREC, Genoa Italy, 2006, pp. 449–54. [Google Scholar]
- 112. Charniak E, Johnson M. Coarse-to-fine n-best parsing and MaxEnt discriminative reranking. In: Proceedings of the 43rd Annual Meeting on Association for Computational Linguistics. New York, NY: Association for Computational Linguistics, 2005, pp. 173–80. [Google Scholar]
- 113. McClosky D. Any Domain Parsing: Automatic Domain Adaptation for Natural Language Parsing. Brown University, Providence, Rhode Island, 2010. [Google Scholar]
- 114. Bunescu RC, Mooney RJ. A shortest path dependency kernel for relation extraction. In: Proceedings of the conference on Human Language Technology and Empirical Methods in Natural Language Processing. Vancouver, Canada, Association for Computational Linguistics, 2005, pp. 724–31. [Google Scholar]
- 115. Miyao Y, Sagae K, Sætre R. et al. Evaluating contributions of natural language parsers to protein–protein interaction extraction. Bioinformatics 2009;25:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sagae K, Tsujii J. Dependency parsing and domain adaptation with LR models and parser ensembles. In: EMNLP-CoNLL, Prague, Czech Republic, 2007, pp. 1044–50. [Google Scholar]
- 117. Bodenreider O. The unified medical language system (UMLS): integrating biomedical terminology. Nucleic Acids Res 2004;32:D267–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Miller GA. WordNet: a lexical database for English. Commun ACM 1995;38:39–41. [Google Scholar]
- 119. McDonald R, Pereira F, Ribarov K. et al. Non-projective dependency parsing using spanning tree algorithms. In: Proceedings of the Conference on Human Language Technology and Empirical Methods in Natural Language Processing. Association for Computational Linguistics, 2005, pp. 523–30. [Google Scholar]
- 120. Riedel S, Chun HW, Takagi T. et al. A markov logic approach to bio-molecular event extraction. In: Proceedings of the Workshop on Current Trends in Biomedical Natural Language Processing: Shared Task. Singapore: Association for Computational Linguistics, 2009, pp. 41–9. [Google Scholar]
- 121. UniPort Consortium. The universal protein resource (UniProt). Nucleic Acids Res 2008;36: D190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu H, Hu ZZ, Zhang J. et al. BioThesaurus: a web-based thesaurus of protein and gene names. Bioinformatics 2006;22:103–5. [DOI] [PubMed] [Google Scholar]
- 123. Pyysalo S, Ginter F, Heimonen J. et al. BioInfer: a corpus for information extraction in the biomedical domain. BMC Bioinformatics 2007;8:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nagel K, Jimeno-Yepes A, Rebholz-Schuhmann D. Annotation of protein residues based on a literature analysis: cross-validation against UniProtKb. BMC Bioinformatics 2009;10:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pyysalo S, Salakoski T, Aubin S. et al. Lexical adaptation of link grammar to the biomedical sublanguage: a comparative evaluation of three approaches. BMC Bioinformatics 2006;7:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sleator DD, Temperley D. Parsing English with a link grammar. Third International Workshop on Parsing Technologies, Tilburg Netherlands, 1993, pp. 1–14. [Google Scholar]
- 127. Huang Y, Lowe HJ, Klein D. et al. Improved identification of noun phrases in clinical radiology reports using a high-performance statistical natural language parser augmented with the UMLS specialist lexicon. J Am Med Inform Assoc 2005;12:275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Schneider G, Hess M, Merlo P. Hybrid long-distance functional dependency parsing. PhD, University of Zürich, Zürich, Switzerland, 2008. [Google Scholar]
- 129. Briscoe T, Carroll J, Watson R. The second release of the RASP system. In: Proceedings of the COLING/ACL on Interactive presentation sessions. Sydney, Australia: Association for Computational Linguistics, 2006, pp. 77–80. [Google Scholar]
- 130. Krallinger M, Morgan A, Smith L. et al. Evaluation of text-mining systems for biology: overview of the Second BioCreative community challenge. Genome Biol 2008;9:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Krallinger M, Vazquez M, Leitner F. et al. The Protein-Protein Interaction tasks of BioCreative III: classification/ranking of articles and linking bio-ontology concepts to full text. BMC Bioinformatics 2011;12:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Krallinger M, Leitner F, Rodriguez-Penagos C. et al. Overview of the protein-protein interaction annotation extraction task of BioCreative II. Genome Biol 2008;9:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Leitner F, Mardis SA, Krallinger M. et al. An overview of BioCreative II. 5. IEEE/ACM Trans Comput Biol Bioinform 2010;7:385–99. [DOI] [PubMed] [Google Scholar]
- 134. Huang M, Ding S, Wang H. et al. Mining physical protein-protein interactions from the literature. Genome Biol 2008;9:S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Tikk D, Solt I, Thomas P. et al. A detailed error analysis of 13 kernel methods for protein–protein interaction extraction. BMC Bioinformatics 2013;14:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bunescu R, Ge R, Kate RJ. et al. Comparative experiments on learning information extractors for proteins and their interactions. Artif Intell Med 2005;33:139–55. [DOI] [PubMed] [Google Scholar]
- 137. Fundel K, Küffner R, Zimmer R. RelEx-Relation extraction using dependency parse trees. Bioinformatics 2007;23:365–71. [DOI] [PubMed] [Google Scholar]
- 138. Ding J, Berleant D, Nettleton D. et al. Mining MEDLINE: abstracts, sentences, or phrases?. In: Pacific Symposium on Biocomputing, Big Island, Hawaii: World Scientific, 2002, pp. 326–37. [DOI] [PubMed] [Google Scholar]
- 139. Nédellec C. Learning language in logic-genic interaction extraction challenge. In: Proceedings of the 4th Learning Language in Logic Workshop (LLL05), Bonn, Germany, 2005. [Google Scholar]
- 140. Giuliano C, Lavelli A, Romano L. Exploiting shallow linguistic information for relation extraction from biomedical literature. In: EACL, Trento Italy, 2006, pp. 401–8. [Google Scholar]
- 141. Vishwanathan S, Smola AJ. Fast kernels for string and tree matching. In: NIPS, Vancouver, Canada, 2002, pp. 569–76. [Google Scholar]
- 142. Collins M, Duffy N. Convolution kernels for natural language. Advances in neural information processing systems, 2001, pp. 625–32. [Google Scholar]
- 143. Moschitti A. Efficient convolution kernels for dependency and constituent syntactic trees. In: Machine Learning: ECML 2006. Berlin Heidelberg: Springer, 2006, pp. 318–29. [Google Scholar]
- 144. Kuboyama T, Hirata K, Kashima H. et al. A spectrum tree kernel. Inform Media Technol 2007;2:292–9. [Google Scholar]
- 145. Erkan G, Özgür A, Radev DR. Semi-supervised classification for extracting protein interaction sentences using dependency parsing. In: EMNLP-CoNLL, Prague, Czech Republic, 2007, pp. 228–37. [Google Scholar]
- 146. Kim S, Yoon J, Yang J. Kernel approaches for genic interaction extraction. Bioinformatics 2008:24;118–26. [DOI] [PubMed] [Google Scholar]
- 147. Moschitti A. A study on convolution kernels for shallow semantic parsing. In: Proceedings of the 42nd Annual Meeting on Association for Computational Linguistics. Association for Computational Linguistics, Barcelona, Spain, 2004, p. 335. [Google Scholar]
- 148. Thomas P, Neves M, Rocktäschel T. et al. WBI-DDI: drug-drug interaction extraction using majority voting. In: Second Joint Conference on Lexical and Computational Semantics (* SEM), Atlanta, GA, 2013, pp. 628–35. [Google Scholar]
- 149. Chen B, Dong X, Jiao D. et al. Chem2Bio2RDF: a semantic framework for linking and data mining chemogenomic and systems chemical biology data. BMC Bioinformatics 2010;11:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Lin D. Dependency-based evaluation of MINIPAR. In: Treebanks. Berlin Heidelberg: Springer, 2003, pp. 317–29. [Google Scholar]
- 151. Lease M, Charniak E. Parsing biomedical literature. In: Natural Language Processing–IJCNLP 2005. Berlin Heidelberg: Springer, 2005, pp. 58–69. [Google Scholar]
- 152. Freund Y, Schapire RE. A decision-theoretic generalization of on-line learning and an application to boosting. J Comput Syst Sci 1997;55:119–39. [Google Scholar]
- 153. Maglott D, Ostell J, Pruitt KD. et al. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res;33:D54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kim S, Thiessen PA, Bolton EE. et al. PubChem substance and compound databases. Nucleic Acids Res 2016;D1:D1202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Elhadad N, Pradhan S, Gorman SL. et al. SemEval-2015 Task 14: analysis of clinical text. In: Proceedings of the 9th International Workshop on Semantic Evaluation (SemEval 2015). Denver, CO: Association for Computational Linguistics, 2015, pp. 303–10. [Google Scholar]
- 156. Lafferty J, McCallum A, Pereira FC. Conditional random fields: probabilistic models for segmenting and labeling sequence data. In: Proceedings of the 18th International Conference on Machine Learning (ICML-01), Williamstown, Massachusetts: 2001, pp. 282–9. [Google Scholar]
- 157. Hakala K. UTU: adapting biomedical event extraction system to disorder attribute detection. In: Proceedings of the 9th International Workshop on Semantic Evaluation (SemEval 2015). Denver, CO: Association for Computational Linguistics, 2015, pp. 375–9. [Google Scholar]
- 158. Gung J, Osborne J, Bethard S. CUAB: supervised learning of disorders and their attributes using relations. In: Proceedings of the 9th International Workshop on Semantic Evaluation (SemEval 2015). Denver, CO: Association for Computational Linguistics, 2015, pp. 417–21. [Google Scholar]
- 159. Huynh N, Ho Q. TeamHCMUS: analysis of clinical text. In: Proceedings of the 9th International Workshop on Semantic Evaluation (SemEval 2015). Denver, CO: Association for Computational Linguistics, 2015, pp. 3704. [Google Scholar]
- 160. Kay M. Algorithm schemata and data structures in syntactic processing. Technical Report CSL80-12, 1980. [Google Scholar]
- 161. Ashburner M, Ball CA, Blake JA. et al. Gene ontology: tool for the unification of biology. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. National Library of Medicine. MeSH. http://www.ncbi.nlm.nih.gov/mesh. [DOI] [PubMed]
- 163. Schuler KK. Verbnet: A Broad-coverage, Comprehensive Verb Lexicon. University of Pennsylvania, Philadelphia, PA, 2005. [Google Scholar]
- 164. Kuhn M, Campillos M, Letunic I. et al. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol 2010;6:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wishart DS, Knox C, Guo AC. et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 2008;36:D901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Vondrasek J, Wlodawer A. HIVdb: a database of the structures of human immunodeficiency virus protease. Proteins 2002;49:429–31. [DOI] [PubMed] [Google Scholar]
- 167. Libin P, Beheydt G, Deforche K. et al. RegaDB: community-driven data management and analysis for infectious diseases. Bioinformatics 2013;29:1477–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Koike A, Takagi T. Gene/protein/family name recognition in biomedical literature. In: Proceedings of BioLink 2004 Workshop: Linking Biological Literature, Ontologies and Databases: Tools for Users, Boston, MA, 2004, p. 56. [Google Scholar]
- 169. Kerrien S, Alam-Faruque Y, Aranda B. et al. IntAct-open source resource for molecular interaction data. Nucleic Acids Res 2007;35:D561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Nijssen S, Kok JN. The gaston tool for frequent subgraph mining. Electron Notes Theor Comput Sci 2005;127:77–87. [Google Scholar]
- 171. Borgelt C, Berthold MR. Mining molecular fragments: finding relevant substructures of molecules. In: Proceedings of 2002 IEEE International Conference on Data Mining. Maebashi City, Japan: IEEE, 2002, pp. 51–8. [Google Scholar]
- 172. Yan X, Han J. gspan: graph-based substructure pattern mining. In: Proceedings. 2002 IEEE International Conference on Data Mining. Maebashi City, Japan: IEEE, 2002, pp. 721–4. [Google Scholar]
- 173. Huan J, Wang W, Prins J. Efficient mining of frequent subgraphs in the presence of isomorphism. In: Data Mining, 2003. ICDM 2003. Third IEEE International Conference on. IEEE, 2003, pp. 549–52. [Google Scholar]
- 174. Clegg AB, Shepherd AJ. Syntactic pattern matching with Graph-Spider and MPL. In: The Proceedings of the Third International Symposium on Semantic Mining in Biomedicine (SMBM 2008), Turku, Finland, 2008, pp. 129–132. [Google Scholar]
- 175. Bikel DM. Design of a multi-lingual, parallel-processing statistical parsing engine. In: Proceedings of the Second International Conference on Human Language Technology Research. San Francisco, CA: Morgan Kaufmann Publishers Inc, 2002, pp. 178–182. [Google Scholar]
- 176. Rimell L, Clark S. Porting a lexicalized-grammar parser to the biomedical domain. J Biomed Inform 2009;42:852–65. [DOI] [PubMed] [Google Scholar]
- 177. Buyko E, Hahn U. Evaluating the impact of alternative dependency graph encodings on solving event extraction tasks. In: Proceedings of the 2010 Conference on Empirical Methods in Natural Language Processing. Boston, MA: Association for Computational Linguistics, 2010, pp. 982–92. [Google Scholar]
- 178. Nivre J, Hall J, Nilsson J. et al. MaltParser: a language-independent system for data-driven dependency parsing. Nat Lang Eng 2007;13:95–135. [Google Scholar]
- 179. Miwa M, Pyysalo S, Hara T. et al. A comparative study of syntactic parsers for event extraction. In: Proceedings of the 2010 Workshop on Biomedical Natural Language Processing. Uppsala, Sweden: Association for Computational Linguistics, 2010, pp. 37–45. [Google Scholar]
- 180. Miwa M, Thompson P, Ananiadou S. Boosting automatic event extraction from the literature using domain adaptation and coreference resolution. Bioinformatics 2012;28:1759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ratkovic Z, Golik W, Warnier P. et al. BioNLP 2011 task bacteria biotope: the Alvis system. In: Proceedings of the BioNLP Shared Task 2011 Workshop. Portland, Oregon: Association for Computational Linguistics, 2011, pp. 102–111. [Google Scholar]
- 182. Karadeniz I, Ozgür A. Detection and categorization of bacteria habitats using shallow linguistic analysis. BMC Bioinformatics 2015;16:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Lavergne T, Grouin C, Zweigenbaum P. The contribution of co-reference resolution to supervised relation detection between bacteria and biotopes entities. BMC Bioinformatics 2015;16:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Žitnik S, Žitnik M, Zupan B. et al. Sieve-based relation extraction of gene regulatory networks from biological literature. BMC Bioinformatics 2015;16:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Miwa M, Pyysalo S, Ohta T. et al. Wide coverage biomedical event extraction using multiple partially overlapping corpora. BMC Bioinformatics 2013;14:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Jiang C, Coenen F, Zito M. A survey of frequent subgraph mining algorithms. Know Eng Rev 2013;28:75–105. [Google Scholar]
- 187. Frantzi KT, Ananiadou S, Tsujii J. The c-value/nc-value method of automatic recognition for multi-word terms. In: Research and Advanced Technology for Digital Libraries. Springer, 1998, pp. 585–604. [Google Scholar]
- 188. Ranu S, Singh AK. Graphsig: a scalable approach to mining significant subgraphs in large graph databases. In: Data Engineering, 2009. ICDE’09. IEEE 25th International Conference on. IEEE, 2009, pp. 844–55. [Google Scholar]
- 189. Kabiljo R, Clegg AB, Shepherd AJ. A realistic assessment of methods for extracting gene/protein interactions from free text. BMC Bioinformatics 2009;10:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Leaman R, Gonzalez G. BANNER: an executable survey of advances in biomedical named entity recognition. In: Pacific Symposium on Biocomputing, Big Island, Hawaii, 2008, pp. 652–63. [PubMed] [Google Scholar]