

Abstract
Objective
Parkinson's disease (PD) presents clinically with several motor subtypes that exhibit variable treatment response and prognosis. Here, we investigated genetic variants for their potential association with PD motor phenotype and progression.
Methods
We screened 10 SNPs, previously associated with PD risk, for association with tremor‐dominant (TD) versus postural‐instability gait disorder (PIGD) motor subtypes. SNPs that correlated with the TD/PIGD ratio in a discovery cohort of 251 PD patients were then evaluated in a multi‐site replication cohort of 559 PD patients. SNPs associated with motor phenotype in both cross‐sectional cohorts were next evaluated for association with (1) rates of motor progression in a longitudinal subgroup of 230 PD patients and (2) brain alpha‐synuclein (SNCA) expression in the GTEx (Genotype‐Tissue Expression project) consortium database.
Results
Genotype at rs356182, near SNCA, correlated with the TD/PIGD ratio in both the discovery (Bonferroni‐corrected P = 0.04) and replication cohorts (P = 0.02). The rs356182 GG genotype was associated with a more tremor‐predominant phenotype and predicted a slower rate of motor progression (1‐point difference in annual rate of UPDRS‐III motor score change, P = 0.01). The rs356182 genotype was associated with SNCA expression in the cerebellum (P = 0.005).
Interpretation
Our study demonstrates that the GG genotype at rs356182 provides molecular definition for a clinically important endophenotype associated with (1) more tremor‐predominant motor phenomenology, (2) slower rates of motor progression, and (3) decreased brain expression of SNCA. Such molecularly defined endophenotyping in PD may benefit both clinical trial design and tailoring of clinical care as we enter the era of precision medicine.
Introduction
Parkinson disease (PD) is a neurodegenerative disease with a diagnosis that remains clinically based on the presence of cardinal motor symptoms of tremor, bradykinesia, rigidity, and postural instability.1 Yet within PD, there is remarkable heterogeneity in terms of symptomatology and rate of progression.2, 3, 4 Ascertaining endophenotypes (i.e., motor phenotypes with a clear genetic connection) linked to differential clinical presentations and rates of progression would benefit patient care and the design of clinical trials.
On clinical grounds, there have been multiple attempts to define motor subtypes of PD.3, 4, 5, 6, 7, 8, 9, 10 Two of the most recognized are tremor‐dominant (TD) and postural instability and gait difficulty (PIGD) subtypes.5 The classification of TD and PIGD is based on the ratio of tremor to axial (nontremor) scores from the motor subscale of the Unified Parkinson Disease Rating Scale (UPDRS).11 This classification of TD and PIGD is not immutable, and up to 78% of individuals may convert from TD to PIGD over time.2, 12 This instability of clinical classification highlights the need for biological and molecular subtyping. As it stands, however, the TD and PIGD motor subtypes do provide a phenomenological classification that is easily understood by most PD clinicians.
The TD and PIGD phenotypes have been associated with different clinical trajectories. The TD subtype has been associated with slower motor progression.13 The PIGD subtype, in contrast, has been associated with increased risk for cognitive impairment12, 14, 15, 16 or dementia,12, 15 depression,5, 17, 18 impulsivity,19 less responsiveness to levodopa,2 poorer quality of life,17 greater disability,5 and greater mortality risk.20, 21 These differences suggest that individuals with the TD subtype overall have a more benign clinical course and may have different biological and molecular underpinnings than the PIGD subtype.
However, the pathophysiologic differences between motor subtypes remain unknown. We sought to ascertain a molecular delineation of PD motor subtypes, and an understanding of why molecularly defined groups might differ in progression. To these ends, we investigated single‐nucleotide polymorphisms (SNPs) previously associated with risk for development of PD and assessed if they are also associated with motor subtype, disease progression, and gene expression.
Methods
Subjects
Two clinical cohorts were used in this study. All participants had a diagnosis of PD by UK PD Society Brain Bank Clinical Diagnostic criteria.1
University of Pennsylvania (UPenn) Cohort
Two hundred and fifty‐one subjects with PD have been recruited to the UPenn Udall Center, a longitudinal observational research cohort, and all available subjects were used for this study. The subjects are followed on an annual basis for the first 4 years, and a biennial basis thereafter. The collection and utilization of the data was approved by the UPenn Institutional Review Board. The baseline study visit data were used for the cross‐sectional analysis in this study, and all completed follow‐up visits (available for 230/251 patients) were included in the longitudinal analysis. Of the 251 subjects, 82 have died, and these subjects, along with three additional subjects who were initially enrolled in the Udall cohort but had insufficient data for determination of TD/PIGD ratio, were included in the secondary analysis as the 85‐subject autopsy cohort.
Replication cohort
A total of 559 subjects with PD were recruited in the replication cohort. These subjects were recruited to observational research cohorts at the Pacific Northwest Udall Center (PANUC) in Seattle and Portland, and the University of Cincinnati Parkinson's disease research centers. The collection and utilization of this data was approved by their respective Institutional Review Boards.
Clinical assessment
In the UPenn cohort, subjects underwent reassessment of their PD with the UPDRS at each study visit. The TD/PIGD ratios were calculated using items from parts II (activities of daily living) and III (motor examination) of the UPDRS (UPDRS‐II and UPDRS‐III), as previously described.5 In brief, the TD/PIGD ratio consists of the average tremor score divided by the average PIGD score. To avoid a non‐numerical result, for ratios with an average PIGD score of 0, the subject's tremor score was substituted as the value of the TD/PIGD ratio. We utilized the TD/PIGD ratio for our analysis, rather than strict categorization as TD or PIGD, in order to capture what in reality is a continuum of phenotypic variation.
In the replication cohort, the more recent version of the UPDRS, the Movement Disorder Society (MDS)–UPDRS,22 was used for motor assessments. Calculations of the TD/PIGD ratio with the MDS‐UPDRS versus the UPDRS have been previously reported.23 We used the items from Part III of the MDS‐UPDRS to obtain average tremor score and average PIGD score for each individual. We then obtained TD/PIGD ratios in the same manner as for the UPenn cohort. For detailed descriptions of the exact items included, please see Data S1.
Genetic analysis
In discovery and replication cohorts, genotypes were ascertained with the NeuroX genotyping array.24 Of the 28 independent single‐nucleotide polymorphisms (SNPs) previously reported to be associated with PD risk,25 the 10 SNPs with the most statistically significant associations were included in our analyses. In some cases, proxy SNPs were substituted for previously‐reported risk SNPs, if the previously reported SNPs were not represented on the NeuroX genotyping array, and the proxy SNP was in strong linkage disequilibrium (LD) with the previously reported SNP. The examined SNPs are presented in Table 1.
Table 1.
SNPs evaluated for association with motor subtype
| PD risk SNP | Neighboring gene | Minor allele frequencies | ||
|---|---|---|---|---|
| Minor allele | UPenn PD cohort | 1000 Genomes | ||
| rs71628662 | GBA‐SYT11 | C | 0.01 | 0.02 |
| rs17649553 | MAPT | T | 0.18 | 0.24 |
| rs34311866 | TMEM175‐GAK‐DGKQ | C | 0.21 | 0.19 |
| rs12637471 | MCCC1 | A | 0.20 | 0.20 |
| rs1955337 | STK39 | T | 0.13 | 0.12 |
| rs6430538 | ACMSD‐TMEM163 | T | 0.49 | 0.50 |
| rs11724635 | BST1 | C | 0.41 | 0.44 |
| rs823118 | RAB7L1‐NUCKS | C | 0.39 | 0.47 |
| rs356182 | SNCA | G | 0.40 | 0.36 |
| rs1077989 | TMEM229B | C | 0.46 | 0.45 |
Ten single‐nucleotide polymorphisms (SNPs) previously associated with increased risk for PD were evaluated in this study. Minor allele frequencies in the UPenn discovery cohort of 251 PD patients versus minor allele frequencies for populations of European ancestry (1000genomes.org, EUR cohort) are shown.
Statistical analysis
The open‐source R statistical package was used for analyses, and R‐scripts are included in the Data S3.
Primary cross‐sectional analyses
Linear regression models were used to assess cross‐sectional associations between genotype at a candidate SNP and the TD/PIGD ratio, with covariates of age, sex, duration of PD, and total levodopa equivalent daily dose (LEDD) included in the models, as indicated in the text. In our screening analysis in the discovery cohort, we made no assumptions about genetic model (major‐allele‐dominant vs. codominant vs. minor‐allele‐dominant), investigating genotypes as categorical variables instead. Significant findings from the screening analysis were then individually assessed, and specific genetic models were applied as indicated in the text. Bonferroni correction was used to adjust for multiple comparisons in our cross‐sectional analysis for a type I error rate of 0.05: 0.05/10 SNPs = 0.005. Two‐tailed corrected P‐values are reported for all analyses.
Among previously reported PD genetic risk SNPs, we chose the ten SNPs with the most statistically significant associations, as indicated by P‐value from the joint‐phase analysis reported by Nalls and coauthors25 for screening in our discovery cohort. SNPs found to significantly associate with TD/PIGD ratio in the discovery cohort were then tested for replication in the replication cohort, using the same model and covariates applied to the discovery cohort. We further performed a subanalysis of clinical site (PANUC‐Seattle, PANUC‐Portland, or University of Cincinnati) as an additional covariate in the replication cohort.
Primary longitudinal analysis
SNPs found to significantly associate with the TD/PIGD ratio in the cross‐sectional analyses were tested for association with motor progression. Specifically, linear mixed‐effects models26 were used to investigate the effect of SNPs on rate of change in UPDRS‐III scores, adjusting for age, sex, baseline UPDRS‐III score, and LEDD as covariates. A random intercept term was included in the linear mixed‐effects model to account for correlations among repeated measures of the UPDRS‐III scores. The fixed effects included SNP, time, interaction between SNP and time, and the covariates mentioned above.
Secondary analyses
For the cross‐sectional analysis, we examined the association of each of the 10 SNPs with other motor outcomes in the discovery cohort using linear regression models. The motor outcomes investigated included tremor subscore, PIGD subscore, and UPDRS‐III score. The linear regression model included age, sex, PD duration, and LEDD as covariates. In addition, SNPs found to correlate significantly with TD/PIGD ratio in the original linear model were further investigated in models omitting covariates, as indicated in the text, to understand whether observed associations were dependent on these covariates. Finally, linear models using log‐transformed values for TD/PIGD ratio were investigated, incorporating the same covariates as in the primary analyses, in order to correct for potential effects of skewing of the distribution on our results.
Similar secondary analyses investigating the effects of including or omitting specific covariates were performed for the longitudinal linear mixed‐effect model analysis as well.
Linear regression models covarying for sex and age at onset were used to investigate the correlation between genotypes and total disease duration within the autopsy cohort.
Analysis of eQTL effects and LD structure at the SNCA locus
To determine whether rs356182 or other PD‐associated variants are located in predicted gene regulatory regions, we visualized the regulatory histone mark histone h3 lysine 27 acetylation (H3K27ac)27, 28, 29 in all available brain regions on the WashU Epigenome Browser (http://epigenomegateway.wustl.edu/).30 To determine if rs356182 is associated with SNCA levels in human brain, we used publicly available data from The Genotype‐Tissue Expression (GTEx) project,31 which has performed RNA sequencing on brain tissue from healthy donors, resulting in genotype and expression phenotype data for ~50–100 normal individuals in multiple different brain regions; information about subjects and RNA quality can be found on the GTEx website (www.gtexportal.org). We queried the association of rs356182 with SNCA levels in frontal cortex (n = 92), cerebellar hemisphere (n = 89), caudate (termed caudate basal ganglia in the GTEx dataset, n = 100), and substantia nigra (n = 56). Finally, to determine the LD structure between rs356182 and other PD‐associated variants, we analyzed genotype data from the 1000 Genomes Phase 1 CEU data set32 using Haploview.33
Results
Minor allele at rs356182, a SNP near SNCA, is associated with a tremor‐predominant phenotype
Ten SNPs that have been previously linked to PD risk were ascertained in 251 PD subjects in our initial single‐site cohort (UPenn Udall cohort, Tables S1, S2). The median age of subjects was 71 years, with a median PD duration of 7 years. 16% of these individuals showed a tremor‐predominant (TD) phenotype, and 68% showed a PIGD phenotype, with the remainder falling in between, an indeterminate phenotype.5
As shown in Table 2, this discovery screen yielded rs356182, near SNCA, as a candidate variant associated with TD/PIGD ratio (corrected P = 0.04 for the GG genotype at rs356182). We evaluated the influence of covariates in this model (Table 3), finding that adjustment for additional covariates minimally affected our results. In contrast, none of the 10 SNPs were significantly associated with motor severity (i.e., UPDRS‐III score).
Table 2.
Association of SNP genotypes with TD/PIGD ratio in UPenn discovery cohort
| PD risk SNP | Genotype | P‐value for association with TD/PIGD ratio |
|---|---|---|
| rs71628662 | A_G | 0.495 |
| rs17649553 | A_G | 0.503 |
| G_G | 0.608 | |
| rs34311866 | A_G | 0.444 |
| G_G | 0.516 | |
| rs12637471 | A_G | 0.767 |
| G_G | 0.540 | |
| rs1955337 | A_C | 0.284 |
| C_C | 0.238 | |
| rs6430538 | A_G | 0.066 |
| G_G | 0.482 | |
| rs11724635 | A_C | 0.111 |
| C_C | 0.192 | |
| rs823118 | A_G | 0.771 |
| G_G | 0.812 | |
| rs356182 | A_G | 0.594 |
| G_G | 0.004 a | |
| rs1077989 | A_C | 0.340 |
| C_C | 0.713 |
Linear regressions were used to evaluate associations between genotypes at 10 candidate SNPs and tremor‐predominant (TD) versus postural instability gait disorder (PIGD) phenotypes. Models were adjusted for age, sex, disease duration, and levodopa equivalent daily dose. P‐values are shown for each genotype in comparison with reference genotype. Only one comparison genotype is shown for rs71628662 as this represents the GBA locus.
indicates P‐value meets Bonferroni‐corrected P ‐value (0.005).
Table 3.
The minor allele at rs356182 near SNCA associates with tremor‐predominant phenotypes in discovery and replication cohorts
| Covariates | Discovery cohort | Replication cohort | ||||||
|---|---|---|---|---|---|---|---|---|
| Co‐dominant | A‐allele dominant | Co‐dominant | A‐allele dominant | |||||
| β | P‐value | β | P‐value | β | P‐value | β | P‐value | |
| Age + sex | 0.55 | 0.029 | 0.56 | 0.015 | 0.31 | 0.128 | 0.42 | 0.020 |
| Age + sex + disease duration | 0.65 | 0.008 | 0.62 | 0.006 | 0.32 | 0.107 | 0.42 | 0.018 |
| Age + sex + disease duration + LEDD | 0.70 | 0.004 | 0.65 | 0.003 | 0.31 | 0.125 | 0.41 | 0.021 |
| Age + sex + disease duration + LEDD + site | 0.31 | 0.125 | 0.42 | 0.018 | ||||
Linear regressions were used to evaluate associations between genotypes at rs356182 and tremor‐predominant (TD) versus postural instability gait disorder (PIGD) phenotypes. TD/PIGD ratios were predicted by genotype under codominant versus major allele (A)‐dominant genetic models, with covariates as indicated. In the codominant model, the coefficient represents the effect size for each additional G allele; in the A‐dominant model, the coefficient represents the effect size for the GG genotype. The discovery cohort consisted of 251 PD patients from the University of Pennsylvania, while the replication cohort consisted of 559 PD patients from multiple clinical sites.
While our initial screen did not assume a specific genetic model, visual inspection of the results suggested a major‐allele (A)‐dominant model for the association between motor phenotype and rs356182 genotype (Fig 1A). Applying a major‐allele‐dominant genetic model to our analysis did not affect the association between rs356182 genotype and motor subtype (corrected P = 0.03), with the minor allele (G) homozygotes demonstrating a more tremor‐predominant phenotype (Fig 1B). Log‐transformation of TD/PIGD ratios (Fig. S1), to account for skew in our distribution, similarly did not affect our results (corrected P = 0.02).
Figure 1.
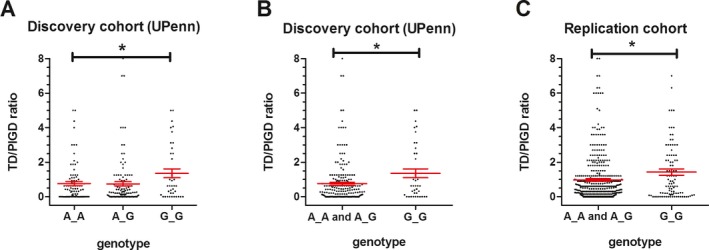
Genotype at rs356182 near SNCA associates with tremor‐predominant phenotype. (A, B) The tremor‐dominant (TD)/postural‐instability‐gait‐difficulty (PIGD) ratio is shown for PD patients with each genotype at rs356182 in the UPenn (discovery) cohort (n = 251) under categorical (Panel A) and major‐allele dominant (Panel B) models. UPDRS scores were used to calculate TD/PIGD ratios, with a cut‐off ratio of >1.5 previously proposed to define the TD motor subtype. (C) The TD/PIGD ratio by rs356182 genotype is shown for PD patients from the 3‐site replication cohort (n = 559) under a major‐allele dominant model. MDS‐UPDRS scores were used to calculate ratios, with a cut‐off ratio of >1.15 previously proposed to define the TD motor subtype. 5/559 (0.9%) of individuals were outliers with TD/PIGD ratios >8 and are not shown; their TD/PIGD ratio values are 13 (AA genotype), 12 (AA genotype), 10 (GG genotype), 9 (AA genotype), and 9 (AG genotype). For both panels, each dot represents one individual, and means +/− SEMs are indicated. * denotes corrected p < 0.05
Stratification by rs356182 genotype into minor allele (G) homozygotes versus other genotypes yielded significant differences in the proportion of patients with tremor‐predominant versus PIGD versus indeterminate motor phenotypes, as expected. In contrast, GG homozygotes did not differ from other rs356182 genotype carriers with respect to age, sex, disease duration, UPDRS‐III score, or LEDD (Table S1).
Replication of association between rs356182 genotype and motor phenotype in a multisite cohort
We next asked if the association between rs356182 genotype and motor phenotype observed in our discovery cohort would replicate in PD cohorts from multiple clinical sites. In the replication cohort, 559 subjects were recruited from three sites, and the demographics of this multi‐site replication cohort were similar to that of the discovery cohort (Table S1). Because only the MDS‐UPDRS motor subscale was uniformly evaluated in all three sites, tremor/PIGD ratios were calculated based only on the relevant items of this subscale (see Data S2.). The GG genotype at rs356182 again associated significantly with a more tremor‐predominant phenotype in the major‐allele‐dominant genetic model (P = 0.02, Fig. 1C). Further adjusting our linear regression model for site within the multi‐site replication cohort did not alter the results (P = 0.02, Table 3). Log transformation of TD/PIGD ratios in the replication cohort also did not affect our results (P = 0.004).
The minor allele at rs356182 is associated with slower motor progression
Clinically, the TD phenotype has been associated with slower rates of motor progression. Having defined a potential genetic determinant of this TD phenotype, we next asked if the minor allele at rs356182 is associated with slower PD motor symptom progression.
To answer this question, we used the 230 individuals in the UPenn discovery cohort who had undergone repeated UPDRS assessments for an average of 4 years (range: 1–7 years, Fig 2A). Genotypic frequencies at rs356182 did not differ in patients with longer versus shorter follow‐up times (Table S2). However, in a linear mixed‐effects model the GG genotype at rs356182 was associated with a slower rate of motor progression (P = 0.01; Fig. 2B). Specifically, the annual rate of increase in UPDRS‐III scores was approximately 1 point per year less in rs356182 GG genotype carriers than in individuals with AG or AA genotypes.
Figure 2.
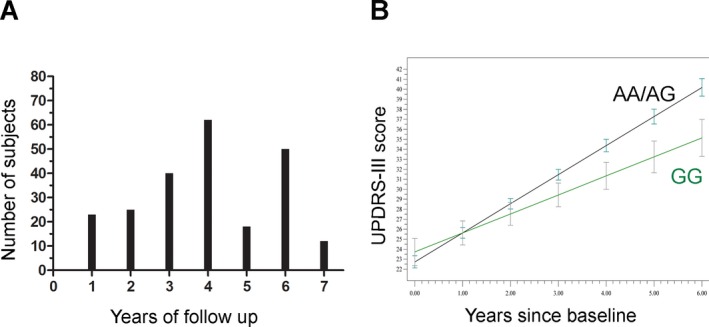
Genotype at rs356182 predicts rate of motor progression. (A) 230 PD patients were followed longitudinally with assessment of motor symptom scores with the UPDRS‐III. The number of patients with various lengths of follow‐up in our longitudinal cohort is shown. (B). Linear mixed‐effect models demonstrate that rs356182 genotype predicts rate of motor progression in these 230 patients. Means +/− SEMs are shown for a model adjusted for age, sex, baseline UPDRS‐III score, and LEDD. Note that individuals with the GG genotype at rs356182 have a 1‐point difference in annual rate of change in UPDRS‐III, so that by year 6, GG genotype carriers have an average UPDRS‐III score 6 points lower than carriers of other genotypes. LEDD, levodopa equivalent daily dose.
Given the observed association of rs356182 genotypes with rate of motor progression, we asked whether the G allele at this SNP was also associated with a longer overall disease duration. Using linear regression models adjusting for sex and age at disease onset, we found that rs356182 genotype correlated with total disease duration in an autopsy cohort of 85 PD patients carriers (P = 0.007, Table 4). Each additional G allele increased the disease duration by 1.9 years.
Table 4.
Overview of the discovery, replication, and autopsy cohorts
| UPenn discovery cohort Median (IQR) | Replication cohort Median (IQR) | Autopsy Cohort Median (IQR) | |
|---|---|---|---|
| Sample size | 251 | 559 | 85 |
|
Sex Male/Female (%) |
70/30 | 70/30 | 78/22 |
| Age (years) | 71 (64–76) | 67 (62–74) | 80 (77–83) |
| Disease duration (years) | 7 (4–11) | 8 (4.5–12) | 12 (9–17) |
| UPDRS‐III total | 22 (14.5–31) | 26 (18–36) | |
| LEDD (mg/day) | 698.5 (400–1000) | 700.8 (400–1210) | |
| Motor subtype (%) | |||
| TD | 16.3 | 28.4 | |
| Indeterminate | 15.5 | 2.7 | |
| PIGD | 68.1 | 68.9 | |
Medians +/− interquartile range (IQR) are shown. UPDRS‐III, motor section of the Unified Parkinson's Disease Rating Scale; LEDD, Levodopa equivalent daily dose. For details on individual items included in motor subtype classification, see Methods and Data S1. 82/85 patients in the autopsy cohort overlapped with the UPenn discovery cohort; 3/85 individuals in the autopsy cohort had insufficient clinical data to determine motor phenotype. For the autopsy cohort, age and disease duration are shown at time of death. For the discovery and replication cohorts, all demographic information shown is from the baseline visit.
The minor allele at rs356182 is associated with lower levels of SNCA expression in the brain
Having established an association between rs356182 and motor phenotype as well as motor progression in PD, we asked whether the genotype at rs356182 is associated with levels of SNCA expression in the brain. The rs356182 polymorphism lies 19 kb to the 3′ end of the SNCA gene and is located in a region displaying the histone mark H3K27ac in brain (Fig. 3A). As H3K27ac has been reported to reliably mark active gene regulatory regions,29, 34 these data suggest that rs356182 genotype might affect SNCA regulation. We therefore hypothesized that the effects of rs356182 genotype on motor phenotype and rate of PD motor progression may reflect genotype‐based differences in levels of SNCA expression in the brain. To investigate this, we analyzed recently released GTEx Consortium data.
Figure 3.
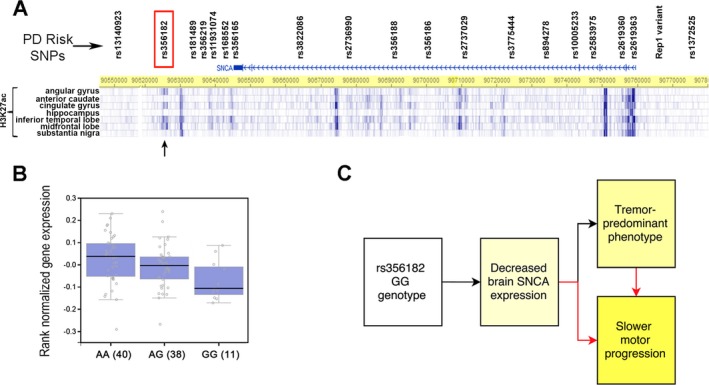
Genotype at rs356182 associates with SNCA expression in the brain and defines a clinically important endophenotype in PD. (A) The genomic positions of rs356182 and other PD‐associated variants in or near the SNCA gene are indicated. The regulatory histone mark H3K27ac is shown in various brain regions. The rs356182 polymorphism lies 19 kb to the 3′ end of the SNCA gene and is located in a region of H3K27ac (arrow), indicative of active regulatory potential in brain. (B) Boxplot showing the association between rs356182 genotype and SNCA expression in the cerebellum in 89 healthy postmortem samples (P = 0.005) from the GTEx Consortium, with GG genotype carriers showing the lowest levels of expression. Medians and interquartile ranges are indicated. (C) Our results are compatible with a model in which rs356182 genotypes influence expression levels of SNCA, with the GG genotype associating with decreased expression. Decreased expression of SNCA then results in both a tremor‐predominant phenotype and slower motor progression, with the slower motor progression reflecting either decreased brain SNCA expression or the more benign motor phenotype.
In postmortem samples from 89 normal individuals from the GTex dataset, the rs356182 genotype was associated with SNCA expression levels in the cerebellum (P = 0.005; Fig. 3B), with GG genotype carriers showing the lowest levels of expression. In contrast, SNCA expression levels did not differ by rs356182 genotype in other brain regions, including the substantia nigra, caudate and frontal cortex.
Discussion
In this study, we evaluated 10 candidate SNPs previously associated with global PD risk for effects on PD motor endophenotype. In a 251‐patient, single‐site discovery cohort, as well as a 559‐patient, multi‐site replication cohort, the GG genotype at rs356182, near the SNCA gene, correlated cross‐sectionally with a more tremor‐predominant phenotype. Moreover, in longitudinal analyses spanning up to 7 years in 230 PD patients, rs356182 genotype predicted the rate of motor progression, with GG genotype carriers exhibiting a slower rate of change in UPDRS‐III scores. Finally, brain expression quantitative trait locus (eQTL) data from 89 healthy individuals demonstrated a relationship between rs356182 genotype and expression levels of SNCA in the cerebellum, with the GG genotype associated with lower levels of expression. Taken together, our data provide molecular support for an important endophenotype in PD that correlates with brain SNCA expression, clinical features, and long‐term disease progression (Fig. 3C).
We found that the rs356182 GG genotype associates with a more tremor‐predominant phenotype in both our discovery and replication cohorts, an association that persists despite these two cohorts capturing motor phenomenology using slightly different scales (UPDRS and the updated MDS‐UPDRS, respectively). In addition, the rs356182 GG genotype associates with a slower rate of motor progression. Previously, the rs356182 genotype was linked to PD risk by genome‐wide association.25 Common variants at the SNCA locus (Fig. 3A) have been reported to associate with age at PD onset. These previously reported variants include the Rep1 microsatellite variant near the SNCA promoter,35 the rs356165 SNP in the SNCA 3′UTR,36 and the rs356219 SNP 3′ to the SNCA gene.37 All three of these common variants demonstrate strong LD with rs356182 (r 2 > 0.7), and, in the case of each of these variants, the genotypes associated with increased risk for PD were also associated with earlier age at onset. We note, however, that “translation” of these genotypic influences into more clinically meaningful terms – influences on motor phenomenology and rate of progression – has been more difficult. For example, association of genotypes with clinically recognizable motor subtypes has rarely been explored, although some examples do exist.6 Moreover, previous reports of associations between common variants at SNCA and rate of progression have yielded mixed results: while the 263 bp Rep1 variant has been reported in one cohort of 232 PD patients to be associated with faster rate of motor progression,38 this result failed to replicate in another cohort of 1098 PD patients, in which the most common 263 bp Rep1 genotype (261/263) associated with slower motor progression.39 We note here that, to the extent that rs356182 is linked to Rep1 (r 2 > 0.7), our results are more in agreement with the findings of the second, larger study.
Our analysis differs in several respects from prior work in this area. First, rather than dichotomizing outcomes into whether or not a given individual has passed a specific threshold of motor impairment, as the two prior analyses of motor progression have done, we used linear mixed‐effects models to estimate the rate of change in UPDRS‐III score for different genotypes. Such an approach, applied to our longitudinal cohort, maximizes statistical power and explicitly models individual change across time, which is arguably a more meaningful and sensitive analysis of disease progression than time to specific outcome. We moreover note that the 1‐point difference in annual rate of motor symptom progression found between carriers of different rs356182 genotypes is not only statistically significant, but also large in effect size, since all‐comer UPDRS‐III progression rates have been previously reported at ~1.5 points per year.13, 40 Indeed, in <5 years, the “average” PD patient carrying the GG genotype at rs356182 would be expected to notice a clinically meaningful difference from carriers of other genotypes at this locus, based on estimates of a 4.5 point change on the UPDRS‐III constituting a moderate clinically important difference.41
Second, we chose to investigate the rs356182 SNP rather than the more complex Rep1 variant. This decision stemmed scientifically from the finding across multiple GWAS that 3′ SNCA variants associate more robustly with risk for PD,25, 37, 42, 43 that they may be at least partially independent of 5′ or promoter variants,44 and that 3′ and 5′ variants may differ in their association with cognitive aspects of PD.45 This decision also reflected real‐world, practical considerations. Specifically, as a biallelic SNP, rs356182 has exactly three genotypes – GG, AG, and AA – and genetic effects can be modeled simply as major‐allele (A)‐dominant, codominant, or minor‐allele (G)‐dominant. In contrast, Rep1 is a polymorphic microsatellite repeat, with variable numbers of repeats present on each allele, generating multiple potential genotypes, which may not fall into clear models of dominance. While it is likely that dominant/recessive models are overly simplistic, and that multiple loci may interact in complicated ways to influence phenotypes, we suggest that the overall goals and uses of a marker be kept in mind. If the goal is to provide a molecular definition of a clinically useful endophenotype, for use in, for example, clinical trial stratification, the practical advantage in defining that endophenotype by a relatively straightforward SNP marker is substantial. We note here that our study neither establishes, nor argues for, causality for the rs356182 SNP itself. Rather, this SNP may serve as a useful marker of a linked genetic regulatory mechanism that underlies the association with PD risk, motor endophenotype, and differential rates of progression.
As evidence links increased expression of SNCA to the pathogenesis of PD,38, 46, 47 we also investigated the relationship of rs356182 genotype to brain SNCA expression. Prior studies have investigated associations of common variants near SNCA with SNCA expression. However, these studies have either (1) investigated eQTL effects in tissues outside of the brain,44, 48 or (2) investigated brain eQTL effects in a fairly limited number of samples.48, 49 Here, we took advantage of the newly released GTEx Consortium data,50 in which 100+ individuals have been genome‐wide genotyped and expression phenotyped across myriad tissues, to arrive at a more definitive answer. Interestingly, in an earlier brain eQTL study of 10 cerebellar samples and 22 substantia nigra samples, the C allele at rs356219 correlated with lower cerebellar SNCA transcript levels, but not with SNCA expression levels in the substantia nigra.48 As the C‐allele at rs356219 is well‐proxied by the G‐allele at rs356182 (R 2 = 0.74, 1000 Genomes),32 our current results are concordant in both direction of effect and brain region in which the eQTL effect is seen. Further investigation of the region‐specific eQTL effect observed, especially as it involves a brain region not thought to be preferentially affected by Lewy body pathology, and was not found in the substantia nigra, frontal cortex, or caudate, would be a valuable addition to the data presented here.
The most unexpected aspect of our findings concerns the direction of association between rs356182 genotype and downstream molecular and clinical phenotypes. Specifically, the minor allele, G, and in particular the GG genotype, at rs356182 is linked in our study with decreased SNCA brain expression, a more tremor‐predominant phenotype, a slower rate of motor progression, and possibly, although this observation is based on a smaller number of individuals, a longer total disease duration (Fig. 3C). Thus, the direction of association across these various phenotypes follows expectations based on our current understanding of the (positive) effects of decreased SNCA expression, and the (favorable) outcomes associated with a more tremor‐predominant phenotype. However, the G allele at rs356182 – linked to “better” phenotypes within PD – is the allele reported to associate with increased risk for developing PD.25
We considered three potential explanations for this finding. First, our current endophenotype findings could be incorrect. However, this is unlikely, given the cohort numbers (n = 89 for brain eQTL analysis, n > 800 for TD/PIGD analysis, n > 200 for motor progression analysis), the strength of each of these associations, and the fact that our analyses largely involved nonoverlapping cohorts. Moreover, other groups have reported seemingly paradoxical directions of association as well. As mentioned previously, the allele associated with increased risk for PD at rs356219 has been reported to correlate with decreased SNCA brain expression,48 and the allele associated with increased risk for PD at Rep1 has been reported to correlate with a slower rate of motor progression in a study of >1000 PD patients.39
A second possibility is that the association between the G allele at rs356182 and increased risk for PD could be incorrect. On the one hand, statistical considerations (large numbers in the meta‐analyzed GWAS, strength of associations reported) argue against this possibility. The fact that the G allele at rs356182 associates with a more tremor‐predominant phenotype, however, raises the possibility that ascertainment bias could affect the reported association between rs356182 genotype and PD risk; individuals with the highly recognizable resting tremor of PD might be more likely to be diagnosed with PD. In this respect, we note that in the only GWAS performed to date on neuropathologically confirmed PD, association between the SNCA locus and PD was absent; the closest SNP (rs13140923) showing any degree of association with PD in this analysis of 484 neuropathologically confirmed cases and 1145 controls was >75kB away from the SNCA gene, demonstrating an R2 of only 0.017 with rs356182.51
A third possibility is that rs356182 genotypes do not exert the same effect on risk for developing PD as they do on risk for more versus less severe PD. This third possibility may at first seem paradoxical; however, the longstanding clinical observation in PD that patients with tremor‐dominant PD have both an earlier age at onset,52 and a more benign disease course,13 is conceptually similar.
The 10 SNPs screened in this study are not the only genetic variants associated with PD risk: one meta‐analysis reported 28 independent risk loci,25 and the 10 SNPs examined here represent the most statistically significant associations by P‐value of these 28 reported loci. We focused on the top 10 SNPs based on statistical power considerations in our discovery cohort. To guard against the possibility of spurious correlation with motor phenotype, we replicated our analysis in an additional cohort. One limitation of our study, however, is that additional PD genetic risk loci not examined here may also influence progression or motor phenomenology; studies in larger patient cohorts looking specifically at these variants would be helpful. Additionally, replication of our current longitudinal finding in additional studies would be a valuable addition to this work; we note in this regard that, based on the results captured in Figure 2, a follow‐up period of ~4 years may be needed to confirm or reject separation of trajectories by rs356182 genotype. Finally, as mentioned previously, the eQTL effect relating rs356182 genotype to SNCA brain expression has only been observed in cerebellum, and only been examined in normal brain tissue. As a consequence, the question of whether there may be PD‐specific eQTL effects, including eQTL effects in additional brain regions, remains to be answered.
In summary, using a screen of 10 SNPs already associated with PD risk, we found an association between rs356182 genotype and multiple different, but related, phenotypes, ranging in scale from molecular effects (brain SNCA expression) to clinical outcomes (motor progression over time), with an intermediate phenotype of clinical symptomatology (TD/PIGD). These genotype‐endophenotype relationships were, moreover, investigated in large and, where possible, nonoverlapping cohorts, decreasing the likelihood that our observations were due to chance. We thus define a specific and clinically important PD endophenotype. We further suggest that such molecularly defined endophenotyping in PD may be of significant benefit for both clinical trial design and tailoring of clinical care as we enter the era of precision medicine.
Author Contributions
CAC performed study design, data analysis, manuscript drafting, and editing. NJ and MDG performed data analysis, manuscript drafting, and editing. DW performed database design and manuscript editing. SXX contributed to statistical consulting and graphical design. YB did data formatting and design; AJE, JQ, KLE, TM, JT, and CPZ involved in database design, data collection, and manuscript editing. VMVD performed database design, genetics analysis, and manuscript editing; ASCP contributed to study design, database design, data analysis, manuscript drafting and editing.
Conflicts of Interest
None.
Supporting information
Data S1. Supplementary listing of exact items included in calculations of tremor‐predominant and postural‐instability‐gait‐disorder ratio.
Data S2. Supplementry methods. UPDRS items used to calculate the TD/PIGD ratio in the UPenn discovery cohort.
Data S3. R‐scripts (statistical analyses).
Figure S1. Genotype at rs356182 near SNCA associates with tremor‐predominant phenotype.
Table S1. Clinical and demographic characteristics of discovery cohort stratified by rs356182 genotype.
Table S2. Genotypes for longitudinal subgroup do not differ between individuals with longer versus shorter follow‐up times.
Acknowledgments
We thank Xiaoyan Han for her help with the statistical programming for Figure 2B. This work was supported by grants from the NIH (U01 NS082134, P50 NS062684, and P50 NS053488, and AG10124). Dr. Chen‐Plotkin also receives research support from the Burroughs Wellcome Fund, the Pechenik Montague Award Fund, and the Benaroya Fund.
References
- 1. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vu TC, Nutt JG, Holford NHG. Progression of motor and nonmotor features of Parkinson's disease and their response to treatment: progression of Parkinson's disease. Br J Clin Pharmacol 2012;74:267–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu P, Feng T, Wang Y, et al. Clinical heterogeneity in patients with early‐stage Parkinson's disease: a cluster analysis. J Zhejiang Univ Sci B 2011;12:694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fereshtehnejad S‐M, Romenets SR, Anang JBM, et al. New clinical subtypes of parkinson disease and their longitudinal progression: a prospective cohort comparison with other phenotypes. JAMA Neurol 2015;72:863. [DOI] [PubMed] [Google Scholar]
- 5. Jankovic J, McDermott M, Carter J, et al. Variable expression of Parkinson's disease A base‐line analysis of the DAT ATOP cohort. Neurology 1990;40:1529–1529. [DOI] [PubMed] [Google Scholar]
- 6. Factor SA, Steenland NK, Higgins DS, et al. Postural instability/gait disturbance in Parkinson's disease has distinct subtypes: an exploratory analysis. J Neurol Neurosurg Psychiatry 2011;82:564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma L‐Y, Chan P, Gu Z‐Q, et al. Heterogeneity among patients with Parkinson's disease: cluster analysis and genetic association. J Neurol Sci 2015;351(1–2):41–45. [DOI] [PubMed] [Google Scholar]
- 8. Flensborg Damholdt M, Shevlin M, Borghammer P, et al. Clinical heterogeneity in Parkinson's disease revisited: a latent profile analysis. Acta Neurol Scand 2012;125:311–318. [DOI] [PubMed] [Google Scholar]
- 9. van Rooden SM, Colas F, Martínez‐Martín P, et al. Clinical subtypes of Parkinson's disease. Mov Disord 2011;26:51–58. [DOI] [PubMed] [Google Scholar]
- 10. Selikhova M, Williams DR, Kempster PA, et al. A clinico‐pathological study of subtypes in Parkinson's disease. Brain J Neurol 2009;132(Pt 11):2947–2957. [DOI] [PubMed] [Google Scholar]
- 11. Fahn S, Elton R; Members of the UPDRS Development Committee . 1987. Unified Parkinson's Disease Rating Scale In: S Fahn, C Marsden, D Caine, M Goldstein, ed. Recent Developments in Parkinson's Disease. Florham Park, NJ, Macmillan Health Care Information. [Google Scholar]
- 12. Alves G, Larsen JP, Emre M, et al. Changes in motor subtype and risk for incident dementia in Parkinson's disease. Mov Disord 2006;21:1123–1130. [DOI] [PubMed] [Google Scholar]
- 13. Jankovic J, Kapadia AS. Functional decline in Parkinson disease. Arch Neurol 2001;58:1611–1615. [DOI] [PubMed] [Google Scholar]
- 14. Verbaan D, Marinus J, Visser M, et al. Cognitive impairment in Parkinson's disease. J Neurol Neurosurg Psychiatry 2007;78:1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burn DJ. Motor subtype and cognitive decline in Parkinson's disease, Parkinson's disease with dementia, and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2006;77:585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldman JG, Weis H, Stebbins G, et al. Clinical differences among mild cognitive impairment subtypes in Parkinson's disease. Mov Disord 2012;27:1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu Y, Guo X‐Y, Wei Q‐Q, et al. Non‐motor symptoms and quality of life in tremor dominant vs postural instability gait disorder Parkinson's disease patients. Acta Neurol Scand 2015;5:330–337. [DOI] [PubMed] [Google Scholar]
- 18. Burn DJ, Landau S, Hindle JV, et al. Parkinson's disease motor subtypes and mood. Mov Disord 2012;27:379–386. [DOI] [PubMed] [Google Scholar]
- 19. Wylie SA, van den Wildenberg W, Ridderinkhof KR, et al. Differential susceptibility to motor impulsivity among functional subtypes of Parkinson's disease. J Neurol Neurosurg Psychiatry 2012;83:1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lo RY, Tanner CM, Albers KB, et al. Clinical features in early Parkinson disease and survival. Arch Neurol 2009;66:1353–1358. [DOI] [PubMed] [Google Scholar]
- 21. Auyeung M, Tsoi TH, Mok V, et al. Ten year survival and outcomes in a prospective cohort of new onset Chinese Parkinson's disease patients. J Neurol Neurosurg Psychiatry 2012;83:607–611. [DOI] [PubMed] [Google Scholar]
- 22. Goetz CG, Tilley BC, Shaftman SR, et al. Movement disorder society‐sponsored revision of the unified parkinson's disease rating scale (MDS‐UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23:2129–2170. [DOI] [PubMed] [Google Scholar]
- 23. Stebbins GT, Goetz CG, Burn DJ, et al. How to identify tremor dominant and postural instability/gait difficulty groups with the movement disorder society unified Parkinson's disease rating scale: Comparison with the unified Parkinson's disease rating scale: PIGD and The MDS‐UPDRS. Mov Disord 2013;28:668–670. [DOI] [PubMed] [Google Scholar]
- 24. Nalls MA, Bras J, Hernandez DG, et al. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging 2015;36:1605.e7–1605.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nalls MA, Pankratz N, Lill CM, et al. Large‐scale meta‐analysis of genome‐wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 2014;46:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laird NM, Ware JH. Random‐effects models for longitudinal data. Biometrics 1982;38:963–974. [PubMed] [Google Scholar]
- 27. Barski A, Cuddapah S, Cui K, et al. High‐resolution profiling of histone methylations in the human genome. Cell 2007;129:823–837. [DOI] [PubMed] [Google Scholar]
- 28. Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007;39:311–318. [DOI] [PubMed] [Google Scholar]
- 29. Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 2010;107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou X, Wang T. Using the Wash U Epigenome Browser to examine genome‐wide sequencing data. Curr. Protoc. Bioinforma. Ed. Board Andreas Baxevanis Al 2012;Chapter 10:Unit10.10. [DOI] [PMC free article] [PubMed]
- 31. GTEx Consortium , Ardlie KG, Deluca DS, Segrè AV, et al. Human genomics. The Genotype‐Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Genomes Project Consortium ; Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265. [DOI] [PubMed] [Google Scholar]
- 34. Rada‐Iglesias A, Bajpai R, Swigut T, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011;470:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hadjigeorgiou GM, Xiromerisiou G, Gourbali V, et al. Association of alpha‐synuclein Rep1 polymorphism and Parkinson's disease: influence of Rep1 on age at onset. Mov Disord 2006;21:534–539. [DOI] [PubMed] [Google Scholar]
- 36. Cardo LF, Coto E, de Mena L, et al. A search for SNCA 3′ UTR variants identified SNP rs356165 as a determinant of disease risk and onset age in Parkinson's disease. J Mol Neurosci MN 2012;47:425–430. [DOI] [PubMed] [Google Scholar]
- 37. Brockmann K, Schulte C, Hauser A‐K, et al. SNCA: major genetic modifier of age at onset of Parkinson's disease. Mov Disord 2013;28:1217–1221. [DOI] [PubMed] [Google Scholar]
- 38. Ritz B, Rhodes SL, Bordelon Y, Bronstein J. α‐Synuclein genetic variants predict faster motor symptom progression in idiopathic parkinson disease. PLoS ONE 2012;7:e36199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Markopoulou K, Biernacka JM, Armasu SM, et al. Does α ‐synuclein have a dual and opposing effect in preclinical vs. clinical Parkinson's disease? Parkinsonism Relat Disord 2014;20:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Louis ED, Tang MX, Cote L, et al. Progression of parkinsonian signs in Parkinson disease. Arch Neurol 1999;56:334–337. [DOI] [PubMed] [Google Scholar]
- 41. Shulman LM, Gruber‐Baldini AL, Anderson KE, et al. The clinically important difference on the Unified Parkinson's Disease Rating Scale. Arch Neurol 2010;67:64–70. [DOI] [PubMed] [Google Scholar]
- 42. Hill‐Burns EM, Wissemann WT, Hamza TH, et al. Identification of a novel Parkinson's disease locus via stratified genome‐wide association study. BMC Genom 2014;15:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCarthy JJ, Linnertz C, Saucier L, et al. The effect of SNCA 3′ region on the levels of SNCA‐112 splicing variant. Neurogenetics 2011;12:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mata IF, Shi M, Agarwal P, et al. SNCA variant associated with Parkinson disease and plasma alpha‐synuclein level. Arch Neurol 2010;67:1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guella I, Evans DM, Szu‐Tu C, et al. α ‐synuclein genetic variability: A biomarker for dementia in Parkinson disease. Ann Neurol 2016;79:991–999. [DOI] [PubMed] [Google Scholar]
- 46. Ross OA, Braithwaite AT, Skipper LM, et al. Genomic investigation of alpha‐synuclein multiplication and parkinsonism. Ann Neurol 2008;63:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singleton AB, Farrer M, Johnson J, et al. α ‐Synuclein locus triplication causes Parkinson's Disease. Science 2003;302:841. [DOI] [PubMed] [Google Scholar]
- 48. Fuchs J, Tichopad A, Golub Y, et al. Genetic variability in the SNCA gene influences alpha‐synuclein levels in the blood and brain. FASEB J 2008;22:1327–1334. [DOI] [PubMed] [Google Scholar]
- 49. Cardo LF, Coto E, de Mena L, et al. Alpha‐synuclein transcript isoforms in three different brain regions from Parkinson's disease and healthy subjects in relation to the SNCA rs356165/rs11931074 polymorphisms. Neurosci Lett 2014;562:45–49. [DOI] [PubMed] [Google Scholar]
- 50. GTEx Consortium , Lonsdale J, Thomas J, Salvatore M, et al. The Genotype‐Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Beecham GW, Dickson DW, Scott WK, et al. PARK10 is a major locus for sporadic neuropathologically confirmed Parkinson disease. Neurology 2015;84:972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Josephs KA, Matsumoto JY, Ahlskog JE. Benign tremulous parkinsonism. Arch Neurol 2006;63:354–357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary listing of exact items included in calculations of tremor‐predominant and postural‐instability‐gait‐disorder ratio.
Data S2. Supplementry methods. UPDRS items used to calculate the TD/PIGD ratio in the UPenn discovery cohort.
Data S3. R‐scripts (statistical analyses).
Figure S1. Genotype at rs356182 near SNCA associates with tremor‐predominant phenotype.
Table S1. Clinical and demographic characteristics of discovery cohort stratified by rs356182 genotype.
Table S2. Genotypes for longitudinal subgroup do not differ between individuals with longer versus shorter follow‐up times.