

Abstract
Background and Purpose
A monoclonal antibody (PF‐00547659) against mucosal addressin cell adhesion molecule (MAdCAM), expressed as both soluble (sMAdCAM) and trans‐membrane (mMAdCAM) target forms, showed over 30‐fold difference in antibody‐target KD between in vitro (Biacore) and clinically derived (KD,in‐vivo) values. Back‐scattering interferometry (BSI) was applied to acquire physiologically relevant KD values which were used to establish in vitro and in vivo correlation (IVIVC).
Experimental Approach
BSI was applied to obtain KD values between PF‐00547659 and recombinant human MAdCAM in buffer or CHO cells and endogenous MAdCAM in human serum or colon tissue. CHO cells and tissue were minimally processed to yield homogenate containing membrane vesicles and soluble proteins. A series of binding affinities in serum with various dilution factors was used to estimate both KD,in‐vivo and target concentrations; MAdCAM concentrations were also measured using LC–MS/MS.
Key Results
BSI measurements revealed low KD values (higher affinity) for sMAdCAM in buffer and serum, yet a 20‐fold higher KD value (lower affinity) for mMAdCAM in CHO, mMAdCAM and sMAdCAM in tissue. BSI predicted KD,in‐vivo in serum was similar to clinically derived KD,in‐vivo, and the BSI‐estimated serum sMAdCAM concentration also matched the measured concentration by LC–MS/MS.
Conclusions and Implications
Our results successfully demonstrated that BSI measurements of physiologically relevant KD values can be used to establish IVIVC, for PF‐00547659 to MAdCAM despite the lack of correlation when using Biacore measured KD and accurately estimates endogenous target concentrations. The application of BSI would greatly enhance successful basic pharmacological research and drug development.
Abbreviations
- BSI
back‐scattering interferometry
- CHO‐rhMAdCAM
CHO cells expressing full length rhMAdCAM
- CHO‐WT
CHO parent cell line that does not express rhMAdCAM
- ECD
extracellular domain
- FIH
first in human
- IBD
inflammatory bowel disease
- ILH
interstitium‐like homogenate
- IOH
interstitium only homogenate
- IVIVC
in vitro and in vivo correlation
- mAb
monoclonal antibody
- MAdCAM
mucosal addressin cell adhesion molecule
- mMAdCAM
membrane‐bound MAdCAM
- rhMAdCAM.Fc
recombinant human MAdCAM‐Fc fusion protein
- RI
refractive index
- sMAdCAM
soluble MAdCAM
- SPR
surface plasmon resonance
- UC
ulcerative colitis
- VRH
vesicle rich homogenate
Table of Links
| TARGETS |
|---|
| Catalytic receptors |
| Integrin α4β7 |
This Table lists key protein targets in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
MAdCAM is an important therapeutic target, expressed as both a soluble MAdCAM (sMAdCAM) and a trans‐membrane MAdCAM (mMAdCAM) protein, mediating either rolling or firm adhesion of lymphocytes via integrin α4β7, to specialized high endothelial vessels (Pullen et al., 2009). A fully human IgG2 monoclonal antibody (mAb) PF‐00547659, designed to bind human MAdCAM, was developed to treat inflammatory bowel disease (IBD) and has been shown to reduce mucosal damage in animal models of colitis (Hokari et al., 2001; Goto et al., 2006; Apostolaki et al., 2008) and be clinically effective in the treatment of ulcerative colitis (UC). In a first‐in‐human (FIH) clinical trial (Martin et al., 2009), it was recognized that, for the binding affinity (KD) of PF‐00547659 to MAdCAM (mAb‐target), there was over 30‐fold difference between the values obtained from Biacore assays and the clinically derived values (KD,in vivo). A high‐affinity mAb (i.e. a smaller KD value) implies a lower concentration and therefore the dose of mAb required for maximal occupancy of the target binding site to affect a physiological response. If the KD value used to predict the FIH starting dose and efficacious doses in patients are based solely on measured Biacore KD, a significant underestimation of the dose could be made, leading to additional costly clinical trials or, in the worst case, to commercial attrition.
The lack of in vitro and in vivo correlation (IVIVC) KD (Schmidt, 2010; Guha, 2011; Brodfuehrer et al., 2014) can be due to a lack of contextual data (target physiology, pathology and micro‐environment) in samples and an assay platform capable of probing the interactome (Araujo et al., 2007). Most in vitro binding assays, such as surface plasmon resonance (SPR, Biacore), are performed in non‐native environments, are restricted to relatively simple matrices such as buffer, use a purified or a recombinant version of the target protein (Karlsson and Lofas, 2002; Ince and Narayanaswamy, 2006) and cannot discriminate binding differences between soluble and membrane‐bound forms of target. Further, these methods may require chemical modification of the target or drug for immobilization or detection (Yan and Marriott, 2003; Wienken et al., 2010; Lohse et al., 2012; Zhang et al., 2014). A new technique, cellular thermal shift assay (CETSA), has been used to study target engagement in clinically relevant samples, (Martinez Molina et al., 2013). While promising, CETSA cannot provide absolute binding affinity of interacting proteins and has limitations related to quantification, specificity and multiple sample processing steps that could change protein conformational structures. While another method, isothermal titration calorimetry, is a label‐free and free‐solution affinity method, it is limited by poor sensitivity and the need for large sample quantities (Velazquez‐Campoy and Freire, 2006; Ababou and Ladbury, 2007). An alternative to these methods is back‐scattering interferometry (BSI), a unique quantitative in vitro, free‐solution and label‐free platform that measures binding affinity at equilibrium with high sensitivity and compatibility with complex matrices (Baksh et al., 2011; Olmsted et al., 2012; Saetear et al., 2015). BSI has been used to quantify interactions between the following pairs – an ion to protein (Bornhop et al., 2007), sugar to lectin (Olmsted et al., 2012), small molecules to membrane proteins in cell‐derived vesicles (Baksh et al., 2011) and proteins to human erythrocyte membrane proteins (Saetear et al., 2015). Most recently, a description of the binding signal observed in label‐free, free‐solution studies with interferometry, referred to as free‐solution response function, has been provided where the signal magnitude correlates with changes in quantifiable intrinsic properties (Bornhop et al., 2016). BSI provides exquisite sensitivity of femtomolar detection limits (Olmsted et al., 2014) and quantifies picomolar KD values (Bornhop et al., 2007). The specificity of the measured binding event is dependent on the use of appropriate reference and control samples. A reference sample is defined as a sample lacking the target but of otherwise identical composition. A control sample is composed of the same test sample matrix and a control molecule that binds to the same receptors/ligands other than the target, thereby isolating the specific target binding interactions. For example when measuring merazoite proteins binding to sialyated receptors on human erythrocytes, cells with receptors devoid of sialyated sugars removed by neuraminidase treatment were used as one control sample, and an orthogonal control was a blocking antibody to the membrane receptor (Saetear et al., 2015). In another example, specificity of binding interactions to CXCR4 on T lymphocytic cells used CXCR4‐positive cells and CXCR4‐negative cells in the binding sample versus the reference sample respectively (Baksh et al., 2011).
For MAdCAM and PF‐00547659 binding, while sMAdCAM protein has been measured in serum and urine of healthy subjects and synovium of osteoarthritis patients (Leung et al., 2004), it has been difficult to measure PF‐00547659‐MAdCAM binding affinities in the native biological environments, especially in tissue where both soluble and membrane‐bound forms coexist. To date, obtaining accurate KD values from tissue has been problematic. Technical challenges include the low abundance of membrane‐bound target ligand, limited accessibility of human samples and the sensitivity constraints of existing assay methodologies (Kastritis et al., 2011). To obtain physiologically relevant drug‐target KD values, it is desirable to perform measurements in complex physiological environments where the target resides in vivo to replicate the numerous factors contributing to in vivo binding affinities. Such factors can include, for example, various protein conformations in different tissues due to pH, pressure and shear force (Di Stasio and De Cristofaro, 2010); other binding partners present in the tissue acting as agonist or inhibitors; or significantly higher concentrations of target ligand or receptor relative to the thermodynamic KD (KD, Table 1). As illustrated in Figure 1 when target concentration is at or below KD, the measured KD is defined by affinity (affinity limited); whereas when target concentration is much higher than KD, the measured KD will be right shifted, and the shifted value is defined by target concentration (concentration limited). Such an increase in the KD as a function of the higher target concentration in the micro‐environment and where the thermodynamic KD remains unchanged is defined as apparent KD (KD,app). KD,app can also be shifted from thermodynamic KD due to multiple forms of the target protein bearing different binding affinities to the drug or competing binding factors, as well as varying concentrations of all the binding forms. Such a shift of the KD,app is described as the apparent integrated KD (KD, app‐int) (Table 1).
Table 1.
Definition of KD types
| In vivo KD types | Abbreviation | Definition | Target ligand concentration (pM) |
|---|---|---|---|
| Thermodynamic KD | KD,{% matrix}, {target form} | Thermodynamic binding affinity (measured with target concentration ≤ true KD) | ≤ Thermodynamic KD |
| KD,{buffer,serum,cells, tissue}, {soluble, membrane} | |||
| Apparent KD | KD, app{% matrix}, {target form} | KD remains the same, though there is an apparent right shift from KD related to higher target concentrations | > > Thermodynamic KD |
| Apparent integrated KD | KD, app‐int {% matrix} | When more than one form of binding partner contributes to the binding affinity | Any |
| In vivo KD | KD,in‐vivo | Model (TMDD) derived in vivo KD based on clinical drug serum concentrations | Endogenous concentration |
Figure 1.
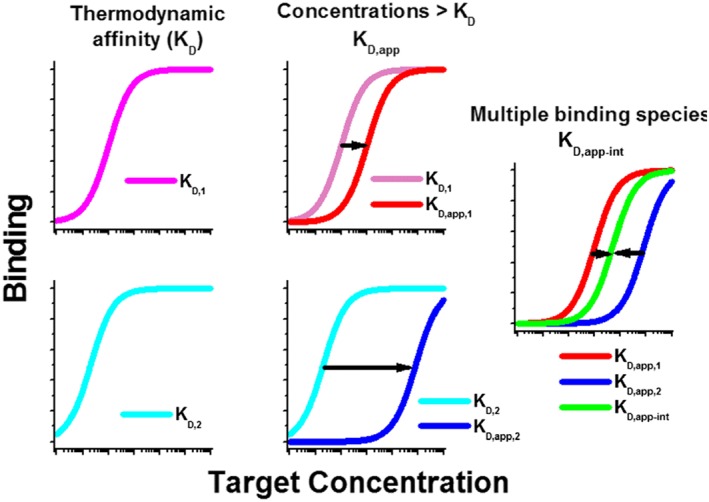
Diagram illustrating differences between thermodynamic KD, KD.app and KD,app‐int. Binding affinity relative to target concentration is plotted. Illustration of when target concentration is at or below KD, the measured KD is defined by affinity (KD,1 and KD,2, affinity limited), whereas when target concentration is much higher than KD, the measured KD will be right shifted, and the shifted value is defined by target concentration (KD,app,1 and KD,app,2, concentration limited). KD,app can also be shifted from thermodynamic KD due to multiple forms of the target ligand or receptor protein bearing different binding affinities to the drug or competing binding factors, as well as varying concentrations of all the binding forms. Such a shift of the KD,app is described as the apparent integrated KD (KD, app‐int) (Table 1).
This study describes the first time BSI measurements were designed and conducted in various biological matrices to analyse the observed IVIVC disparity based on Biacore values. Human serum or tissue homogenate samples were used as binding matrices where the in vivo micro‐environment was retained. Using these samples coupled with the use of BSI, a more complete system‐wide view of the drug‐target interaction with all of its biological/physiological complexity was provided. Binding affinities were determined in buffer with recombinant sMAdCAM ligand, in diluted serum where endogenous sMAdCAM is present, in CHO cell homogenates with recombinant mMAdCAM and in tissue homogenates where both sMAdCAM and mMAdCAM forms coexist or were physically separated. The total MAdCAM concentrations in different matrices were measured using LC–MS/MS. To our knowledge, this is the first report of an in vitro system which can provide physiologically relevant KD measurements, establishing IVIVC, taking into account and maintaining the native environment throughout the assay.
Methods
Human tissue samples and antibodies
Human colon tissue samples (from UC patients and healthy individuals) were obtained from the Cleveland Clinic Tissue Inventory with appropriate informed consent, for use of samples for gene, protein and genomic studies and de‐identified, so patient identification could not be linked to tissue samples. Samples met the criteria of ‘research exempt from IRB review’ due to the redundant nature of tissue collected otherwise normally discarded. Soluble recombinant human MAdCAM‐IgG1‐Fc fusion protein (rhMAdCAM.Fc) was obtained from R&D Systems, MN; pooled human serum (6–8 donors) from Bioreclamation IVT, NY; CHO cells expressing full length rhMAdCAM (CHO‐rhMAdCAM, mMAdCAM), CHO parent cell line that does not express rhMAdCAM (CHO‐WT) and PF‐00547659, a fully human anti‐MAdCAM IgG2 mAb were generated by Pfizer as previously described (Pullen et al., 2009).
CHO cell and colon tissue vesicle rich homogenates (VRH), colon tissue interstitium‐like homogenate (ILH) and colon tissue interstitium only homogenate (IOH) preparation and characterization
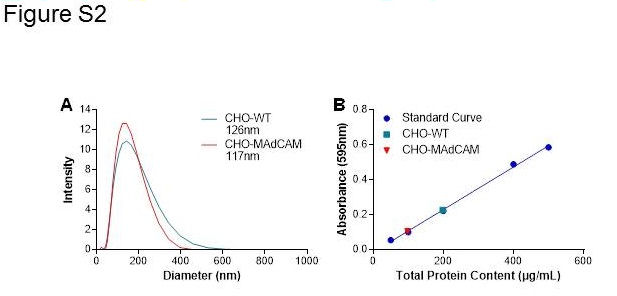
CHO cell vesicle rich homogenates (VRH) and human colon tissue VRH were prepared from CHO‐WT, CHO‐rhMAdCAM and colon tissues from patients diagnosed with UC. A cell pellet containing approximately 5 × 106 CHO cells, or homogenate from approximately 50 mg of tissue homogenized by hand using a mortar and pestle, was resuspended in 3 mL PBS containing 2× protease inhibitors (Roche cOmplete ULTRA Tablets) and probe sonicated on ice using a Qsonica sonicator at a power setting resulting in cavitation (approximately 40% amplitude) for 90 s (for CHO cells) or 2 min (for colon homogenate) in a pulsed manner (5 s on, 1 s off). Samples were centrifuged at 9000 × g for 1 h at 4°C. The supernatant containing VRH was collected for immediate experimentation. Vesicle particle size was characterized using dynamic light scattering at 8°C, using a Malvern Zetasizer Nano ZS (Malvern, 2012) (Figures [Link], [Link]). A Bradford assay (Bradford, 1976) quantified the total protein concentration of VRH samples (Figures [Link], [Link]).
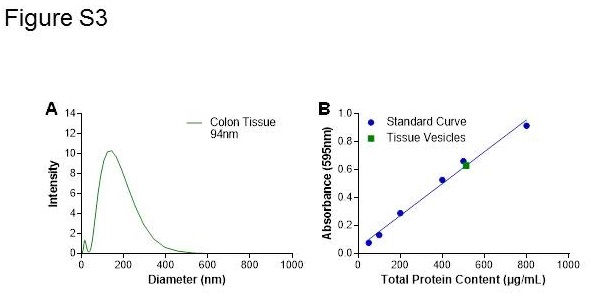
To prepare interstitium‐like homogenate (ILH) samples, human colon tissue from a healthy volunteer was lightly homogenized in 1:4 (w/v) of PBS with 1X protease inhibitor (ThermoFisher, prod#78 430, no EDTA) using a Bullet Blender Storm. The sample was centrifuged at 2000 × g for 10 min. Supernatant was collected and snap frozen in liquid nitrogen for further experimentation. The ILH sample (composed of a mostly aqueous soluble fraction) was further depleted of any membrane fraction yielding interstitium only homogenate (IOH) consisting of tissue‐derived sMAdCAM, by high speed centrifugation step (Karp, 2005) at 100 000 × g for 1 h at 4°C and the resulting supernatant collected.
Sample preparation for BSI binding experiments
Binding samples were prepared by mixing a series of concentrations of PF‐00547659 or an isotype control mAb in PBS over a range of 1 pM to 2 nM with a fixed concentration of MAdCAM protein containing sample (Figures 2 and S3). Binding isotherm assays under equilibrium conditions were conducted to estimate KD of PF‐00547659 to rhMAdCAM.Fc in buffer, KD and KD,app in a series of diluted human serum; KD in CHO cells, tissue VRH and IOH; and KD,app‐int in tissue VRH with added ILH.
Figure 2.
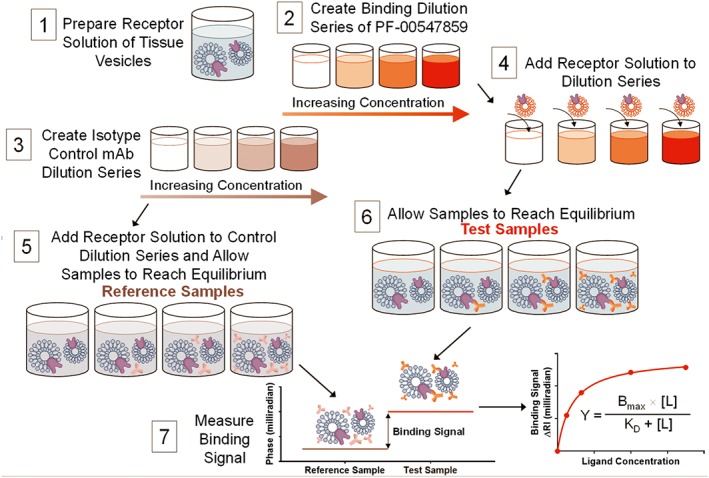
Tissue‐based assay protocol diagram. An illustration of the experimental procedure to measure the binding affinity in human colon tissue VRH. Matching the test sample and reference sample RI by splitting it into two aliquots, it is possible to perform RI measurements at <10−6 RIU sensitivity in the presence of significant matrix complexity. 1) Tissue VRH samples are prepared as described in methods. 2) A set of samples with varying concentrations of PF‐00547659 are prepared in PBS. 3) A set of samples with varying concentrations of an isotype control mAb, using the same concentrations as PF‐00547659 series, are prepared in PBS. Either samples from 2) or 3) are mixed with tissue VRH producing 4) and 6) test samples or 5) reference samples, and the samples were allowed to reach equilibrium. Samples were measured in the same manner sequentially starting with the reference and then the binding samples with increasing concentrations of mAb and fixed target protein, up to the highest concentration of mAb (1 nM). This step‐wise procedure constitutes a single binding assay run producing a saturation isotherm. 7) The binding signal was calculated as the difference between the sample and reference signals at the same mAb concentration and represents the amount of specific binding between mAb and target protein occurring within the sample because all other binding events are cancelled by the reference sample. 8) The binding signal (δ RI in milliradians) was then plotted versus concentration of mAb.
Each sample type was prepared at twice the final concentration used in the BSI instrument as follows: a) rhMAdCAM.Fc was prepared in PBS at 20 pM; b) CHO‐rhMAdCAM VRH and CHO‐WT VRH were diluted in PBS to a concentration of 4 μg·mL−1 total protein; c) human serum was diluted with PBS to make a 0.2, 20, 50, 70 or 100% serum solution; d) colon tissue VRH was diluted in PBS to a concentration of 4 μg·mL−1 total protein in the presence or absence of 50% ILH; e) 80% IOH. Prepared samples were mixed 1:1 with either PF‐00547659 or when applicable isotype control mAb dilution series to result in a set of samples with mAb concentrations ranging from 0.5 pM to 1 nM (Figures 2 and S3). To vary the ratio of mMAdCAM to sMAdCAM, tissue VRH at 4 μg·mL was also diluted in 95% ILH resulting in a range of PF‐00547659 from 0.5 pM to 1 nM with 2 μg·mL−1 VRH in 87.5% ILH. Samples were incubated at room temperature for 1 h to allow binding to reach equilibrium prior to measurement in BSI. A pictorial representation of control, reference and test samples as well as when isotype control mAb was used are shown in Figures 3, 4, 5 and Table S1.
Figure 3.
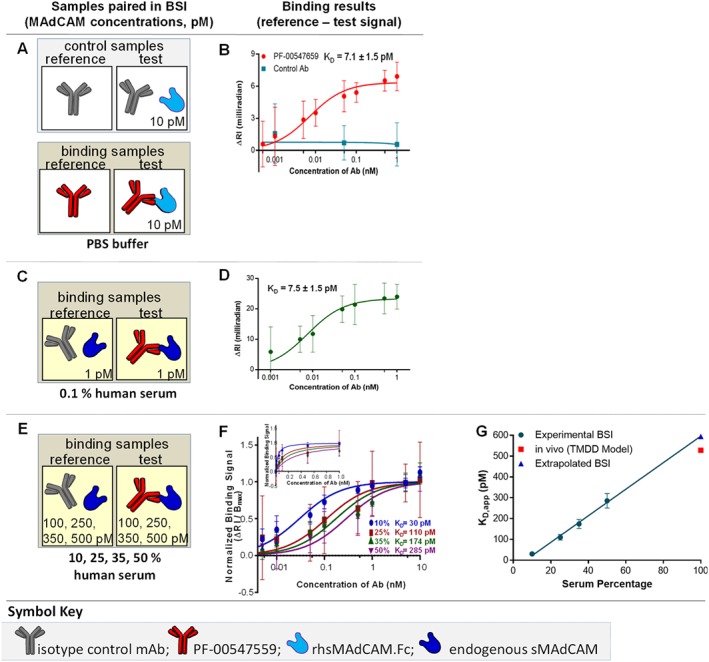
sMAdCAM‐binding assays – thermodynamic KD in buffer, KD in human serum, KD,app{100%serum}. (A) Binding interaction samples are paired as reference and test samples in control or binding samples with (B) resulting saturation binding isotherm used to quantify the thermodynamic KD of PF‐00547659 to rhMAdCAM.Fc fusion protein in PBS. Measuring the binding interaction of PF‐00547659 to endogenous sMAdCAM in 0.1% healthy human serum, (C) binding interaction samples are paired and (D) corresponding saturation binding isotherm used to quantify the thermodynamic KD. Measuring interactions with increasing human serum in 10, 25, 35 and 50% serum using the same (E) binding interaction samples are paired and (F) overlay of saturation binding isotherms of PF‐00547659 to serum sMAdCAM. (G) Plot of KD,app versus serum percentage to determine the KD,app{100%serum}. Error bars for all plots represent the standard deviations of replicate trials over five consecutive experiments performed on different days (n = 4–5).
Figure 4.
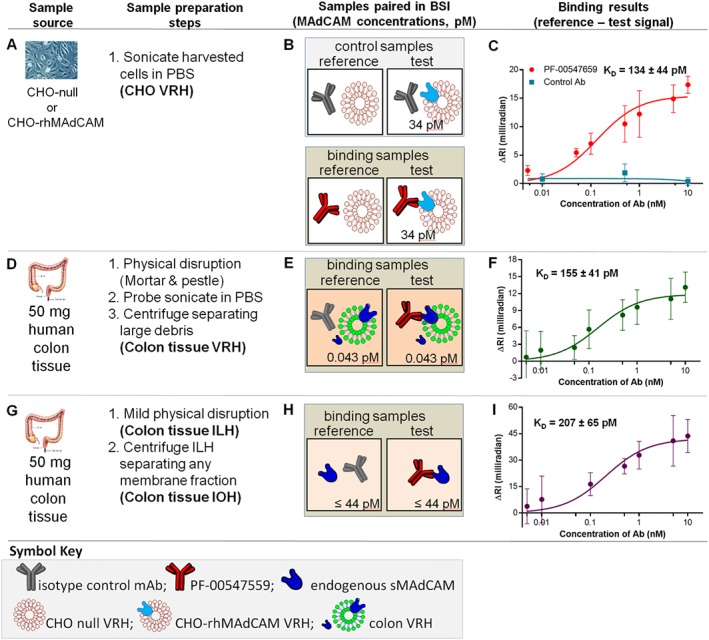
mMAdCAM‐binding assays – thermodynamic KD in CHO cell VRH, human colon VRH and tissue IOH. (A) CHO‐WT VRH and CHO‐rhMAdCAM VRH are prepared by sonication in PBS, (B) binding interaction samples are paired and (C) saturation binding isotherm used to quantify the thermodynamic KD{CHO,mMAdCAM} of PF‐00547659 to mMAdCAM expressed in CHO cells. (D) Human (UC) colon tissue physically disrupted, sonicated and centrifuged to produce tissue VRH sample, (E) binding interaction samples are paired and (F) saturation binding isotherm used to quantify the thermodynamic KD{issue,mMAdCAM} of PF‐00547659 to tissue mMAdCAM in tissue. (G) Human (UC) colon tissue mildly physically disrupted and centrifuged to produce tissue IOH sample, (H) binding interaction samples are paired with (I) saturation binding isotherm used to quantify the thermodynamic KD{tissue,sMAdCAM} of PF‐00547659 to sMAdCAM in tissue. Error bars for all plots represent the standard deviations of replicate trials over five consecutive experiments performed on different days (n = 4–5).
Figure 5.
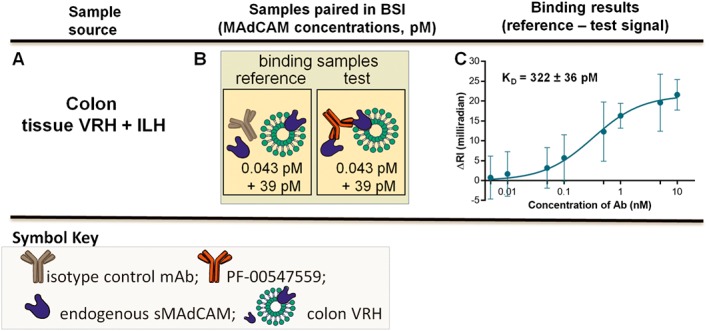
Binding assays combining VRH and ILH – KD,app‐int in tissue VRH and ILH. (A) Tissue VRH is mixed with tissue ILH, (B) binding interaction samples are paired and (C) saturation binding isotherm used to quantify the KD,app‐int{issueVRH + 87.5%ILH} of PF‐00547659 to tissue mMAdCAM and tissue sMAdCAM. Error bars for all plots represent the standard deviations of replicate trials over five consecutive experiments performed on different days (n = 5).
BSI measurements
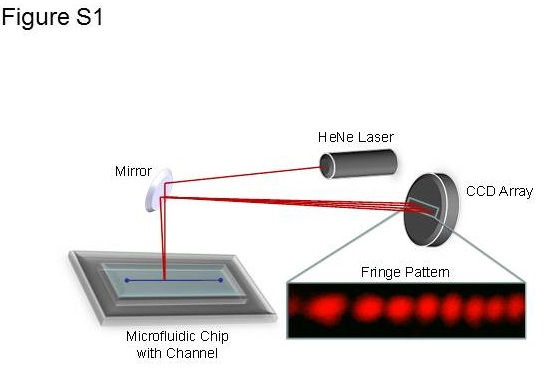
BSI, a universal sensor and refractive index (RI) detector, was employed as previously described (Kussrow et al., 2012; Bornhop et al., 2016) (Figure S4). Briefly, the instrument is composed of a helium‐neon laser (Melles Griot, 25‐LHP‐121‐249), a microfluidic chip (Micronit) and a charge‐coupled device camera acting as a linear array detector (Ames Photonics). The glass chip, containing isotropically etched flow channels with a semicircular cross section (210 × 100 μm) that, upon proper illumination with the laser, creates an optical resonance phenomenon. As a result, BSI has a long effective optical path‐length through the sample leading to high RI sensitivity. As described in Swinney et al., (2000) the parts of the chip that form the cavity are those where the laser impinges, reflects and refracts from the front surface, the channel walls and the back surface of the chip. An optical quality finish is required for all surfaces (~1 nm smoothness), particularly the channel surface. The high‐contrast backscattered interferometric fringe pattern, created by impinging the laser onto the chip that is directed onto the camera, which in concert with a Fourier analysis programme, is used to measure the positional shift in the fringes. The relative RI changes between the test and reference samples (fringe shift, not wavelength) are compared to give a specific binding signal, in this case, the PF‐00547659‐MAdCAM protein molecular interaction. BSI does so in a manner that is quantitative (Schmidt, 2010; Bornhop et al., 2016) and directly proportional to concentration of the measured species. In experiments where target alone cannot be removed from the matrix (serum and tissue VRH), an isotype control mAb with the same concentration range was used in place of PF‐00547659, and these samples serve as the control samples.
To measure the binding signal in a given sample (all binding partners were untethered and free in solution), 1 μL of the sample was injected into the channel by pipetting the sample onto the entrance well of the chip, applying a vacuum for a controlled time period (ca. 145 ms) to deliver the sample into the channel of the chip, and allowing the samples to reach temperature and pressure equilibrium (~10 s). The phase value is then measured for 20 s. After the first assay run, the channel is thoroughly rinsed with buffer ensuring the phase returns to the initial starting point for the buffer, and the measurements for the entire concentration series repeated for five complete trials. Multiple assay trials are conducted for each KD measurement containing approximately 10 points on the isotherm, each sample point being run in triplicate, and the full isotherm repeated over 4–5 days.
Extrapolation to KD,app in 100% serum
To estimate K D,app of PF‐00547659 to endogenous sMAdCAM in 100% serum, KD values were plotted versus a series of human serum concentrations at 10, 25, 35 and 50% (Figure 2E–G). 50% serum was the highest matrix used due to matrix stickiness.
Calculations of sMAdCAM concentration in serum
sMAdCAM concentration in human serum was calculated using the following equation:
Where KD,app is the extrapolated KD,app at 100% serum and KD is at 0.1% human serum where at this dilution, the target concentration is minimal.
Measurement of hMAdCAM protein concentration in biological samples by LC–MS/MS
An immunoaffinity LC–MS/MS assay was employed for the quantification of human MAdCAM in serum, CHO VRH, colon tissue VRH and ILH. The workflow for sample processing involved protein precipitation and/or denaturation. Protocols entailed the subsequent reduction of disulphide bonds using 5 mM (serum) or 10 mM (tissue) dithiothreitol treatment at 57°C for 40 min and alkylation of cysteine using 10 mM (serum) or 20 mM (tissue) iodoacetamide at RT in the dark for 40 min prior to trypsin digestion. The assays target a proteotypic peptide from the extracellular domain (ECD) of MAdCAM which was enriched online using an anti‐peptide antibody prior to LC–MS/MS, akin to a methodology described previously (Neubert et al., 2010). Further details are provided in the supporting information.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are shown as means ± SD, unless otherwise stated. The BSI data were analysed as follows. The binding signal was calculated as the difference between the sample and reference signals at the same mAb concentration and represents the specific binding between mAb and target protein occurring within the sample because all other binding events are canceled by the reference sample. The binding signal (delta RI in milliradians) was then plotted versus concentration of mAb. Affinity was determined by fitting the plot with a single‐site saturation binding curve using an unweighted fit (minimization of the sum‐of‐squares of the distances of the points from the curve) by GraphPad Prism 6 software. To estimate the KD,app at 100% serum, data obtained with lower concentrations of serum were fitted with a linear regression model using GraphPad Prism software, and the KD,app at 100% serum was extrapolated (Chang et al., 1975; Lowe et al., 2009).
Results
KD in buffer
Previously published Biacore methods used PF‐00547659 immobilized to a CM5 biosensor chip and rhMAdCAM.Fc in solution (Pullen et al., 2009). BSI‐binding isotherm assays, used 10 pM of rhMAdCAM.Fc (the same as Biacore), and mAb PF‐00547659 both untethered and free in buffer, yielding a KD of 7.1 (±1.5) pM (Figure 3A–B and Table 2). The isotype control mAb showed minimal binding signal to 10 pM of rhMAdCAM.Fc. The assay meets the requisite condition for obtaining an accurate or thermodynamic KD measurement by having target concentrations at or well below the KD (Chang et al., 1975; Lowe et al., 2009). This result compares well with Biacore values for the same protein (KD, 16.1 pM). The small discrepancy between these two values is likely to be attributable to differences in assay conditions, with the free‐solution method minimizing perturbation to the interacting species (Jung et al., 2008; Hulme and Trevethick, 2010; Olmsted et al., 2012).
Table 2.
Summary of affinity values. Compilation of the measured affinities between PF‐00547659 and the various MAdCAM forms, MAdCAM concentrations and goodness of fit for the binding assays
| KD{matrix},{MAdCAM form} | MAdCAM concentration (pM) | KD (pM) | CV (%) | N | R2 | |
|---|---|---|---|---|---|---|
| BSI experimentally measured values | KD{buffer},{rhMAdCAM.Fc} | 10 | 7.1 | 22 | 5 | 0.97 |
| KD{0.1% serum},{s} | 1.0 | 7.5 | 20 | 5 | 0.98 | |
| KD,app{10% serum},{s} | 100 | 30 | 25 | 5 | 0.96 | |
| KD,app {25% serum},{s} | 250 | 110 | 38 | 5 | 0.92 | |
| KD,app {35% serum},{s} | 350 | 174 | 38 | 5 | 0.93 | |
| KD,app {50% serum},{s} | 500 | 285 | 36 | 5 | 0.94 | |
| KD{CHO VRH},{m} | 34 | 134 | 33 | 5 | 0.95 | |
| KD{tissue,VRH},{m} a | 0.043 | 155 | 27 | 5 | 0.97 | |
| KD{tissue,IOH},{s} | ≤44b | 207 | 31 | 4 | 0.97 | |
| KD,app‐int{tissueVRH + 87.5%ILH},{m+s} | 0.043 + 39 | 322 | 11 | 4 | 0.99 | |
| Calculated value | KD,app{100% serum},{s} | – | 596 | – | – | – |
| Other methods | KD{buffer},{rhMAdCAM.Fc} (Biacore) | – | 16.1c | – | – | – |
| K D,in‐vivo (TMDD Model) | – | 528d | – | – | – |
KD in serum
The KD of PF‐00547659 to 1 pM endogenous sMAdCAM was measured using 0.1% healthy human serum pool (Table 2), yielding a value of 7.5 (±1.5) pM (Figure 3C–D) which was in close agreement to the KD value in buffer. Importantly, these determinations were performed in a near native environment, on endogenous MAdCAM in human serum diluted with only PBS. A reference sample included an identical sample matrix with the isotype control mAb.
In vitro KD,app extrapolated to 100% serum correlates with the in vivo apparent serum KD (KD,in‐vivo)
Affinity measurements of PF‐00547659 with increasing amounts of pooled human serum (10, 25, 35 and 50%) containing sMAdCAM were conducted and yielded increasing serum KD,app values of 30, 110, 174, and 285 pM respectively (Figure 3E–F). It has been shown that KD,app can be right shifted and can be obtained using a positive linear relationship with target concentration described by Chang et al. (Chang et al., 1975) when target concentration is significantly higher than KD. Indeed, by plotting these KD,app values versus serum percentages, we obtained a linear relationship with an unweighted correlation coefficient (R2) of 0.993 (Figure 3G). Extrapolating this plot to 100% serum resulted in an estimated KD,app{100% serum} of 596 pM, which correlates with clinically derived KD,in‐vivo from target‐mediated drug disposition modelling of mAb serum concentrations as 528 pM (Martin et al., 2009).
Calculated sMAdCAM concentration in serum
By using the extrapolated KD,app at 100% serum and KD{0.1% serum}, the calculated sMAdCAM concentration is 1177 pM.
KD in CHO VRH and KD,app‐int in colon VRH
To obtain samples that closely resemble the in vivo micro‐environment of cells or human colon tissue, we prepared VRH that includes the cell membrane, soluble proteins and cytoskeleton components. VRH was prepared using a variation of the protocol to create red blood cell ‘ghosts’ (Schwoch and Passow, 1973) and previously employed with BSI (Baksh et al., 2011). This relatively simple procedure involves physical disruption of tissue only, homogenization, probe sonication which disrupts the cell membranes and breaks them into smaller fragments that reassemble into small unilamellar vesicles retaining the composition and fluidity of native membranes and centrifugation of large debris.
We quantified the binding of PF‐00547659 and the isotype control mAb to MAdCAM containing CHO VRH (Figure 4A–C). The isotype control mAb showed minimum binding signal. Both thermodynamic KD values for mAb‐CHO‐rhMAdCAM VRH (134 pM) and mAb‐colon tissue VRH (155 pM, Figure 4D–F) were similar to each other but surprisingly were approximately 20‐fold higher (weaker affinity) than the KD in buffer (7.1 pM) and human serum (7.5 pM), indicating a different binding interaction of mAb‐sMAdCAM versus mAb‐mMAdCAM.
KD{tissue,sMAdCAM} in IOH
To obtain sMAdCAM from the tissue interstitial space, it was necessary to separate sMAdCAM from any mMAdCAM. Consequently, an ILH sample was generated from healthy human colon tissue using a mild ‘tissue elution’ (Wiig and Swartz, 2012). A further centrifugation step was applied to ILH to separate any membrane fraction and harvest a colon IOH. Surprisingly, the KD{tissue,IOH} value was found to be 207 pM (Figure 4G–I).
KD,app‐int in sample combining VRH and tissue ILH
Combining colon tissue VRH with ILH yielded a sample with a different ratio of mMAdCAM and sMAdCAM where these two forms coexist and KD,app‐int was measured to be 322 pM (Figure 5A–C).
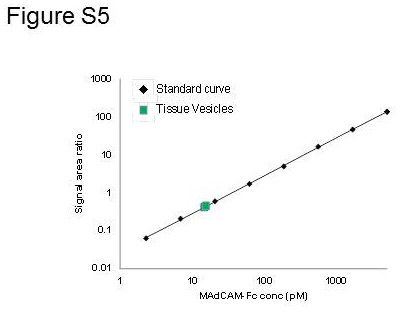
Measured MAdCAM concentrations in serum, CHO and colon tissue VRH and ILH by LC–MS/MS
Total MAdCAM concentrations in various samples were measured by LC–MS/MS Table (S4, Figure S4). The concentration of MAdCAM in 100% pooled human serum sample was 999 pM. The concentration of CHO‐rhMAdCAM VRH was 1720 pM at a total protein concentration of 100 μg·mL−1 that was further diluted 50‐fold to a final MAdCAM concentration of 34 pM in BSI experiments. Total MAdCAM concentrations of VRH and ILH samples in BSI experiment were measured by LC–MS/MS at 0.043 and 39 pM respectively. Correcting for dilutions prior to LC–MS/MS, in 100% colon, total endogenous MAdCAM concentration was 900 pM, and sMAdCAM concentration was 220 pM. Assuming ILH contains mostly sMAdCAM, mMAdCAM concentrations would be calculated at 680 pM.
Discussion
In vitro methods measuring physiologically relevant drug‐target binding interactions that consistently and accurately predict KD,in vivo are critically important to enable early discovery bioengineering and affinity optimization efforts. Clinically, they provide confidence that drug candidates provide the required degree of target coverage for efficacy in both serum and at the site of disease during clinical trials and that efficacy can be achieved with a range of doses. The MAdCAM therapeutic mAb PF‐00547659 lacks IVIVC with an over 30‐fold difference in mAb‐target binding affinity between in vitro SPR KD and clinically derived KD. While clinical efficacy of PF‐00547659 in UC has been demonstrated (Reinisch et al., 2015), it is also noteworthy that PF‐00547659 failed to meet the primary efficacy endpoint in a recent study in Crohn's Disease (Sandborn et al., 2015). Further study is required to understand these results. However, the affinity of PF‐00547659 to MAdCAM as measured by Biacore may be misleading in light of the present BSI results.
Comparable PF‐00547659 KD values for soluble rhMAdCAM.Fc in buffer [Biacore 16.1 pM (Pullen et al., 2009) versus BSI 7.1 pM] and for endogenous sMAdCAM in serum (7.5 pM) were observed. The BSI method also allowed experimentation up to 50% human serum, a level of matrix complexity that cannot be achieved using SPR; enabling quantitative binding determinations on native soluble target in a system more representative of the endogenous environment, with sMAdCAM levels up to 70 times higher than KD. Of note is that BSI extrapolated KD,app in 100% serum is estimated without prior knowledge of MAdCAM concentrations, is similar to clinically derived KD,app and demonstrates the validity of applying BSI to IVIVC.
When target concentrations are unknown in serum or tissue, BSI can be used to estimate KD,app and also target concentrations, presently unattainable by other technologies. As BSI in vitro KD,app in serum closely correlates with in vivo‐derived KD,app, the results demonstrate the right shift of KD,app from thermodynamic KD (resulting in lower apparent affinity) is a result of higher MAdCAM concentrations in serum (999 pM), and thus, KD,app is target concentration limited. This right shift of KD,in‐vivo may also be caused by factors other than high target concentrations, including avidity from higher target density (Rudnick and Adams, 2009) and binding to non‐target proteins up‐regulated in disease pathology. Only by testing binding affinities at high serum levels, one can measure the physiologically relevant KD that ultimately defines clinical efficacy.
Although IVIVC was established using KD measurements in serum, quantifying drug‐target affinity in tissue samples remains a challenge. When predicting efficacy, it is common practice to assume that KD in tissue is equal to KD in serum which remains to be proven. For the target MAdCAM, it is present as a soluble form in serum only and exists as both soluble and membrane forms in the tissue interstitial space and both forms are thought to play an important role in IBD pathology. We set out to measure the KD of PF‐00547659 to sMAdCAM in tissue using colon IOH, KD of mMAdCAM using CHO and tissue VRH and KD,app‐int by combining VRH with ILH. Minimal sample preparation steps of colon tissue and use of only PBS buffer allowed near native target conformation, ratios to other components and micro environment. Surprisingly, compared with KD{0.1% serum} of 7.5 pM, a weaker mAb affinity was observed for sMAdCAM in IOH with KD{tissue,IOH} value of 207 pM, a similarly weak affinity of PF‐00547659 for both recombinant and native mMAdCAM, KD{CHO,VRH} of 135 pM and KD{tissue,VRH} 155 pM. Combining VRH and ILH yielded a further right shifted KD,app‐int of 322 pM. As soluble rhMAdCAM.Fc and CHO mMAdCAM proteins have the same ECD amino acid sequence, we postulate that the transmembrane anchoring and conformation restrictions of the membrane (Yu et al., 2012) are significant contributors to the decreased PF‐00547659 affinity compared with sMAdCAM in serum (Schiller and Fassler, 2013). The underlying physiological mechanism that results in a near 30‐fold difference in sMAdCAM KD between serum and colon is not well understood and requires further investigation. However, this is the first report to disclose different binding affinities of a mAb to its target in serum and tissue, demonstrating that tissue affinity measurements are critical in predicting therapeutic benefit and efficacy. The lower target affinity in colon, as measured by BSI, may contribute to the lack of clinically observed efficacy in Crohn's disease.
The further rightward shift of KD,app‐int from KD for soluble and membrane MAdCAM in tissue is perplexing because the concentration of MAdCAM (<44 pM) in the sample is below the KD of both forms. Thus this shift is not due to the high target concentration. Measuring KD in samples with increasing concentrations of ILH and/or VRH combines sMAdCAM and mMAdCAM at different ratios and would provide information for a modelling effort to calculate KD,app‐int at 100% tissue homogenate. Values from such samples may help explain the rightward KD shift.
To properly measure a binding affinity using BSI, the binding sample must be compared with an RI‐matched reference sample and well‐selected controls used to ensure the readout is reporting a specific molecular interaction. While it is straightforward to select a reference sample for interactions in buffer, in the case of serum and tissues, it can be extremely challenging to obtain an unaltered reference sample devoid of only the target. In this study, many attempts were conducted to remove only sMAdCAM from serum samples by affinity chromatography, but they failed to provide an RI‐matched reference serum sample (data not shown). However, we are confident that the binding measured in this study is specific to the target since 1) we acquired similar KD values of PF‐00547659 to sMAdCAM as recombinant protein (7.1 pM) or endogenous in serum (7.5 pM), and 2) we measured similar KD values of PF‐00547659 to mMAdCAM in CHO VRH (134 pM) with CHO‐WT VRH used as control where specificity is clearly demonstrated, and in colon VRH (155 pM) with an isotype‐matched control mAb used as a reference.
With increasing biological matrix complexity in these BSI experiments, the CV% of the data obtained increased from approximately 20% in buffer and very diluted serum [similar to Biacore (Rich et al., 2009)] to 38 and 33% when using 25–50% serum and colon VRH respectively (Table 2). Additionally, we also noted that with 25% serum, the signal to noise ratio (S/N) was ~3.8 at saturation, and a larger absolute signal with improved S/N would be more desirable to more accurately quantify the affinity. Even with the relatively high variability, the results illustrate a reasonably accurate KD value for endogenous target residing in the serum or tissue environment.
The sensitivity of BSI is demonstrated when correcting for dilutions for VRH (0.005% of the total tissue content), the target concentration was determined by LC–MS/MS to be 43 fM. Within the 7.5 nL probe volume of BSI, we calculated there are only approximately 200 MAdCAM molecules present when measuring the binding signals. Typically any tissue samples are precious, necessitating an assay that can constrain sample consumption. The BSI‐based method uniquely fulfills this requirement. For example, the entire binding assay (five replicate determinations, on 10 different concentrations measured in triplicate) required a total of about 27 μg of tissue sample or 400 μL of serum. Capitalizing on the compatibility of BSI with microfluidic sample preparation and introduction methods, the prospect of further sample volume reduction and increased throughput is under investigation (Chin et al., 2011; Zec et al., 2012).
Employing BSI allows for capturing both the biodiversity of target environments, the complexity of binding scenarios, and represents a major advance in in vitro protein–protein interaction (e.g. binding) assays, allowing establishment of IVIVC, clinically relevant results to be obtained rapidly on real‐world samples, in a volume‐constrained format and at endogenously expressed levels of protein. To our knowledge, this is the first time such an approach has been used to successfully interrogate binding interactions on human tissue samples, quantifying the affinity for the target in a physiologically relevant environment and at the same time estimating the endogenous target concentrations. It is appreciated that this report involves measurements of a single drug‐target pair. The limitations of applying BSI await further assessment with additional drug targets. For example, it is possible that using an isotype control mAb as both control and reference may not be appropriate if the target binds other components in the matrix other than the drug. In such a case, different target depletion methods may be developed to define the binding specificity. We also anticipate that other drug candidates may have different affinities to target forms in various organs due to variable target concentrations, locations, binding partners and the potential of confounding drug‐induced changes. Our study and future ones could provide a physiome‐based approach for advancing pharmacology, with the potential to improve clinical translation for new drug candidates. The in vitro method described above can significantly expedite the discovery process by providing physiologically relevant drug‐target binding affinities and target concentrations, without the necessity of in vivo animal studies, to obtain a preliminary estimate of a safe starting FIH dose, and efficacious doses required in patients.
Author contributions
M.W., A.K.K., D.J.B. and D.O.H. conceptualized the project, designed and analysed experimental data; A.K.K. performed the BSI measurements; M.F.O. conducted MAdCAM measurements by LC–MS/MS; J.R.C. provided the target concentration calculation and modelling discussions; C.S.L. provided IVIVC translation and human contextual literature; and M.W., A.K.K., M.F.O., J.R.C., C.S.L., D.J.B. and D.O.H. contributed to writing and review of the manuscript.
Conflict of interest
D.J.B. and A.K. have a financial interest in a company that is commercializing BSI. Other authors declare that they have no competing financial interests apart from their employment with Pfizer Inc.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Characterization of CHO cell vesicles. Representative results for DLS (A) and Bradford Assay (B) of vesicle solutions made from CHO cells.
Figure S2 Characterization of human colon tissue VRH. Representatives results for DLS (A) and Bradford Assay (B) of VRH made from colon tissue. Polydispersity index was measured and if greater than 0.28 by DLS, the VRH sample was probe sonicated again on ice for 90 s in a pulsed manner (5 s on, 1 s off), followed by centrifugation and characterization of the sample by DLS and Bradford as described.
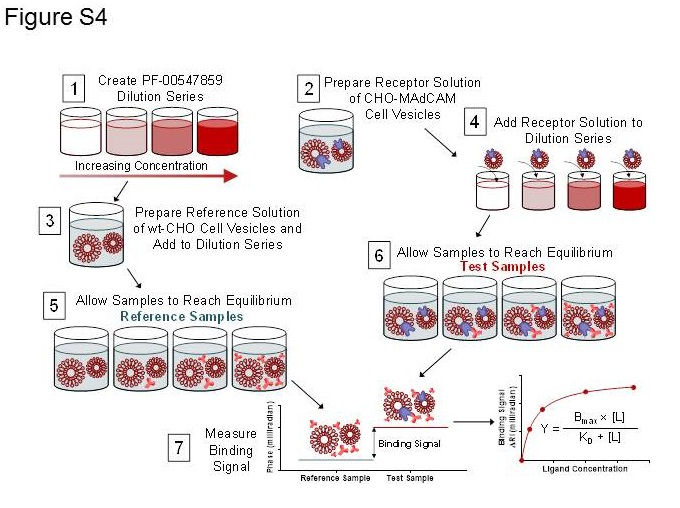
Figure S3 Cell based assay protocol diagram. Illustration of the experimental procedure to measure the binding affinity in CHO cell vesicles.
Figure S4 Block diagram of a Backscattering Interferometer.
Table S1 Components of the reference and test samples. A pictorial representation of the reference and test samples used in the work‐flow.
Table S2 MadCAM sample concentrations. Tabulation of sample concentrations measured by LC–MS/MS and used in BSI.
Supporting info item
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgements
The authors would like to thank the following Pfizer colleagues: Kathleen M. Shields for providing antibodies and cell lines; Anson K. Abraham for discussion in IVIVC; Nick Pullen and Hannah Jones for discussion of the MAdCAM programme; Steve Martin and Alaa Ahmad for providing clinical data and discussion; and Hendrik Neubert for MAdCAM LC–MS/MS discussions.
This work was funded by Pfizer Inc and the National Science Foundation Grant (CHE‐1307899) awarded to D.J. Bornhop.
Wang, M. , Kussrow, A. K. , Ocana, M. F. , Chabot, J. R. , Lepsy, C. S. , Bornhop, D. J. , and O'Hara, D. M. (2017) Physiologically relevant binding affinity quantification of monoclonal antibody PF‐00547659 to mucosal addressin cell adhesion molecule for in vitro in vivo correlation. British Journal of Pharmacology, 174: 70–81. doi: 10.1111/bph.13654.
Contributor Information
Darryl J Bornhop, Email: darryl.bornhop@vanderbilt.edu.
Denise M O'Hara, Email: denise.o'hara@pfizer.com.
References
- Ababou A, Ladbury JE (2007). Survey of the year 2005: literature on applications of isothermal titration calorimetry. J Mol Recognit 20: 4–14. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolaki M, Manoloukos M, Roulis M, Wurbel MA, Muller W, Papadakis KA et al. (2008). Role of beta7 integrin and the chemokine/chemokine receptor pair CCL25/CCR9 in modeled TNF‐dependent Crohn's disease. Gastroenterology 134: 2025–2035. [DOI] [PubMed] [Google Scholar]
- Araujo RP, Liotta LA, Petricoin EF (2007). Proteins, drug targets and the mechanisms they control: the simple truth about complex networks. Nat Rev Drug Discov 6: 871–880. [DOI] [PubMed] [Google Scholar]
- Baksh MM, Kussrow AK, Mileni M, Finn MG, Bornhop DJ (2011). Label‐free quantification of membrane‐ligand interactions using backscattering interferometry. Nat Biotechnol 29: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhop DJ, Kammer MN, Kussrow A, Flowers RA 2nd, Meiler J (2016). Origin and prediction of free‐solution interaction studies performed label‐free. Proc Natl Acad Sci U S A 113: E1595–E1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhop DJ, Latham JC, Kussrow A, Markov DA, Jones RD, Sorensen HS (2007). Free‐solution, label‐free molecular interactions studied by back‐scattering interferometry. Science 317: 1732–1736. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976). Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein–dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Brodfuehrer J, Rankin A, Edmonds J, Keegan S, Andreyeva T, Lawrence‐Henderson R et al. (2014). Quantitative analysis of target coverage and germinal center response by a CXCL13 neutralizing antibody in a T‐dependent mouse immunization model. Pharm Res 31: 635–648. [DOI] [PubMed] [Google Scholar]
- Chang KJ, Jacobs S, Cuatrecasas P (1975). Quantitative aspects of hormone‐receptor interactions of high affinity – effect of receptor concentration and measurement of dissociation‐constants of labeled and unlabeled hormones. Biochim Biophys Acta 406: 294–303. [DOI] [PubMed] [Google Scholar]
- Chin CD, Laksanasopin T, Cheung YK, Steinmiller D, Linder V, Parsa H et al. (2011). Microfluidics‐based diagnostics of infectious diseases in the developing world. Nat Med 17: 1015–U1138. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasio E, De Cristofaro R (2010). The effect of shear stress on protein conformation: physical forces operating on biochemical systems: The case of von Willebrand factor. Biophys Chem 153: 1–8. [DOI] [PubMed] [Google Scholar]
- Goto A, Arimura Y, Shinomura Y, Imai K, Hinoda Y (2006). Antisense therapy of MAdCAM‐1 for trinitrobenzenesulfonic acid‐induced murine colitis. Inflamm Bowel Dis 12: 758–765. [DOI] [PubMed] [Google Scholar]
- Guha M (2011). PARP inhibitors stumble in breast cancer. Nat Biotechnol 29: 373–374. [DOI] [PubMed] [Google Scholar]
- Hokari R, Kato S, Matsuzaki K, Iwai A, Kawaguchi A, Nagao S et al. (2001). Involvement of mucosal addressin cell adhesion molecule‐1 (MAdCAM‐1) in the pathogenesis of granulomatous colitis in rats. Clin Exp Immunol 126: 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme EC, Trevethick MA (2010). Ligand binding assays at equilibrium: validation and interpretation. Br J Pharmacol 161: 1219–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince R, Narayanaswamy R (2006). Analysis of the performance of interferometry, surface plasmon resonance and luminescence as biosensors and chemosensors. Anal Chim Acta 569: 1–20. [Google Scholar]
- Jung HS, Yang T, Lasagna MD, Shi JJ, Reinhart GD, Cremer PS (2008). Impact of hapten presentation on antibody binding at lipid membrane interfaces. Biophys J 94: 3094–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson OP, Lofas S (2002). Flow‐mediated on‐surface reconstitution of G‐protein coupled receptors for applications in surface plasmon resonance biosensors. Anal Biochem 300: 132–138. [DOI] [PubMed] [Google Scholar]
- Karp G (2005). Cell and molecular biology: Concepts and experiments, 4th edn. Hoboken, NJ: Von Hoffman press. [Google Scholar]
- Kastritis PL, Moal IH, Hwang H, Weng Z, Bates PA, Bonvin AM et al. (2011). A structure‐based benchmark for protein–protein binding affinity. Protein Sci 20: 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussrow A, Enders CS, Bornhop DJ (2012). Interferometric methods for label‐free molecular interaction studies. Anal Chem 84: 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E, Lehnert KB, Kanwar JR, Yang Y, Mon Y, McNeil HP et al. (2004). Bioassay detects soluble MAdCAM‐1 in body fluids. Immunol Cell Biol 82: 400–409. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Nuber S, Hoffmann C (2012). Fluorescence/bioluminescence resonance energy transfer techniques to study G‐protein‐coupled receptor activation and signaling. Pharmacol Rev 64: 299–336. [DOI] [PubMed] [Google Scholar]
- Lowe PJ, Tannenbaum S, Wu K, Lloyd P, Sims J (2009). On setting the first dose in man: quantitating biotherapeutic drug‐target binding through pharmacokinetic and pharmacodynamic models. Basic Clin Pharmacol Toxicol 106: 195–209. [DOI] [PubMed] [Google Scholar]
- Malvern (2012). Zetasizer Nano Series User Manual MAN0498–1‐0. Malvern Instruments Ltd.: Worchestershire, UK. [Google Scholar]
- Martin SW, Magnusson MO, Weatherley BC, Matthews IT, Burgess B, Niezychowski W (2009). Mechanistic population pharmacokinetics (PK) model of PF‐00547659, a fully human IgG2 anti‐MAdCAM antibody, in ulcerative colitis patients: results of a first in human (FIH) study In: American Gastroenterological Association Digestive Disease Week. Chicago, IL: Digestive Disease Week (DDW). [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C et al. (2013). Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341: 84–87. [DOI] [PubMed] [Google Scholar]
- Neubert H, Gale J, Muirhead D (2010). Online high‐flow peptide immunoaffinity enrichment and nanoflow LC–MS/MS: assay development for total salivary pepsin/pepsinogen. Clin Chem 56: 1413–1423. [DOI] [PubMed] [Google Scholar]
- Olmsted IR, Kussrow A, Bornhop DJ (2012). Comparison of free‐solution and surface‐immobilized molecular interactions using a single platform. Anal Chem 84: 10817–10822. [DOI] [PubMed] [Google Scholar]
- Olmsted IR, Hassanein M, Kussrow A, Hoeksema M, Li M, Massion PP et al. (2014). Toward rapid, high‐sensitivity, volume‐constrained biomarker quantification and validation using backscattering interferometry. Anal Chem 86: 7566–7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N, Molloy E, Carter D, Syntin P, Clemo F, Finco‐Kent D et al. (2009). Pharmacological characterization of PF‐00547659, an anti‐human MAdCAM monoclonal antibody. Br J Pharmacol 157: 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinisch W, Sanborn B, Danese S, Cataldi F, Hebuterne X, Alzberg B et al. (2015). A randomized, multicenter double‐blind, placebo‐controlled study of the safety and efficacy of anti‐MAdCAM antibody PF‐00547659 (PF) in patients with moderate to severe ulcerative colitis: results of the TURANDOT study. Gastroenterology 148: 1. [Google Scholar]
- Rich RL, Papalia GA, Flynn PJ, Furneisen J, Quinn J, Klein JS et al. (2009). A global benchmark study using affinity‐based biosensors. Anal Biochem 386: 194–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick SI, Adams GP (2009). Affinity and avidity in antibody‐based tumor targeting. Cancer Biother Radiopharm 24: 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetear P, Perrin AJ, Bartholdson SJ, Wanaguru M, Kussrow A, Bornhop DJ et al. (2015). Quantification of Plasmodium‐host protein interactions on intact, unmodified erythrocytes by back‐scattering interferometry. Malar J 14: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandborn W, Lee SD, Tarabar D, Louis E, Klopocka M, Klaus J et al. (2015). Anti‐MAdCAM‐1 antibody (PF‐00547659) for active refractory Crohn's disease: results of the OPERA study. Gastroenterology 148: 1. [Google Scholar]
- Schiller HB, Fassler R (2013). Mechanosensitivity and compositional dynamics of cell‐matrix adhesions. EMBO Rep 14: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C (2010). GSK/Sirtris compounds dogged by assay artifacts. Nat Biotechnol 28: 185–186. [DOI] [PubMed] [Google Scholar]
- Schwoch G, Passow H (1973). Preparation and properties of human erythrocyte‐ghosts. Mol Cell Biochem 2: 197–218. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney K, Markov D, Bornhop DJ (2000). Chip‐scale universal detection based on backscatter interferometry. Anal Chem 72: 2690–2695. [DOI] [PubMed] [Google Scholar]
- Velazquez‐Campoy A, Freire E (2006). Isothermal titration calorimetry to determine association constants for high‐affinity ligands. Nat Protoc 1: 186–191. [DOI] [PubMed] [Google Scholar]
- Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S (2010). Protein‐binding assays in biological liquids using microscale thermophoresis. Nat Commun 1. [DOI] [PubMed] [Google Scholar]
- Wiig H, Swartz MA (2012). Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol Rev 92: 1005–1060. [DOI] [PubMed] [Google Scholar]
- Yan YL, Marriott G (2003). Analysis of protein interactions using fluorescence technologies. Curr Opin Chem Biol 7: 635–640. [DOI] [PubMed] [Google Scholar]
- Yu YM, Zhu JH, Mi LZ, Walz T, Sun H, Chen JF et al. (2012). Structural specializations of alpha(4)beta(7), an integrin that mediates rolling adhesion. J Cell Biol 196: 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zec H, Rane TD, Wang TH (2012). Microfluidic platform for on‐demand generation of spatially indexed combinatorial droplets. Lab Chip 12: 3055–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Duhr S, Baaske P, Laue E (2014). Microscale thermophoresis for the assessment of nuclear protein‐binding affinities. Methods Mol Biol 1094: 269–276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Characterization of CHO cell vesicles. Representative results for DLS (A) and Bradford Assay (B) of vesicle solutions made from CHO cells.
Figure S2 Characterization of human colon tissue VRH. Representatives results for DLS (A) and Bradford Assay (B) of VRH made from colon tissue. Polydispersity index was measured and if greater than 0.28 by DLS, the VRH sample was probe sonicated again on ice for 90 s in a pulsed manner (5 s on, 1 s off), followed by centrifugation and characterization of the sample by DLS and Bradford as described.
Figure S3 Cell based assay protocol diagram. Illustration of the experimental procedure to measure the binding affinity in CHO cell vesicles.
Figure S4 Block diagram of a Backscattering Interferometer.
Table S1 Components of the reference and test samples. A pictorial representation of the reference and test samples used in the work‐flow.
Table S2 MadCAM sample concentrations. Tabulation of sample concentrations measured by LC–MS/MS and used in BSI.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item