

Abstract
Objective
Mitochondrial dysfunction plays a key role in the pathophysiology of neurodegenerative disorders such as ataxia and Parkinson's disease. We describe an extended Belgian pedigree where seven individuals presented with adult‐onset cerebellar ataxia, axonal peripheral ataxic neuropathy, and tremor, in variable combination with parkinsonism, seizures, cognitive decline, and ophthalmoplegia. We sought to identify the underlying molecular etiology and characterize the mitochondrial pathophysiology of this neurological syndrome.
Methods
Clinical, neurophysiological, and neuroradiological evaluations were conducted. Patient muscle and cultured fibroblasts underwent extensive analyses to assess mitochondrial function. Genetic studies including genome‐wide sequencing were conducted.
Results
Hallmarks of mitochondrial dysfunction were present in patients’ tissues including ultrastructural anomalies of mitochondria, mosaic cytochrome c oxidase deficiency, and multiple mtDNA deletions. We identified a splice acceptor variant in POLG2, c.970‐1G>C, segregating with disease in this family and associated with a concomitant decrease in levels of POLG2 protein in patient cells.
Interpretation
This work extends the clinical spectrum of POLG2 deficiency to include an overwhelming, adult‐onset neurological syndrome that includes cerebellar syndrome, peripheral neuropathy, tremor, and parkinsonism. We therefore suggest to include POLG2 sequencing in the evaluation of ataxia and sensory neuropathy in adults, especially when it is accompanied by tremor or parkinsonism with white matter disease. The demonstration that deletions of mtDNA resulting from autosomal‐dominant POLG2 variant lead to a monogenic neurodegenerative multicomponent syndrome provides further evidence for a major role of mitochondrial dysfunction in the pathomechanism of nonsyndromic forms of the component neurodegenerative disorders.
Introduction
Mitochondrial dysfunction caused by disturbances of mtDNA integrity gives rise to a diverse array of neurological, neuromuscular, and multisystem disorders. The clonal expansion of multiple mtDNA deletions is typically associated with late‐onset primary mitochondrial disease characterized by autosomal‐dominant progressive external ophthalmoplegia (adPEO) as the predominant clinical feature.1 MtDNA is replicated by the DNA polymerase gamma complex, which is composed of a monomeric catalytic subunit encoded by POLG and a dimeric accessory subunit encoded by POLG2. Pathogenic POLG variants are a common cause of primary mitochondrial disorders typically resulting in adPEO, but can also lead to a phenotypic spectrum manifesting variably across brain, muscle, and/or liver (OMIM 157640, 258450, 203700, 613662, and 607459).
Few families have been reported to have disease due to variants in POLG2. The first reported case of POLG2 disease was documented in a patient with adult‐onset autosomal dominant PEO in whom multiple mtDNA deletions were detected.2 The second identified case of POLG2 disease was a patient who developed ptosis at age 30, but not ophthalmoplegia, and proximal and distal myopathy in her late forties. Muscle biopsy showed clonally expanded multiple mtDNA deletions.3 This family included a brother who possessed the same POLG2 variant and showed no signs of disease at age 57, indicating possible incomplete penetrance. Subsequently, three cases with pediatric‐onset autosomal dominant disease with varying phenotypes have been reported: a young adult with disease onset of adPEO in late teens and two unrelated infants presenting with hypotonia, seizures, and liver disease.4 With only a small number of cases reported, it is clearly evident that POLG2‐related disorders display a spectrum of clinical phenotypes with a wide range of age of onset.
We describe a severe neurological syndrome in a large family with cerebellar ataxia, axonal sensory ataxic neuropathy, and tremor. Some subjects also presented with parkinsonism, seizures, cognitive decline, and ophthalmoplegia. Most died within 15 years of onset of clinical symptoms of disease. Genetic studies revealed a POLG2 splice acceptor variant segregating with disease, leading to loss of POLG2 protein in patient cells. This work extends the clinical spectrum observed with POLG2 disease and lends further support for the role of the accumulation of mtDNA deletions in neurodegeneration.
Materials and Methods
Muscle histopathology and mtDNA analyses
Cryostat sections (10 μm) were cut from transversely orientated muscle blocks and subjected to standard histological and histochemical analysis including COX, succinate dehydrogenase (SDH), and sequential COX‐SDH oxidative enzyme reactions. Large‐scale mtDNA rearrangements within the major arc were screened by long‐range PCR to amplify a ~13.0 kb product in wild‐type mtDNA (primers: NC_012920.1 chrM:129‐110, chrM:3965‐3984). The level of deleted mtDNA in individual muscle fibers isolated by laser microcapture was determined by quantitative real‐time PCR using the ABI PRISM Step One real‐time PCR System (Life Technologies, Paisley, UK) to confirm the presence of clonally expanded mtDNA deletions.5
Whole exome sequencing and bioinformatic analyses
Whole exome sequencing was performed using Illumina paired‐end precapture libraries constructed according to manufacturer's protocol. Sequence data were aligned, variants were called by GATK, and custom Perl scripts were used to annotate variants as previously described.6 Multiple metrics for prediction of potential functional consequences of variants were applied: CADD,7 Genomic Evolutionary Rate Profiling (GERP),8 and PhyloP. The Exome Aggregation Consortium (ExAC) Cohort was used as reference population data (http://exac.broadinstitute.org, accessed January, 2016). All genetic alleles studied were annotated in reference to POLG2 NM_007215.
Western blotting
Fibroblasts were grown in DMEM High Glucose with L‐Glutamine and Sodium pyruvate (Hyclone, ThermoFisherScientific) with 15% FBS. Crude mitochondrial fraction was prepared from 1 × 107 cells following Graham & Rickwood method.9 Mitochondrial fractions were lysed in RIPA buffer containing 50 mm Tris, pH 8.0, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Triton X‐100, 0.1% SDS, 5 mg/mL leupeptin, 2 mg/mL aprotinin, and 0.5 mmol/L PMSF. Thirty milligram of mitochondrial lysates were resolved on SDS‐polyacrylamide gel electrophoresis and transferred to a 0.45‐mm PVDF membrane (Millipore). Western blotting was performed as previously described.4
Reverse Transcription Quantitative PCR
RNA extraction was performed using TRIzol and purified using PureLink RNA minikit (Life Technologies). First‐strand cDNA synthesis was done using SuperScript III First‐Strand Synthesis System for RT‐PCR (Life Technologies) and oligodT. Quantitative RT‐PCR experiments were performed using StepOne Plus RT‐PCR system (Applied Biosystems). POLG2 oligonucleotides: forward 5′AAACCCTGTGGAACCTAGGAG3′ and reverse 5′AGCATGCCTCGGTCTAGGTC3′. Each sample was run in triplicate and was normalized to GAPDH.
Membrane potential
Fibroblasts were grown on glass chamber slides and treated with 30 ng/mL rotenone for 4 h at 37°C, or the same volume of solvent. 5,5′,6,6′‐tetraethylbenzimidazolyl‐carbocyanine iodide (JC‐1, Life Sciences) of 5 μg/mL was added, and the cells were incubated for 30 min at 37°C. Membrane potential was determined by the ratio of red/green fluorescence in eight random 400× microscopic fields and expressed as mean ± standard deviation (SD).
Consent for the study was obtained from all subjects and a local ethics review board approved the study.
Results
Clinical presentation
Seven native Belgian individuals from two sibships belonging to the same extended family presented with a neurological disorder involving cerebellar and sensory ataxia with tremor (Fig. 1). Initial signs of disease rose to clinical attention between 50 and 77 years of age and in most cases resulted in loss of life in less than 15 years from onset (Table 1). Peripheral neuropathy developed in all individuals either concomitant with the cerebellar syndrome or manifesting up to 6 years postonset of disease. Additional features of disease in some individuals included epilepsy, cognitive decline, ophthalmoplegia, hearing loss, and parkinsonism. An overview of clinical and paraclinical signs is presented in Table 1 and is summarized as follows.
Figure 1.
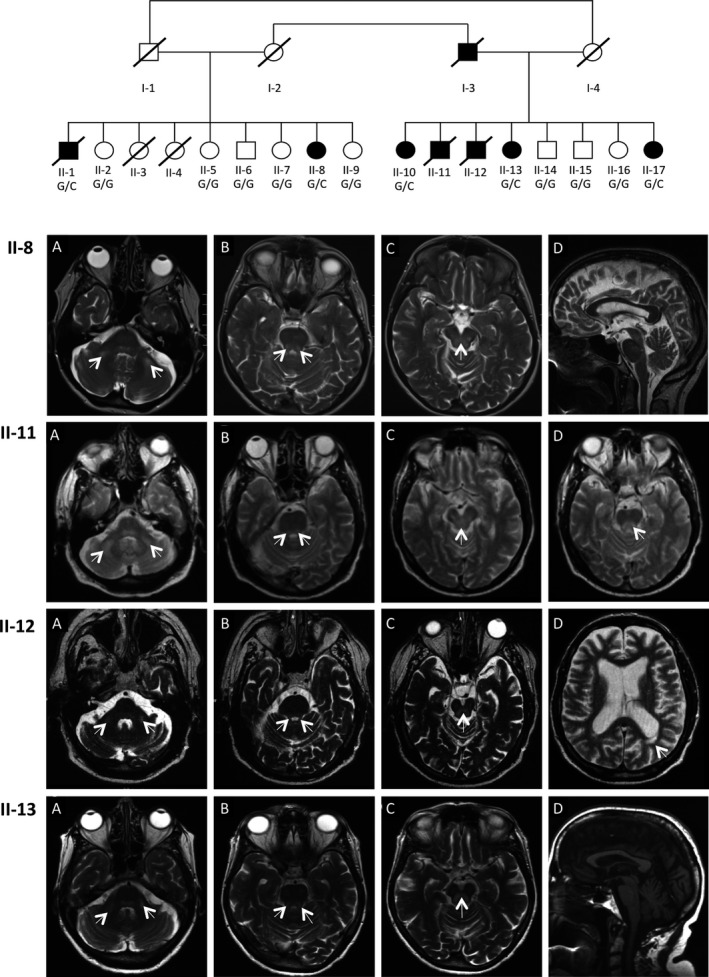
Pedigree and MRIs for family segregating POLG2 c.970‐1G>C variant. Affected individuals are represented as black symbols, whereas unaffected are open symbols. The genotype for the POLG2 c.970‐1G>C variant is indicated for individuals who were genotyped. Brain MRIs reveal abnormalities of the cerebellum and brain stem and white matter lesions. Axial T2‐weighted images revealed bilateral hyperintense lesions of (A) middle cerebellar peduncles, (B) upper cerebellar peduncles, and (C) periaqueductal grey matter in patients II‐8, II‐11, II‐12, and II‐13. Also shown are the following: II‐8 (D) Sagittal T2 image demonstrating mild supra and infratentorial atrophylenticular nuclei; II‐11 (D) Axial T2 image showing hypersignal of medial longitudinal fasciculus; II‐12 (D) Axial T2 image showing important hemispheric atrophy and periventricular white matter lesion; and II‐13 (D) Sagittal T1 image demonstrating moderate pontocerebellar and supratentorial atrophy.
Table 1.
An overview of clinical characteristics
| Subject ID | |||||||
|---|---|---|---|---|---|---|---|
| (II‐1) | (II‐8) | (II‐10) | (II‐11) | (II‐12) | (II‐13) | (II‐17) | |
| POLG2 c.970‐1G>C | + | + | + | NA | NA | + | + |
| Gender | M | F | F | M | M | F | F |
| Age at onset | 53 | 51 | 77 | 61 | 60 | 61 | 56 |
| Age at death | 68 | Alive 69 | Alive 83 | 69 | 68 | 77 | Alive68 |
| Cerebellar syndrome, age at onset | 50 | 52 | 77 | 61 | 60 | 61 | 56 |
| Postural and action tremor | + | + | 77 | 61 | 63 | 63 | 56 |
| Head tremor | 60 | 55 | 77 | − | >66 | 65 | 58 |
| Cerebellar ataxia | + | + | 77 | 61 | 60 | 61 | 58 |
| Dysmetria | − | 67 | 63 | 71 | 58 | ||
| Peripheral nervous system, age at onset | 50 | 52 | 80 | 67 | 60 | 61 | 57 |
| Sensory loss | + | + | 80 | 67 | 63 | 67 | 57 |
| Areflexia, deep tendon | + | + | − | 67 | 63 | 67 | 57 |
| Sensory ataxia | + | + | − | 67 | 63 | 61 | 57 |
| Neuropathic pain | − | − | − | − | 60 | − | − |
| Parkinsonism, age at onset | − | − | 77 | − | − | 65 | 57 |
| Bradykinesia | − | − | 77 | − | − | 65 | 57 |
| Rest tremor | − | − | 77 | − | − | − | 59 |
| Rigidity | − | − | 77 | − | − | 74 | 69 |
| Postural Instability | − | − | 77 | − | − | 67 | 57 |
| Foot dystonia | − | − | 77 | − | − | 71 | 59 |
| Epileptic seizures | + | + | − | 69 | 63 | − | 59 |
| Myoclonus | − | − | − | 69 | 63 | − | − |
| Cognitive impairment | + | − | − | 68 | >66 | 61 | 57 |
| Ophthalmoplegia | − | − | − | − | − | 61 | 59 |
| Ptosis | − | − | − | − | − | − | 58 |
| Hearing loss | − | − | − | − | 64 | 67 | − |
| Nerve conduction studies | |||||||
| Sensory axonal neuropathy | + | + | − | + | + | + | + |
| Motor axonal neuropathy | + | − | − | + | − | − | − |
| MRI hypersignals of brain stem, cerebellum, hemispheric white matter | + | + | − | + | + | + | + |
| Muscle biopsy: mosaic COX defect | + | + | + | NA | NA | NA | + |
| Multiple mtDNA deletions | + | + | + | NA | NA | NA | + |
NA, Not Available.
Cerebellar ataxia, sensory neuropathy, and action tremor
Patient II‐1, presented at age 53 with gait disturbances. He experienced both cerebellar and sensory ataxia, progressing within 6 years to a loss of ambulation. Additionally, he experienced head tremor followed closely by an arm tremor that was both intentional and resting. Electrophysiological studies, comprised of recordings of nerve conduction velocities (NCV), electromyography (EMG), and somesthesic‐evoked potentials (SEPs) indicated an axonal sensorimotor neuropathy in the lower limbs, and a pure sensory neuropathy of upper limbs. SEPs indicated a near‐absent spinal response and a very low‐amplitude cortical response. Patients II‐8, II‐11, II‐12, II‐13, and II‐17 showed similar clinical signs and courses with details shown in Table 1. Available results from electrophysiology studies for all subjects are shown in Table S1.
Parkinsonism
One subject, II‐10, displayed a significantly different clinical presentation and clinical course compared to the rest of the kinship. Her disease presented at age 77 years, two decades later than most others in this family, when she received a diagnosis of asymmetrical Parkinson's disease based on the onset of a resting tremor of the left hand, mild left‐sided rigidity, generalized akinesia (right<left), hypomimia, head tremor, mild reduction in stride length, reduced arm swing, and mild dystonic movements of the left foot. While mild tandem ataxia was observed, notably, patient II‐10 had no sensory complaint at the onset of symptoms and NCV and brain MRI were normal. In a 5‐year follow‐up exam, only moderate reduction in pallesthesia of lower limbs was noted, whereas NCV and MRI were not repeated. This subject's parkinsonian symptoms improved with levodopa treatment.
Two additional patients displayed parkinsonism in combination with ataxia and peripheral neuropathy as described above. Patients II‐13 and II‐17 had bilateral bradykinesia, reduced postural reflexes, and facilitory paratonia without resting tremor or cogwheel rigidity at the onset of their illness. In contrast to subject II‐10, these parkinsonian symptoms did not respond to levodopa.
Seizures and cognitive impairment
Five patients (II‐1, II‐11, II‐12, II‐13, and II‐17) developed progressive cognitive impairment of the frontal‐subcortical subtype leading to profound dementia in two of them (III, 11 and III, 17). The three males II‐1, II‐11, and II‐12 had a rapidly progressive cognitive decline followed by generalized tonic‐clonic seizures a few months before death in status epilepticus at the end of their seventh decade. Two females II‐8 and II‐17 had isolated episodes of generalized tonic‐clonic seizures a few years after the onset of their symptoms.
Ophthalmologic manifestations
Two patients displayed ophthalmological signs. Patient II‐17 showed classic hallmarks of PEO with ptosis but no nystagmus, at age 59. By the age of 68, oculomotor examination revealed ophthalmoplegia with moderate limitation of amplitude and marked slowness of horizontal and vertical saccades and pursuit movements. Patient II‐13 had moderate ophthalmoparesis without ptosis at 72. It is noteworthy that the age of onset of this patient's ophthalmologic dysfunction was beyond the age of death of most affected subjects in this family.
Brain MRI reveals cortical and cerebellar atrophy with cerebellar and brainstem hypersignals
Affected individuals displayed cerebellar and brainstem abnormalities on T2‐Weighted and T2 FLAIR images with focal hyperintense lesions of middle and upper cerebellar peduncles, posterior area of the pons and mesencephalon (medial longitudinal fasciculus), and the periaqueductal grey matter. They also exhibited various degrees of multifocal hyperintense hemispheric white matter lesions, cortical atrophy, and pontocerebellar atrophy (Fig. 1). Patient II‐8 had a normal MRI at the onset of symptoms. However, a second MRI performed 6 years later showed the appearance of T2 and FLAIR diffuse hypersignals at the level of middle cerebellar peduncles and midbrain and cerebellar cortical atrophy, similar to those observed in other affected family members.
Patient muscle shows signatures of mitochondrial dysfunction including multiple mtDNA deletions
Skeletal muscle biopsies from four affected individuals were examined for evidence of mitochondrial dysfunction. Electron microscopy ultrastructural studies revealed several anomalies including subsarcolemmal accumulation of abnormal mitochondria with paracrystalline inclusions (Fig. 2A). Histopathological studies exhibited evidence of subsarcolemmal mitochondrial accumulation as ragged‐blue fibers following SDH enzyme histochemistry, whereas sequential COX‐SDH histochemistry revealed a mosaic pattern of COX deficiency, with preserved SDH activity, in all four patients (Fig. 2B). Interestingly, there was variability in severity of the associated COX defect with 25% COX‐deficient fibers and 5% ragged‐red fibers observed in patient II‐8, whereas patient II‐10 showed milder changes with only 2% COX‐deficient fibers and 1% ragged‐red fibers (Fig. 2B), correlating with disease severity.
Figure 2.
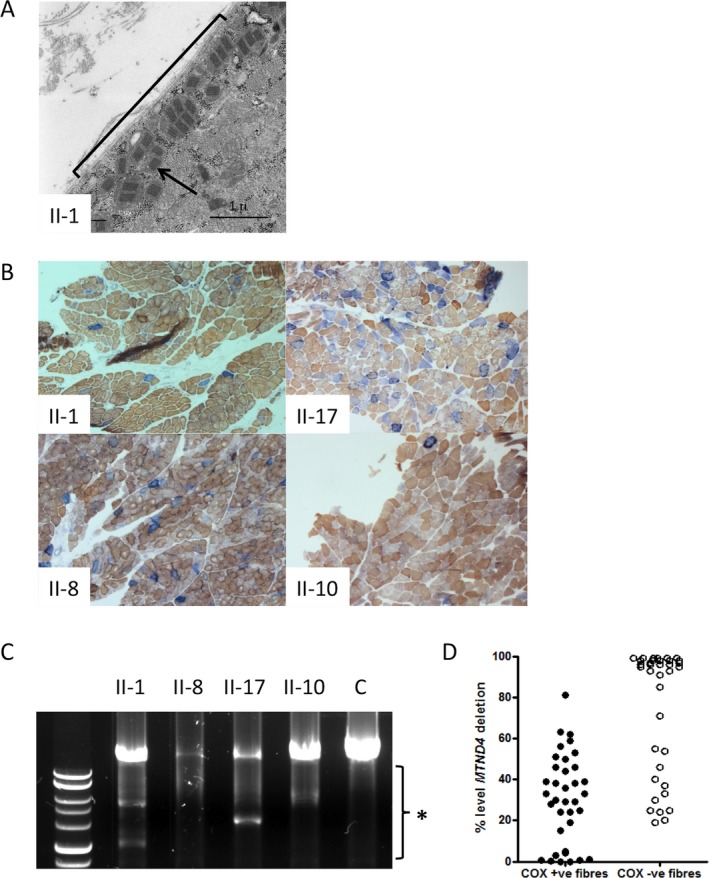
Electron microscopy, histochemistry, and mtDNA analyses in patient muscle show signs of mitochondrial dysfunction. (A) Electron microscopy of Patient II‐1 skeletal muscle reveals subsarcolemmal accumulation of abnormal mitochondria with paracrystalline inclusions. The general region of subsarcolemnal accumulation is indicated with a black bracket and a black arrow points to one of the paracrystalline inclusions. (B) Sequential COX succinate dehydrogenase (SDH) histochemistry demonstrates a mosaic distribution of COX‐deficient muscle fibers (blue) among fibers exhibiting normal COX activity (brown). Illustrated are the images for Patients II‐1, II‐17, II‐8, and II‐10. (C) Long‐range PCR across the major mtDNA shows evidence of variable mtDNA deletions in muscle from patients II‐1, II‐17, II‐8, and II‐10. The black bracket indicates the region of the gel where deletion fragments segregate. (D) Quantitative, single‐fiber real‐time PCR reveals the majority—but not all—of COX‐deficient fibers contain high levels of a clonally expanded mtDNA deletion involving the MTND4 gene (36 COX‐positive and 36 COX‐deficient fibers laser captured from the biopsy material of patients II‐1, II‐8, and II‐17).
Molecular studies revealed that muscle from all four patients studied showed evidence of multiple mtDNA deletions (Fig. 2C). Single muscle fibers were investigated to characterize the presence of clonally expanded mtDNA deletions, demonstrating that the majority of COX‐deficient fibers revealed very high levels (>80% mutated mtDNA) of clonally expanded mtDNA deletion, whereas all COX‐positive reacting fibers apart from one had levels of mtDNA deletion below this level (Fig. 2D).
Genetic studies reveal POLG2 c.970‐1G>C heterozygous variant segregates with disease
Whole‐exome sequencing identified a heterozygous splice variant, POLG2 c.970‐1G>C, in affected individual II‐17. Variants in POLG2 are known to cause multiple mtDNA deletions which was one of the molecular phenotypes observed in muscle biopsy from our patients. The POLG2 c.970‐1 G>C variant has not been previously reported in the POLG2 disease literature and is not present in ExAC database. Sequencing of family members confirmed POLG2 c.970‐1G>C segregated with disease in this pedigree (Fig. 1).
POLG2 displays reduced RNA and protein levels in patient fibroblasts
The pathogenic potential of the POLG2 variant was tested through measuring the levels of POLG2 RNA and protein in patient cells. The POLG2 c.970‐1G>C variant is located in the intron 4 splice acceptor site and predicted to cause skipping of exon 5. Improper splicing of a transcript may trigger nonsense‐mediated decay or nonsense‐associated splicing and result in reduced levels of mRNA and protein. Reduced amounts of POLG2 mRNA were detected in both patient fibroblast cell lines tested (Fig. 3A). POLG2 protein levels in patient cells were 40% lower than levels in healthy control cells (Fig. 3B and C). To our knowledge this is the first example of a POLG2 variant causing reduction in protein levels. This drastic reduction in POLG2 protein is likely the cause of the mtDNA deletions detected (Fig. 2C and D) and the pathological symptoms.
Figure 3.
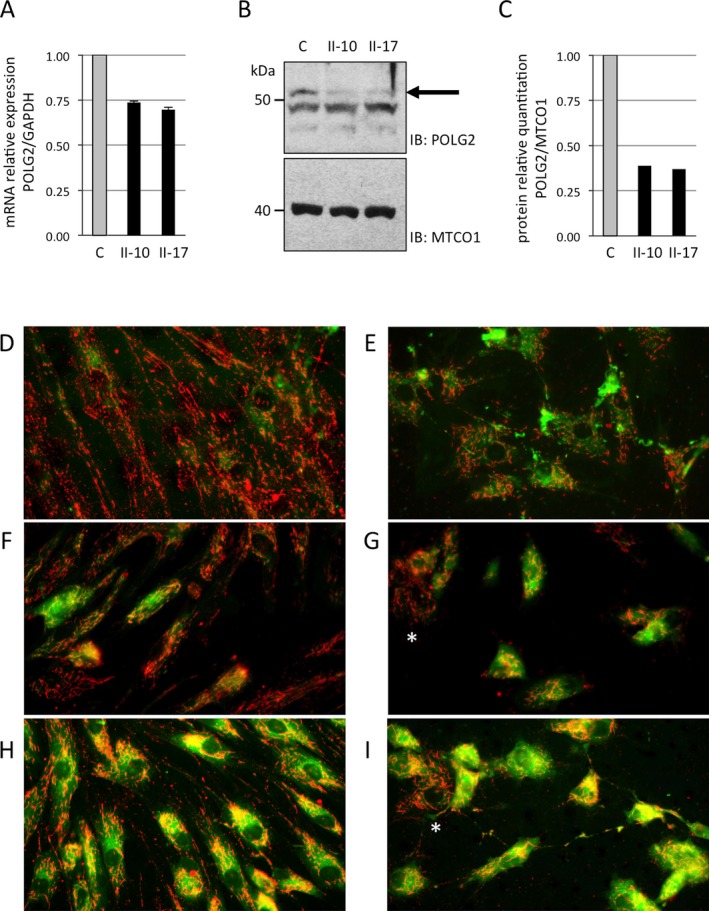
POLG2 mRNA and protein levels and mitochondrial membrane potential are decreased in patient fibroblasts. The level of POLG2 RNA and protein was measured in control (C) and patient cells (II‐10 and II‐17). (A) Quantitative real‐time PCR experiments were performed for human GAPDH and POLG2 and expressed as a relative quantitation of POLG2/GAPDH. (B) Mitochondrial lysates were analyzed by Western blotting using a rabbit polyclonal antisera raised against the recombinant human POLG2 protein. POLG2 is represented by the top band visible migrating at 55 kilodaltons (kDa), while two additional lower bands represent nonspecific binding. The black arrow points to the POLG2 band to help distinguish it from the nonspecific bands. (C) Relative quantitation of protein was measured by comparing the intensity of POLG2 and MTCO1 bands in controls and patients. D‐I. JC‐1 fluorescence studies of patient fibroblasts show reduced mitochondrial membrane potential. Imaging visualized mitochondria with inactive (green) or active (red) membrane potential. Membrane potential was assessed in untreated (D, F, H) and rotenone‐treated (E, G, I) fibroblast cells. Control fibroblasts are shown in D, E; Patient II‐1 fibroblast in F, G; Patient II‐17 fibroblast in H, I. A mosaic staining pattern of individual cells is present in patient fibroblasts, with some cells displaying predominantly red staining indicative of active mitochondria in these cells (these cells are marked with a white asterisk).
Patient fibroblasts show deficiencies in mitochondrial membrane potential
Patient fibroblasts demonstrate a substantial reduction in mitochondrial membrane potential (Fig. 3D–I). Membrane potential was 2.18 ± 0.44 in controls (mean ± SD), whereas patients showed a substantial reduction to 1.45 ± 0.34 in II‐1, 1.71 ± 0.34 in II‐10, and 1.61 ± 0.31 in II‐17. Notably, these results reveal a reduction in mitochondrial membrane potential concordant with severity of patient clinical presentation. Inhibition of complex I activity depresses membrane potential and causes intracellular movement of the mitochondria toward the nucleus, thus further exposing bioenergetics defects. Upon addition of rotenone, a complex I inhibitor, mitochondrial membrane potential shifted down in controls and showed even greater deficits in patients’ cells. Notably, JC‐1 staining showed variability in the extent of green to red across individual cells, roughly correlating with severity of disease (Fig. 3D–I). This mosaic pattern of staining across cells has been previously observed in primary mitochondrial disease patients with combined OXPHOS deficiency due to an underlying molecular etiology that results in a variable mutational load in mtDNA across patient cells.10 To our knowledge this is the first demonstration of this staining pattern in cells from patients with a POLG2 pathogenic variant as well as the first documentation of this pattern in any patient with multiple mtDNA deletions.
Variable expressivity and the possibility of reduced penetrance
The four founders in this pedigree were not available for participation in this study as all were deceased. The mother of the first sibship (I‐2) and the father of the second sibship (I‐3) were siblings (Fig. 1). Likewise, the father of the first sibship (I‐1) and mother of the second sibship (I‐4) were siblings. Of these four individuals only one, I‐3, was noted by surviving family members as having a neurological disorder. This individual received a diagnosis of Parkinson's disease at the age of 70 and died at the age of 79. This makes I‐3 the likeliest carrier of the POLG2 c.970‐1G>C variant among the four founders, which obligates I‐2 as a carrier as well. Individual I‐2 reportedly died in ‘older age’ without signs of neurological disease, raising the possibility of incomplete penetrance of this variant. However, all members of the second generation of the pedigree who were available for genotyping have genotypes that correspond to full penetrance of the variant, this included eight individuals who were asymptomatic and did not possess the variant. Additionally, the oldest age of disease onset in this family is 77 and at the most recent clinic visit this subject II‐10, at age 82, was the oldest symptomatic person in this study and manifested the mildest form of disease with parkinsonism, cerebellar ataxia, and sensory loss. This suggests that variable expressivity may present with this variant, as is not uncommon with POLG disease‐causing variants. Low levels of inherited heteroplasmic mutations of mtDNA appear to accelerate the accumulation of somatic mutation in mtDNA11 and this may influence variable ages of onset of clinical presentation across individuals carrying the same mutation.
Discussion
We describe a neurological syndrome comprised of cerebellar ataxia, axonal sensory ataxic neuropathy, and tremor in combination with epileptic seizures, cognitive decline, parkinsonism, and ophthalmoplegia. Brain imaging studies indicated cerebellar and brainstem atrophy. Genetic studies identified a heterozygous POLG2 splice acceptor variant, c.970‐1G>C, segregating with disease that caused diminished levels of POLG2 mRNA and protein in patient tissue. To our knowledge this is the first example of a POLG2 variant causing reduction in protein levels. The clinical signs in this family are distinct from previously reported cases of POLG2 deficiency and the syndrome described here extends the POLG2‐related phenotypic spectrum.
Recent microscopy reveals that POLG2 expression and subcellular localization is critical for mtDNA replication.12 Autosomal dominant nonsynonymous point mutations in POLG2 have previously been shown to cause late‐onset PEO with mtDNA deletions.2, 13 While total reduction in POLG2 is embryonic lethal, 14 a reduction in POLG2 by siRNA causes a 50% reduction in mtDNA in only 6 days.15 Mutations that affect splicing and expression in POLG2 have previously been described by Walter et al. 2010.3 In that patient, a heterozygous 24 bp in‐frame insertion was identified that caused skipping of exon 7 and degradation of the POLG2 message affecting overall POLG2 expression. This reduction in POLG2 expression was associated with late‐onset ptosis and myopathy. Thus, the c.970‐1G>C splice acceptor variant in POLG2 we identified that causes reduction in POLG2 expression is consistent with previous POLG2 mutations causing mtDNA deletions and late‐onset mitochondrial disease.
Like POLG2 mutations, other genetic causes of primary multiple mtDNA deletion syndromes most often present as adPEO with myopathy, but also manifest a broad range of age of onset with variable phenotypes and expressivity. The neurological syndrome described here shares some similarities in clinical presentation with a POLG‐related syndrome, sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO).16 SANDO may be distinguished from this disorder by age of onset as it typically starts in childhood or early adulthood and by the inclusion of dysarthria in the SANDO clinical presentation. Likewise, variants in another member of the mtDNA replication complex, C10orf2, can lead to an ataxia‐neuropathy syndrome that includes dementia.17 Parkinsonism is a clinical feature of the neurological syndrome described here that is not commonly reported in conjunction with multiple mtDNA deletions, however, variants in POLG, 18, 19 C10orf2, 20 OPA1, 21 and MPV17 22 have been associated with neurological syndromes that include parkinsonism as a component.
A thorough consideration of diseases included in the differential diagnosis is particularly relevant given the demonstrated variable expressivity of this variant and others leading to similar phenotypes. Among additional neurological syndromes that overlap with this disorder, but do not have a primary mitochondrial etiology, a special attention should be paid to Fragile X‐associated tremor/ataxia syndrome (FXTAS), as it includes a similar age of onset with progressive intention tremor, parkinsonism, cognitive decline, and generalized cortical atrophy and hypersignals in the middle cerebellar peduncles on MRI.23 Taking into consideration these similarities, we suggest considering POLG2 deficiency in the differential diagnosis for cases of ataxia and sensory neuropathy in adults, especially when it is accompanied by tremor or parkinsonism with white matter disease.
The mtDNA deletions identified in the patients’ tissues in this study result from faulty replication of the mitochondrial genome due to the germline transmission of a genetic variant, POLG2 c.970‐1G>C, that likely results in reduced efficacy of the mtDNA polymerase. mtDNA deletions may also occur in the tissues of individuals without a primary mitochondrial disorder through DNA damage induced by ROS coupled with improper repair24 or through faulty replication.25 As a result, mtDNA deletions along with a corresponding mosaic COX deficiency pattern and single‐cell clonal expansion of mtDNA deletions have also been observed in aged tissues of healthy individuals as well as in tissues from patients with neurodegeneration that is not due to a primary mitochondrial disorder. A relatively high level of deleted mtDNA has been observed in substantia nigra neurons from both aged controls and individuals with Parkinson's disease (67% of COX‐deficient fibers).26 Some extent of mutant mtDNA in a cell can seemingly be tolerated without an apparent functional consequence. This has been studied through multiple modes and end points and generally a mutational load of 60–90% of mtDNA molecules within a single cell must be mutated before functional consequences are appreciated.27 In this study, approximately 25% of patient muscle fibers tested were COX deficient and of these individual fibers typically showed >80% mutant mtDNA molecules. The germline transmission of a pathogenic variant in a gene essential for proper maintenance of the mitochondrial genome leads to the accumulation of somatic mtDNA deletions which may result in the same clinical end points, such as Parkinson's disease, as manifest in individuals who do not possess a germline variant but rather have accumulated somatic mtDNA deletions for other reasons (DNA damage induced by ROS or faulty replication). These studies converge around the fundamental concept of accumulation of somatic mtDNA deletions playing a crucial role in the neurological decline observed in brain aging and in Parkinson's disease, regardless of etiology.
Author Contributions
Conception and design of the study: LVM and PEB. Acquisition and analyses of data: PEB, AB, VA, BDP, RVC, RWT, ELB, LH, KC, GS, MG, AR, GD, PL, PH, JJM, JB, MMH, and FGD. Drafting the manuscript or figures: PEB, LVM, WCC, JP, RWT, BDP, AB, and VA.
Conflict of Interest
The authors declare no competing interests.
Supporting information
Table S1. Electrophysiological studies: conduction studies on sensory and motor nerves.
Acknowledgments
We thank the family for their participation in this study.
[The copyright line for this article was changed on 21 January 2017 after original online publication]
Contributor Information
Lionel Van Maldergem, Email: vmald@hotmail.com.
Penelope E. Bonnen, Email: pbonnen@bcm.edu
References
- 1. Sommerville EW, Chinnery PF, Gorman GS, et al. Adult‐onset Mendelian PEO associated with mitochondrial disease. J Neuromusc Dis 2014;1:119–133. [PubMed] [Google Scholar]
- 2. Longley MJ, Clark S, Yu Wai Man C, et al. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet 2006;78:1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walter MC, Czermin B, Muller‐Ziermann S, et al. Late‐onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2 . J Neurol 2010;257:1517–1523. [DOI] [PubMed] [Google Scholar]
- 4. Young MJ, Longley MJ, Li FY, et al. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum Mol Genet 2011;20:3052–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He L, Chinnery PF, Durham SE, et al. Detection and quantification of mitochondrial DNA deletions in individual cells by real‐time PCR. Nucleic Acids Res 2002;30:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonnen PE, Yarham JW, Besse A, et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet 2013;93:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davydov EV, Goode DL, Sirota M, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 2010;6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Graham J, Rickwood D. Subcellular fractionation: a Practical Approach. In J Graham, D (eds) Rickwood. Oxford: Oxford University Press, 1997. [Google Scholar]
- 10. De Paepe B, Smet J, Vanlander A, et al. Fluorescence imaging of mitochondria in cultured skin fibroblasts: a useful method for the detection of oxidative phosphorylation defects. Pediatr Res 2012;72:232–240. [DOI] [PubMed] [Google Scholar]
- 11. Ross JM, Stewart JB, Hagstrom E, et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013;501:412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lewis SC, Uchiyama LF, Nunnari J. ER‐mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016; 353: aaf5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Craig K, Young MJ, Blakely EL, et al. A p. R369G POLG2 mutation associated with adPEO and multiple mtDNA deletions causes decreased affinity between polymerase gamma subunits. Mitochondrion 2012;12:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Humble MM, Young MJ, Foley JF, et al. Polg2 is essential for mammalian embryogenesis and is required for mtDNA maintenance. Hum Mol Genet 2013;22:1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Re M, Sembongi H, He J, et al. The accessory subunit of mitochondrial DNA polymerase gamma determines the DNA content of mitochondrial nucleoids in human cultured cells. Nucleic Acids Res 2009;37:5701–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fadic R, Russell JA, Vedanarayanan VV, et al. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology 1997;49:239–245. [DOI] [PubMed] [Google Scholar]
- 17. Echaniz‐Laguna A, Chanson JB, Wilhelm JM, et al. A novel variation in the Twinkle linker region causing late‐onset dementia. Neurogenetics 2010;11:21–25. [DOI] [PubMed] [Google Scholar]
- 18. Bandettini di Poggio M, Nesti C, Bruno C, et al. Dopamine‐agonist responsive Parkinsonism in a patient with the SANDO syndrome caused by POLG mutation. BMC Med Genet 2013;14:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 2004;364:875–882. [DOI] [PubMed] [Google Scholar]
- 20. Baloh RH, Salavaggione E, Milbrandt J, et al. Familial parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch Neurol 2007;64:998–1000. [DOI] [PubMed] [Google Scholar]
- 21. Carelli V, Musumeci O, Caporali L, et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol 2015;78:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garone C, Rubio JC, Calvo SE, et al. MPV17 Mutations Causing Adult‐Onset Multisystemic Disorder With Multiple Mitochondrial DNA Deletions. Arch Neurol 2012;69:1648–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berry‐Kravis E, Abrams L, Coffey SM, et al. Fragile X‐associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord 2007;22:2018–2030. [DOI] [PubMed] [Google Scholar]
- 24. Krishnan KJ, Reeve AK, Samuels DC, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet 2008;40:275–279. [DOI] [PubMed] [Google Scholar]
- 25. Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 2012;13:659–671. [DOI] [PubMed] [Google Scholar]
- 26. Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006;38:515–517. [DOI] [PubMed] [Google Scholar]
- 27. Keogh MJ, Chinnery PF. Mitochondrial DNA mutations in neurodegeneration. Biochim Biophys Acta 2015;1847:1401–1411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Electrophysiological studies: conduction studies on sensory and motor nerves.