

Abstract
Preventing and treating Alzheimer’s disease require understanding the aggregation of amyloid beta 1-42 (Aβ1-42) to give oligomers, protofibrils, and fibrils. Here we describe footprinting of Aβ1-42 by hydroxyl radical-based fast photochemical oxidation of proteins (FPOP) and mass spectrometry (MS) to monitor the time-course of Aβ1-42 aggregation. We resolved five distinct stages characterized by two sigmoidal behaviors, showing the time-dependent transitions of monomers–paranuclei–protofibrils-fibrillar aggregates. Kinetic modeling allows deciphering of the amounts and interconversion of the dominant Aβ1-42 species. Moreover, the irreversible footprinting probe provides insights into the kinetics of oligomerization and subsequent fibrillar growth by allowing the conformational changes of Aβ-1-42 at sub-regional and even amino-acid-residue levels to be revealed. The middle domain of Aβ1-42 plays a major role in aggregation whereas the N-terminus retains most of its solvent-accessibility during aggregation, and the hydrophobic C-terminus is involved to an intermediate extent. This approach affords an in-situ, real-time monitoring of the solvent accessibility of Aβ1-42 at various stages of oligomerization, and provides new insights on site-specific aggregation of Aβ1-42 for a sample state beyond the capabilities of most other biophysical methods.
Graphical abstract
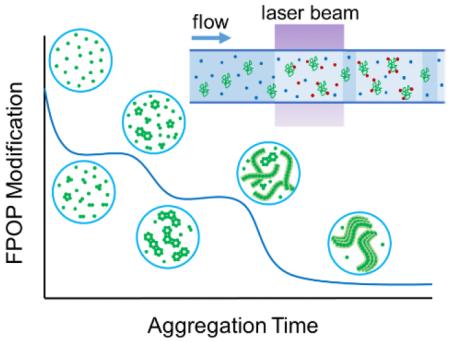
1. Introduction
The aggregation of the amyloid beta (Aβ) into oligomers and fibrils is a key process associated with Alzheimer’s disease (AD). Among Aβ isoforms that present in AD, Aβ1-42 is generally considered to be the most pathogenic.1 Extensive effort has focused on characterizing the conformation, size, and shape of Aβ aggregates (e.g., as dimers, pentamers, dodecamers2-7 and fibrils8-13), and the outcomes show a complex picture of Aβ1-42 aggregation. Although soluble oligomers are thought to be the most critical players in the pathology of AD, large aggregates and fibrils may also be toxic.1,14 Unfortunately, we know little of their rates and extents of formation.
The most common approach in methodology for characterizing structure is multiphase NMR and X-ray crystallography, providing an essential, high resolution picture of the final state of Aβ fibrils while addressing some intermediates or oligomerized states that can be retained under non-physiologically relevant conditions. Owing to the vast heterogeneity and high aggregation propensity, however, the low molecular weight (low-MW) Aβ oligomers are not amenable to NMR or X-ray crystallography. Along a similar vein, atomic force microscopy (AFM) visualizes morphology of the aggregates but provide no site specificity. More recently, Bowers and coworkers15 implemented high resolution AFM to provide insight into Aβ1-42 and Aβ1-40 oligomers formed at early stage. The results with respect to the morphology is clear, but the process used to evaporate the droplet of Aβ solution before the visualization is not free of ambiguity.
Fluorescence may be the most widely adopted approach to follow Aβ aggregation, offering a solution medium that affords some physiological relevance. The signature measurement for amyloid formation is the fluorescence-based Thioflavin T (ThT) labeling assay that shows two regions or stages of aggregation separated by a sigmoid. Recently, Frieden and coworkers.16 labeled Aβ with tetramethylrhodamine (TMR) as a novel fluorescent reporter for oligomerization. This method reports more details, showing a lag and growth phases in amyloid formation. Although those fluorescence-based methods inform on the states of aggregation, only low structural resolution data is available. Despite their ease of use, they require either adding a dye molecule, which affects the accuracy in measuring amyloid-ligand interactions because anti-amyloid compounds such as polyphenols with strong absorption and fluorescence properties can significantly bias the fluorescence readout,17 or pre-modifying Aβ, a perilous approach given that addition or subtraction of one amino acid changes significantly the properties of Aβ.
Mass spectrometry is now playing a role in understanding Aβ oligomerization, principally via measurements of the ion mobility of the gas-phase species.3 Most ion-mobility work reports on small oligomers and short Aβ fragments, whereas the full protein and its very large soluble oligomers (n ~ 100) likely hold the secret of its debilitating role in the human brain. Contemporaneously with the development of the TMR fluorescence method, we applied MS-based pulsed hydrogen-deuterium exchange (HDX)18 and obtained similar results as Frieden and coworkers except we could obtain regional information on aggregation. Although pulsed HDX avoids the problem of pre-modifying Aβ, the application of sophisticated proteomics for downstream protein analysis must be constrained to minimize the back-exchange resulted from hydrogen/deuterium scrambling.19 As an alternative, Axelsen and coworkers20 applied a synchrotron-based hydroxyl radical footprinting to study Aβ-1-40 fibril and compared the outcome with those of multiple NMR structural studies. Despite all these efforts, the approaches currently applied do not fully address, however, the intermediate conformational transitions of Aβ1-42 during aggregation. Thus, the transient aggregates of oligomerization remain insufficiently characterized, and our tools to characterize the oligomerization are temporarily exhausted.
In this report, we describe a new platform based on fast hydroxyl-radical footprinting (FPOP) and mass spectrometry to follow the aggregation of Aβ1-42, which is regarded to be the most pathologically relevant Aβ isoform in AD. Unlike Aβ1-40, the folding and assembly of Aβ1-42 are highly complicated and multi-step process,2,3,6,7,13,14,21. Here, we seek an informative, time-dependent approach that not only follows oligomerization but also can be extended to test the effects of other proteins, lipids, and potential drugs on the monomer/soluble-oligomer transitions. The cutting-edge approach, building on proteomics measurements and MS-based footprinting, utilizes hydroxyl radical-mediated irreversible reactions22 initiated on a FPOP platform23 to footprint Aβ1-42 as it undergoes oligomerization. FPOP allows various Aβ1-42 oligomeric species to be footprinted rapidly and irreversibly on amino-acid side chains. To locate the modified regions, we implemented Lys-N rapid digestion and LC-MS/MS to characterize Aβ1-42 fragments. Our hypothesis is that FPOP modifications sensitively respond to the solvent accessibility changes of Aβ1-42 upon its self-association. This approach uses downstream sample processing independent of protein conformational changes occurring post footprinting to report changes on the Aβ1-42 side chains at the global, peptide regional, and even amino-acid levels.
2. Results and Discussion
2.1 Aggregation of Aβ1-42 at the global (protein) level
Given that Aβ1-42 aggregation is time-sensitive and continues after footprinting, it is helpful to utilize an irreversible footprinting “snapshot” to capture accurately the oligomerization intermediates. In our triplicate experiments, formation of Aβ1-42 aggregates begins with incubating Aβ1-42 monomers in PBS buffer for various times up to 48 h. Footprinting of the protein occurs in a flow system after irradiation by a laser pulse that initiates the footprinting following the incubation (Figure S1). MS and MS/MS analysis of intact Aβ1-42 labeled by hydroxyl radical reports the location of oxidative labeling at solvent-accessible Aβ1-42 side chains to give variously labeled proteins (Figure 1).
Figure 1.

Mass spectra showing extents of FPOP modification for intact Aβ1-42 (5+ charged) as a function of incubation time: (a) Control: Aβ1-42 monomer with all reagents, including H2O2, flowed through the FPOP tubing but without laser irradiation, (b) extensively hydroxyl radical-modified, unstructured Aβ1-42 monomers, (c-e) decreasing FPOP modification extents of Aβ1-42 aggregates, reflecting increasing structural protection to FPOP modification, (f) minimal Aβ1-42 modification, reflecting solvent-inaccessible, highly ordered core structure of the fibrillar aggregates that resist FPOP modification.
Owing to its intrinsically disordered structure, 77% of unfolded Aβ1-42 monomers undergo modification (Figure 1b) at short times. As Aβ1-42 gains some conformational order by folding into higher-order oligomers, it loses solvent accessibility, gains protection, and undergoes correspondingly decreased FPOP modification (Figure 1c-e). At the longest incubation times, Aβ1-42 becomes highly resistant to modification, indicating formation of soluble mature aggregates, presumably with beta-sheet bundles (Figure 1f).10,24 The FPOP modification pattern for various charge states of Aβ1-42 produced in the electrospray ionization are consistent with a relative error of less than 2% for the calculated extent of modification as seen in the mass spectra of various charge states.
We calculated the fraction modified of the full-length Aβ1-42 and its digested peptide (as discussed later) from the ratio of the signal of the modified peptide (Iox) to that of the total amount of the modified and unmodified peptides (I) (Eq. 1), as described in the experimental section,
| Eq.1 |
By adopting this metric, we can pursue the change in the amount of modified species caused by Aβ1-42 conformational change and aggregation independent of the intrinsic reactivity or reaction kinetics of the peptide with hydroxyl radical. The use of the above metric for quantifying the level of modification is not unusual25. Here the fraction unmodified is directly inferred as (1 - fraction modified), and for a given peptide or residue it increases monotonically (because it is the product of the fraction unmodified at each reaction site) as the reactivity of the overall polypeptide or of a specific residue decreases owing to Aβ1-42 association and its concomitant increase in protection. The sensitivity of this metric to increasing reactivity diminishes, however, as the percent unmodified becomes very small. For small proteins like Aβ1-42 that undergo aggregation and become protected and, more importantly, for amino-acid residues, the relative amount of the unmodified species remains relatively high; thus, this concern is minor under the conditions of our footprinting.
Another possible concern is that the above method underestimates the extent of modification when multiple oxidative substitutions occur. A solution to this concern is a metric in which the intensities of oxidized species are weighted by the number of oxygens.26 This is the case for processing hydrogen-deuterium exchange (HDX) data of proteins where the signal intensity of a given deuterated peptide is multiplied by the number of deuteriums installed (i.e., a centroid is calculated). This solution can obfuscate behavior when some residues increase in reactivity and others decrease, as happens in HDX,27 and has the potential to magnify uncertainties arising from signals with low signal-to-noise ratios.28 Similar problems pertain to weighing the intensities by the number of modifications (centroid calculations) in hydroxyl radical footprinting, especially when the number of OH substitution is large (Figure 1b, c, d). Furthermore, such a metric is difficult to apply for the modifications less common than OH substitution. We tested whether both metrics reveal the aggregation characteristics (Figure S5) for the N-terminal peptide, which undergoes the most oxidative modification, and found no apparent differences. Therefore, we chose the simpler, more precise metric (Eq. 1) that does not compromise the accuracy in quantification yet sufficiently provides insights in reporting the conformational change of Aβ1-42 as it associates.
We see that the oxidation patterns of Aβ1-42 at various incubation times in the FPOP experiment follow a Poisson-distribution;29 that is, the signal of unmodified Aβ1-42 is the most intense, followed by those corresponding to the oxidized species with the mass shifts of +16, +32, +48 Da etc. in a sequentially descending manner. The patterns of various extents of oxidation follow this manner as the overall oxidation level decreasing owning to the folding of Aβ1-42.
2.2 Structural Rationalization of the Multiple Kinetic Phases
The hydroxyl radicals are comparably sized as water molecules, allowing them to modify solvent-accessible side chains and report on the transient states of soluble aggregates. We chose to model the Aβ1-42 aggregation kinetics, observed at 25 °C (Figure 2a), on the basis of the nucleation-autocatalytic growth mechanism proposed by Watsky, Finke, and coworkers30 with modifications that address the transient stages observed in the present case.3,31 This modeling approach is perhaps the simplest model that accounts for the various stages of aggregation, and we chose it in the spirit of Occam's razor. Two nucleation and two autocatalytic reactions, as shown in Scheme 1, were used to describe the time-dependent aggregation of Aβ1-42, one more stage than seen by fluorescence and pulsed HDX.
Figure 2.

Characterization of Aβ1-42 aggregation on the global (full-polypeptide) level by a kinetic simulation: a) points represents experimental data (10 μM, 25 °C, pH 7.4, no agitation), and the solid curve is a model fit based on two autocatalytic reactions. The inset in figure (a) shows a comparison of the fitting with and without the consideration of the 2-min FPOP dead time. b) Time-dependent concentrations of the various Aβ1-42 species from the solution to the differential equations of the fitted model in a). M-monomer; D-dimer; D*-product of 1st catalytic reaction, D**-product of 2nd catalytic reaction. For each species, the concentration is of monomeric Aβ1-42 equivalents.
Scheme 1.


Proposed kinetic scheme for Aβ1-42 aggregation.
With kinetic modeling (see experimental section) based on Scheme 1 and support from other published work,3,14,31 we can follow the aggregation through five stages: AB, BC, CD, DE, and EF (Figure 2a), instead of the four observed previously by pulsed HDX and fluorescence.16,18 In the modeling, our assumption is that the monomer concentration at time zero is 10 μM, which is the original concentration upon diluting Aβ1-42 into the PBS buffer, and the solution at that time is free of oligomers.
In Figure 2a, the first stage corresponds to (1) Aβ1-42 monomers assembling rapidly to form dimers and small oligomers. As the monomers continue to oligomerize, their solvent accessibility and concomitant FPOP reactivity decrease in a fast, exponential-like manner (AB). (2) The early-formed small oligomers cooperatively nucleate to provide paranuclei-like “seeds” for subsequent aggregation (transition BC). During this time, the oxidation extent cannot significantly change because the monomers have almost ceased to disappear. (3) As the concentration of “seeds” reaches a critical threshold, larger oligomers form in an autocatalytic fashion (transition CD).32 (4) Lateral oligomers likely with the beta-strand structure associate, elongate into large aggregates/protofibrils presumably with in-register beta sheets (transition DE),31,33 and (5) large aggregates/protofibrils further associate and catalyze the reorganization of other Aβ1-42 species into mature fibrils presumably with the well-defined beta-sheet structure (transition EF).10 The additional stage resolved by FPOP is represented by transition from D to F, which leads to the formation of presumably lateral protofibrillar and then to fibrillar aggregates, whereas the transition is a single growth phase according to the pulsed HDX and fluorescence platforms.
We chose Scheme 1 to support the observation that there are additional stages reported in the aggregation kinetic curve while applying the principle of parsimony in modeling. The monomer to dimer transition (reaction A-B) is illuminated by the work of Frieden and coworkers16, in which early changes in their kinetic curves with different starting Aβ concentrations behaved consistently with a model of dimer and trimer formation. Here we have not deployed the trimer formation component because we argue that the amount of additional protection afforded by the trimer over the dimer is not sufficiently large to justify the additional parameters that the trimer would require in the scheme. Visual inspection of the kinetic curves shows two steps; each step is characteristic of a slow nucleation phase (reaction B-C and D-E) followed by a rapid autocatalytic growth phase (reaction C-D and E-F). Given there are two steps, we invoked the Finke-Watzky two-step mechanism twice in succession, which might be described as double autocatalytic.34
The use of the Finke-Watzky two-step mechanism does have limitations. It does not account for the equilibrium between the Aβ1-42 monomer and other species.35 An alternative and still minimal model that addresses this issue is by Crespo et al36. In addition, nucleation and autocatalysis models augmented with fibril growth and fragmentation fail to match correctly the lag time scaling as a function of monomer concentration for a number of fibrilizing protein systems.37
The time for the each Aβ1-42 solution plug passing through the FPOP workflow is 2 min (i.e., from start of the syringe pump to irradiation). Thus, for each experimental time point, the FPOP fraction modified quantifies an assemble average of Aβ1-42 conformations over that 2 min in addition to the reported aggregation time. Although this “dead time” in FPOP will have a minor influence on the samples with long-time incubation, it may affect the early-stage characterization of the curve as the monomer will aggregate to some extent in this short period of time. To test whether this affects the fit, we repeated the simulation by shifting the actual time of aggregation by 2 min as a maximum and extrapolated the curve to the actual start point. The overall fitting of the aggregation curve is not significantly affected by considering the FPOP dead time. In a zoom-in view of the first 60 min (inset in Figure 2a), the “-2 min” on the time axis of the solid grey curve is regarded as the actual starting point of the aggregation that modeled in the testing trial. The outcomes overlap well with the curve for which the 2 min was neglected (dashed blue) even for the early aggregation stage as one can think it as the corrected aggregation curve is right-shifted by 2 min. According to the kinetics revealed by TMR fluorescence, which does provide clearer time resolution for the first a few minutes of Aβ1-42 aggregation, the initial exponential decrease in fluorescence intensity representing the monomer-dimer-trimer transition proposed happens over 30-45 min for the Aβ1-42 with concentrations ranging from 0.5 μM up to 4 μM and with stirring the solution16 (not done here). Although the initial phase is not well defined, pulsed HDX shows a burst tendency of increasing protection over 30 min for Aβ1-42 of 50 μM also without any stirring.18 These observations, taken together, suggest that it is unlikely that the dead time from the FPOP labeling has a substantial influence on the aggregation kinetics. If a burst phase occurred so rapidly in the 2 min prior to laser irradiation to initiate FPOP such that we no longer can explain transition AB, the kinetics will need to be addressed with a model accommodating the additional conformation change in the burst phase as well as transition AB tandem to it.
Although we calculated the reaction rate constants for each stage by solving the differential-equation for each reaction in Scheme 1, we are not quoting them because they do not directly relate to the actual rate constants. Each rate constant in the model is condensed and simplified as a summary of many underlying reaction steps.30 The “dimer” species, for example, is expected to be made up of many other species, each forming with its own rate constant. Others noted that nucleation rate constants for the lag phase are unreliable.37 Reliable rate constants associated with the growth phases require representation of the maximum slopes during the growth phase, which are not present in the data. Nevertheless, we can specify the species concentrations as a function of the time obtained from the model and describe the disappearance of Aβ1-42 monomers (M) and the formation of various oligomers including paranuclei (D), protofibrils (D*) and fibrils (D**) (Figure 2b). In the experiment, the initial concentration of Aβ1-42 monomers is 10 μM. In the early oligomerization, ~30% of the monomers rapidly assemble into low-MW oligomers (solid black curve), followed by a lag phase during which low-MW oligomers slowly accumulate until the concentration is sufficient to catalyze (seed) the formation of larger oligomers (dashed black curve). As the amount of large aggregates/protofibrils reaches a concentration corresponding to ~80% consumption of the original monomer equivalents (dashed grey curve), a structural reorganization occurs to deplete the monomers and form rapidly mature fibrillar aggregates (solid grey curve). At the end of the aggregation, equilibrium is nearly achieved for fibrils and persistent, leftover monomers (~10%). At this stage, the rapid second autocatalytic reaction almost shuts down the first autocatalytic reaction, and, as a result, the monomer concentration becomes relatively stable.
A previous study shows that Aβ1-42 and Aβ1-40 have distinct aggregation pathways.6 The assembly of Aβ1-42 involves formation of several distinct transient structures to form paranuclei that assemble further to form superstructures similar to early protofibrils, whereas Aβ1-40 mainly forms low-MW oligomers at a much slower rate than that of Aβ1-42 at similar concentration.3,18 We have preliminary results from the FPOP study of Aβ1-40 under the same conditions (10 μM, pH 7.4, 25 °C, no agitation) as for Aβ1-42. This proteoform shows extensive but relatively constant FPOP modification over the aggregation time scale we investigated (0 to ~48 h) (Figure S2a), indicating that it is considerably less reactive than Aβ1-42 in term of the aggregation propensity. Further, we examined the aggregation of Aβ1-42 at 37 °C (Figure S2b). The higher temperature accelerates Aβ1-42 aggregation substantially without showing any lag or intermediate phases. Within the first 8 h of incubation, the fraction modified in footprinting decreases sharply and then levels off, suggesting either the nucleation process happens so rapidly that no apparent lag phase is observed or the oligomerization pathway changed from a self-catalytic seeding mechanism.
2.3 High-Resolution Footprinting View of Oligomerization
The FPOP approach permits a high resolution view of the oligomerization down to even the amino-acid level for some residues. To interrogate the conformational changes at a regional level, we proteolyzed the irreversibly footprinted protein and analyzed the resulting peptides by LC-MS/MS. Normally, this is a straightforward for soluble proteins, but Aβ1-42 and its hydrophobic C-terminal proteolytic fragments continue to associate during the digestion, resulting in appreciable resistance to long-term proteolysis, a loss in mass spectral signals, and a possible bias towards those regions that are less prone to association.18,24 These compounded issues motivate a kinetic study that targets high structural resolution by effective and rapid digestion of Aβ1-42 aggregates. We used Lys-N protease under optimized digestion conditions to address this issue successfully. Taking advantage of Lys-N maintaining its high enzymatic activity in 6 M urea,38 we reconstituted a post-labeling aliquot of Aβ1-42 in 6 M urea and incubated it with Lys-N (enzyme: protein ratio of 1:10 (w/w)) at 45 °C to digest the Aβ1-42 as quickly as 30 min. The high-concentration of urea present in the digestion not only affords rapid proteolysis by denaturing Aβ1-42, but minimizes non-covalent association of the hydrophobic peptides. More importantly, Lys-N cleaves Aβ1-42 at the amino side of lysine residues, leaving the hydrophilic residue K28 on the highly hydrophobic C-terminal proteolytic fragment, thereby increasing the sensitivity by two orders of magnitude for the peptide covering this region compared to the corresponding peptide formed when trypsin is used (data not shown), improving quantification and accuracy.
2.4 Aggregation of Aβ1-42 at the Peptide and Residue Levels
We characterized further the conformational changes in aggregation for the regions represented by the three peptides resulting from Lys-N digestion (N-terminal region 1-15, middle domain 16-27, C-terminal region 28-42). We simulated the kinetics for each region by using the model described above (Figure 3), both independently and constrained by the outcome of the global (protein)-level rate constants. As discussed above, we expect regions that undergo conformational changes and serve as oligomerization interfaces to exclude solvent and show decreased oxidative modification. The different scales of fraction modified for each peptide is related to the distinct reactivities of the corresponding Aβ1-42 sub-regions. The reactivity is a function of: 1) the intrinsic reactivity of those sub-regions as contributed by the amino acid residues with respect to its primary sequence, and 2) the protection from the secondary and/or ternary structure of Aβ1-42 for that sub-region. Given that FPOP modification of the N-terminal peptide does not change significantly as a function of aggregation time, this region must remain structurally flexible with little self-association and little loss of solvent accessibility as Aβ1-42 associates. This is in accord with solid state-NMR data, indicating that the N-terminal region remains disordered in various Aβ1-42 oligomers and fibrils.2,9 In contrast, the extent of FPOP modification significant decreases (protection increases) over time for the middle domain peptide 16-27 and the C-terminal peptide 28-42 (6 and 2.5 times decreases in rate from Aβ1-42 monomers to fibrils, respectively). This indicates convincingly the importance of those two regions in Aβ1-42 aggregation whereby the central region must play a prominent role and serve as the self-association interface to drive aggregation. This is key experimental evidence that the middle region is a nucleation interface, as suggested by molecular dynamics studies.33,39
Figure 3.

Time-dependent FPOP labeling of Aβ1-42 incubated at 25 °C, pH 7.4, no agitation and kinetic simulations for Lys-N digested Aβ1-42 peptides: oligomerization of (a) N-terminal region 1-15, (b) middle region 16-27, and (c) C-terminal region 28-42. Solid curves are simulations that afford rates constants for each peptide treated independently, whereas the dashed curves are simulations constrained by global rates.
The peptide-level results are better modelled by allowing the rate constants for each peptide region to be independent (Figure 3, solid curves). When the modeling is restricted by the rate constants from fitting for the whole protein, we find relatively pronounced discrepancies between the results and the simulated curves (Figure 3, dashed curves). Assuming the discrepancy is not solely contributed by any structural bias in the digestion, we suggest that regions of Aβ1-42 act nearly independently in the oligomerization of Aβ1-42 and that the local (regional) rates are not predictable from the kinetic results for the whole protein. Note that in the pulsed HDX, the “half lives” of the peptide-level transitions are also different18 and show that the center and C-terminal regions are more important in the oligomerization than is the N-terminus.
Furthermore, measuring the modification at the amino-acid level allows characterizing aggregation tendencies for Aβ1-42 at that level at least for some of the amino-acid residues (Figure 4). The oxidative modifications on the residue level were assigned by using the product-ion spectra of the modified peptide precursor ion. Their aggregation curves are generated based on measuring the ratio of the signal abundance from that specific oxidized residues to the total amount of the peptide observed that contained that residue (see experimental section). The modification for H6, H13, F19, F20 and M35 are well resolved chromatographically with quantifiable extent of oxidation at better than a signal-to-noise ratio of 10:1. We also identified FPOP modifications on H12, L17, V18, V39/V40/I41. The extent of modification for those residues, however, is very low (less than 1%) and not well sampled; thus, in the present case not quantified.
Figure 4.

FPOP results for Aβ1-42 amino acid residues and kinetic simulations: (a) H6 showing little participation in the oligomerization, (b) H13 showing slightly increasing modification initially and then level off, (c) and (d) F19/F20 and M35 showing large changes of solvent accessibility along with oligomerization (FPOP modifications on F19 and F20 were summed to improve accuracy in quantification).
Residues H6 and H13 show an overall constant extent of FPOP modification during aggregation, indicating that few conformational changes occur for these two residues (Figure 4a and 4b). Interestingly, H13, despite showing little change in the fraction modified, shows a clear trend of an initial increase in FPOP modification within the first 30 min, suggesting this site becomes relatively solvent-exposed and could be a critical nucleation site either to promote or delay the next transition. Note that the absolute FPOP modification extents for residues of the same type are only related to the surrounding steric environment and protection. The modification extent of H6 is 40% and of H13 is 19%, whereas that of H12 is too low to characterize accurately. These results suggest that, although the N-terminal region remains structurally disordered during aggregation, the solvent accessibilities of these residues represent different microenvironments. H6 is the most solvent-exposed among the three, followed by H13 and then H12. For F19 and F20 we summed the FPOP modifications before plotting those values. These two residues show a decrease in FPOP modification extent by 4.5 times in going from monomers to fibrils (Figure 4c), and M35 from the C-terminal region showed a similar decrease of 2.5 times (Figure 4d). For the middle domain of Aβ1-42 represented by peptide 16-27, the difference in modification largely occurred on F19/F20, because not only is phenylalanine highly reactive to hydroxyl radical compared to other nearby residues40, but their high hydrophobicity potentially drives the oligomerization. We envision that the ability of FPOP to reveal these site-specific aggregation can be applied to understand interactions of Aβ with other molecules, especially drug candidates that affect oligomerization.
2.5 Results from Complementary Biophysical Tools
Many available biophysical tools provide complementary results in tracking Aβ assembly. We are limiting our comparison to those that can follow the time course of aggregation under physiologically relevant condition. We are not considering the many approaches that view Aβ as its initial, largely low-MW oligomers (e.g., solution NMR) or in its final aggregated state (e.g., solid state NMR). Among the methods tracking aggregation, ThT dye fluorescence is regularly used to follow amyloid formation. The dye undergoes a large enhancement in fluorescence upon binding to amyloid fibrils, making it a particularly powerful and convenient tool. A typical ThT fluorescence curve reports a single sigmoid including a lag phase and a rapid growth phase to form fibrils. Because ThT primarily interacts with β-sheet structure, this method is less suitable to study the amorphous oligomers lacking well-defined β-strand structure. ThT does not give site-specific information on the oligomerization. Furthermore, because ThT bears structural similarities to many amyloid inhibitors, it is possible that ThT can influence the fibril structure and the formation kinetics.17
The recent development of TMR, as an alternative fluorescence dye, affords a more informative sigmoidal curve. In this experimental design, Aβ was pre-modified to carry an extra lysine residue that was covalently linked to the TMR molecule at the N-terminus of Aβ.16 Unlike in the ThT assay, fluorescence quenching due to the proximity of the TMR molecules was detected as Aβ associates. This assay characterized the early-stage oligomer formation by showing an initial exponential drop in TMR fluorescence intensity before the emergence of the first lag phase. This, like all the fluorescence methods, however, suffers from the need to modify the Aβ, a polypeptide whose aggregation is highly sensitive to its length and size (consider the difference between Aβ1-42 and Aβ1-40).
We found similar results with pulsed HDX as with TMR. In the pulsed HDX method, the hydrogen on Aβ backbone amide is in exchange with the deuterium in the solvent, and the level of deuterium-uptake within a certain short time window reflects changes in the conformation of the polypeptide backbone.18 This approach, although yielding regional information, is not highly sensitive.
More recently, using high resolution AFM, Buratto, Bowers, and co-workers15 found that Aβ1-42 hexamer- and dodecamer-sized structure become dominant in as short as 5 min, and the density of large spherical aggregates termed preprotofibrils grows considerably by 20 min. This result brings new insight, but it may be confounded by surface effects because a subset of protein aggregates may be preferentially deposited on the surface for AFM and the interactions between the protein and the sample surface can affect aggregate morphology and formation kinetics.41,42 Therefore, comparisons of solution measurements by fluorescence and MS footprinting with measurements from using sample deposition will require careful interpretation. The former approaches map co-populated species in a mixture, and report Aβ1-42 conformation quantitatively as an assemble average of structural protection in bulk solution without pointing to individual oligomers. The latter provides a visualization of the oligomer morphology at certain oligomeric orders.
As we discussed above, the initial burst phase of the early oligomerization is represented by an exponential decrease in fluorescence intensity in the TMR assay and a rapid increase in the structural protection in pulsed HDX experiment. This phase occurs over a time scale of 30-60 min, with our outcome from the FPOP experiment falling in the same range. Considering the vast complexity and high sensitivity of Aβ1-42 oligomerization towards external conditions, experimental phenomena observed in various approaches are not necessarily proportionate in terms of the time scale, and it is sensible for us to set our sights on the general trends.
More importantly, caution is needed to correlate the “phases” observed using various available tools. The molecular-level mechanisms of the probes used in each method discussed above, including FPOP, differ significantly. The aggregation of Aβ, especially of Aβ1-42 under study, is a process highly sensitive to buffer conditions, concentration, temperature, pH, agitation etc. Therefore, the observed phases uncovered by these methods can be different. For example, using an equivalent Aβ1-42 system, the growth phase indicated by ThT fluorescence comes later than that observed in the TMR assay.16 In addition, we do not expect the fluorescence and HDX data to fit into Scheme 1 because we invoked the double-catalytic reactions to address the five distinct stages revealed uniquely by FPOP. In the FPOP approach, the footprinting reagent, here a hydroxyl radical, is comparably sized to water molecules and directly modifies the solvent-accessible amino-acid side chains of Aβ1-42. The probe is sensitive to subtle conformational changes involving the transient states, especially those in which Aβ1-42 side chains are involved (e.g., several protofibrillar filaments twist to form bundles of mature fibrils, in which case the polypeptide backbone stays relatively unchanged.10,11).
3. Conclusions
FPOP footprinting provides new, in-depth insights into Aβ1-42 conformational changes and its aggregation by affording structural resolution even down to the amino-acid residue level. It not only defines the early oligomerization stages but also reports subtle conformational changes that occur after early beta-strand formation, allowing the intermediate transitions to be effectively resolved to reveal the multi-step nature of oligomerization. We chose to model the aggregation curve by two autocatalytic reactions based on a modified Finke-Watzky mechanism, being attracted by the simplicity of this model. The successful fit of the observed aggregation kinetics to two-sigmoid model provides more insight on the molecular mechanisms involved in Aβ1-42 self-assembly.
The FPOP approach overcomes most of the weaknesses of other methodologies used to study the time-dependent amyloid formation by affording a direct, real-time, fast, and precise measurement of the solvent accessibility of Aβ. Moreover, applications of the platform can be extended to address the effects of pH, concentration, agitation, and Aβ modification on aggregation. We expect this measurement strategy to guide the design of optimal compounds that inhibit Aβ aggregation and/or toxicity, and to find utility for the evaluation of other amyloidogenic proteins.
4. Experimental
Materials
Synthetic wild type human amyloid beta 1-42 (Aβ1-42) was from AnaSpec (San Jose, CA). Lys-N protease was from Seikagaku Corporation (Tokyo, Japan). Hexafluoroisopropanol (HFIP), sodium hydroxide (NaOH), L-glutamine, L-methionine, catalase, hydrogen peroxide (H2O2), phosphate buffer saline (PBS, 10 mM phosphate, 138 mM NaCl, 2.7 mM KCl), urea, formic acid and trifluoroacetic acid were from Sigma Aldrich (St. Louis, MO).
Formation of Aβ1-42 aggregates
The purchased Aβ1-42 was dissolved in HFIP at 0.1 mM and incubated at room temperature for 1 h to disrupt any pre-existing aggregates. The resulting solution was then aliquoted into tubes, and the HFIP was evaporated in a fume hood. After solvent evaporation, a clear film of Aβ1-42 remained at the bottom of the tube. HFIP treatment was then repeated two more times. Samples were then frozen in −80 °C for future experiments. Prior to incubation for aggregate formation, HFIP-pretreated Aβ1-42 was dissolved in 3 mM NaOH (pH 11.7) and incubated without stirring at room temperature for 3 min followed by sonication for 1 min. Aggregation was initiated upon diluting the Aβ1-42 NaOH solution by 20 fold (v/v) with PBS buffer (pH 7.4). The final Aβ1-42 concentration after dilution was 10 μM. Different extents of aggregation were achieved by varying the incubation time from 0 to 48 h in PBS buffer (25 °C or 37 °C). For each time point, the incubation and analysis was done in triplicate.
FPOP labeling of Aβ1-42
At the various incubation times, Aβ1-42 was immediately submitted to FPOP labeling, as previously described for other proteins.29 Briefly, a KrF excimer laser (GAM Laser Inc., Orlando, FL, USA) generated a laser beam with an excitation wavelength of 248 nM and initiated H2O2 photolysis to give hydroxyl radicals. The Aβ1-42 in PBS was mixed with 20 mM glutamine (scavenger) and 20 mM H2O2 just prior to injection into the flow tubing for FPOP. The flow rate was adjusted according to the width of the laser irradiation window to ensure 20% irradiation-excluded volume and to minimize repeated laser exposure. The total time for one sample to pass through the silica tubing was ~ 2 min. After laser irradiation, the sample was collected in a tube containing 10 mM catalase and 20 mM methionine to decompose leftover H2O2 and prevent oxidation-artifacts during storage. For each aggregation time point, Aβ1-42 was incubated independently in triplicate and subjected to FPOP. In addition, control samples of Aβ1-42 were handled in the same manner in triplicate, but not laser-irradiated. Each FPOP-labeled sample was transferred into aliquots for intact Aβ1-42 characterization and enzymatic digestion, respectively.
Proteolysis
Urea was added to each 20 μL aliquot of the FPOP-labeled Aβ1-42 sample to give a final concentration of 6 M. Lys-N was then added with an enzyme to protein ratio of 1:10 (w/w). The concentrated urea facilitated rapid digestion and prevented hydrophobic peptides from self-assembly during digestion. Samples were incubated at 45 °C for 30 min. The digestion was then quenched by adding trifluoroacetic acid to a final concentration of 1% (by volume).
Mass spectrometry
For intact Aβ1-42 characterization, 20 μL of the FPOP-labeled sample was directly submitted to an Agilent 1100/1200 separation system at a flow rate of 200 μL before being admitted to a MaXis 4G quadrupole-time-of-flight (Q-ToF) mass spectrometer (Bruker Daltonics, Billerica, MA). Solvent A was water + 0.1% formic acid, and solvent B was 80% acetonitrile + 0.1% formic acid. The gradient started from 5% B and increased to 15% B in 0.3 min, ramped to 50% B in 5.2 min, increased to 100 % B in 0.5 min, held at 100 % B for 0.5 min, returned to 5% B in 0.1 min and equilibrated at 5 % B for 2 min. The mass spectrometer was operated in the positive-ion electrospray ionization mode at a mass resolving power of 30,000 (m/z 400).
For peptide and residue level analysis, 5 μL digested sample was submitted to LC-MS/MS analysis. Samples were pre-concentrated on an Acclaim PepMap C18 column (Thermo Scientific, 100 μm × 2 cm, 5 μm, 100 Å) and desalted for 15 min before elution. Separation was performed on a 15 cm custom-packed C18 column (Magic, 75 μm × 15 cm, 5 μm, 200 Å) maintained at 65 °C by using a Nano UltiMate 3000 Rapid Separation system (Dionex, Co.). Solvent A and B were the same as above. Peptides were eluted at a flow rate of 700 nL/min with the following gradient: 2% B to 20% in 1 min, ramped to 70% B in 10 min, increased to 90% in 1 min, held at 90% for 3 min, returned to 2% B in 1 min, and equilibrated at 2% B for 4 min. LC separation was directly coupled to online detection using a Q Exactive Plus hybrid quadrupole orbitrap mass spectrometer with a Nanospray Flex ion source (Thermo Fisher, Santa Clara, CA). The top ten abundant ions seen in the mass spectrum were subjected to higher energy collision dissociation (HCD) for identification and characterization of possible FPOP modifications. The mass resolving power was 70,000 (at m/z 400) for MS1 and 17,500 (m/z 400) for MS/MS.
Data analysis
Whole protein-level analysis with the custom program afforded the fraction of unmodified Aβ1-42 after FPOP for all samples. Signal intensities for the modified (Iox) and unmodified species (I) were integrated from the raw data files, either with the custom program for the intact Aβ1-42 or Thermo Xcalibur for the digested peptide and residue-level analysis. The extent of modification was calculated by using the following equation, as previously described,29
| Eq.1 |
For analysis of Aβ1-42 digested peptides, product-ion spectra obtained with the orbitrap mass spectrometer were searched for peptide identification by using Mascot (Matrix Science, London, UK) software. Masses of unmodified peptides and assigned modifications validated by manual inspection were input into an inclusion list to afford better sampling and identification of FPOP modifications.
Modification sites on the peptide were assigned on the basis of product-ion (MS/MS) spectra (SI Figure 4). In a few cases, the location of a modification to a single residue was not possible owing to limited fragmentation information from MS/MS or to the presence of interference from co-elution of peptide isomers. In that case, the modification was indicated to occur on a set of possible residues. For any specific residues, the fraction modified was calculated by the following equation as the ratio of the signal of each peptide modified at that residue (ΣIox on residue X) to the total intensity of modified and unmodified peptide signal spanning this residues.43
| Eq.2 |
Kinetic Modeling
The fraction of FPOP modified molecules was modeled on the basis of Scheme 1 by using six rate constants as model parameters (see “Fitting Parameters” in SI). The overall FPOP fraction modified F(t) was computed as shown in Eq. 2 from the species concentrations ([D], [D*], [D**]), which varied with time, and the species fraction-modified (F0, ΔFD, ΔFD*, ΔFD**) that are also model parameters. F0 is the fraction modified for Aβ1-42 monomer. The monomer concentration [M] is implicated by the experimental starting concentration of the monomer MT.
| Eq.3 |
The model curve fractions modified F(t) were calculated by solving differential equations (see “Fitting Parameters” in SI) representing each reaction in Scheme 1 with are time-dependent species concentrations and the postulated species fractions modified (F0, ΔFD, ΔFD*, ΔFD**). The quantities of ΔFD, ΔFD*, ΔFD** are properties (in term of fraction modified) of the proposed Aβ1-42 species in various oligomeric states (monomeric Aβ1-42 as the reference). Note that the definition of the fraction modified (Eq. 1) should not be interpreted in terms of intrinsic reactivities with the hydroxyl radical; that is, the differences in the fraction modified signaled the presence of different Aβ1-42 species with a characteristic protection level.
The process for determining the model parameters and the kinetic curve was implemented in Mathcad v.14.0 M020 (Parametric Technology Corp.). The "Nonlinear Quasi-Newton" mode was used in the "Minimize" function in a search for the solution model parameters. In each trial, the postulated normalized rate constants were converted to their physical values by multiplication by the initial physical rate constants to accommodate the numerically large range of rate constants. The adaptive step-size fourth order Runge-Kutta "Rkadapt" function was then used to solve the system of ordinary differential equations corresponding to Scheme 1 with initial conditions that all concentrations were zero except [M] = MT = 10 μM.
Generally, all ten model parameters were varied in a search to minimize the root-mean of the squares of the residuals between experiment data and model curve as shown in Figure 2 for Aβ1-42 and the solid curves shown in Figure 3 and Figure 4. For the dashed curves in Figure 3 and Figure 4, all rate constants were fixed to the values obtained from the model whose curve is shown in Figure 2 for Aβ1-42 while the four postulated species fractions modified were varied.
Supplementary Material
Acknowledgements
We thank Dr. Kwasi G. Mawuenyega for providing Lys-N protease and Dr. Ying Zhang and Dr. Mawuenyega for helpful discussions and grants from NIH NIGMS (Grant 8P41 GM103422), BrightFocus Foundation (Grant A2014270S), and the Graduate School of WU.
Footnotes
Contents include schmatic description of FPOP, results on Aβ1-40, representative LC/MS and MS/MS data, comparison of metrics for data processing, and modeling details including data for Figure 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Klein WL, Stine WB, Jr., Teplow DB. Neurobiol. Aging. 2004;25:569. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- (2).Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Nat. Struct. Mol. Biol. 2010;17:561. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea JE, Ruotolo BT, Robinson CV, Bowers MT. Nat Chem. 2009;1:326. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Carulla N, Zhou M, Arimon M, Gairí M, Giralt E, Robinson CV, Dobson CM. Proceedings of the National Academy of Sciences. 2009;106:7828. doi: 10.1073/pnas.0812227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hashimoto T, Adams KW, Fan Z, McLean PJ, Hyman BT. J. Biol. Chem. 2011;286:27081. doi: 10.1074/jbc.M111.257378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete NV, Cote S, De Simone A, Doig AJ, Faller P, Garcia A, Laio A, Li MS, Melchionna S, Mousseau N, Mu Y, Paravastu A, Pasquali S, Rosenman DJ, Strodel B, Tarus B, Viles JH, Zhang T, Wang C, Derreumaux P. Chem. Rev. 2015;115:3518. doi: 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Parbhu A, Lin H, Thimm J, Lal R. Peptides. 2002;23:1265. doi: 10.1016/s0196-9781(02)00061-x. [DOI] [PubMed] [Google Scholar]
- (8).Bertini I, Gonnelli L, Luchinat C, Mao J, Nesi A. J. Am. Chem. Soc. 2011;133:16013. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- (9).Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Nat. Struct. Mol. Biol. 2015;22:499. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. Proc Natl Acad Sci U S A. 2005;102:17342. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Cell. 2013;154:1257. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Selkoe DJ. Nat. Cell Biol. 2004;6:1054. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- (13).Schmidt M, Sachse C, Richter W, Xu C, Fändrich M, Grigorieff N. Proceedings of the National Academy of Sciences. 2009;106:19813. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Proc Natl Acad Sci U S A. 2003;100:330. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Economou NJ, Giammona MJ, Do TD, Zheng X, Teplow DB, Buratto SK, Bowers MT. J. Am. Chem. Soc. 2016;138:1772. doi: 10.1021/jacs.5b11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Garai K, Frieden C. Proc Natl Acad Sci U S A. 2013;110:3321. doi: 10.1073/pnas.1222478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hudson SA, Ecroyd H, Kee TW, Carver JA. FEBS J. 2009;276:5960. doi: 10.1111/j.1742-4658.2009.07307.x. [DOI] [PubMed] [Google Scholar]
- (18).Zhang Y, Rempel DL, Zhang J, Sharma AK, Mirica LM, Gross ML. Proc Natl Acad Sci U S A. 2013;110:14604. doi: 10.1073/pnas.1309175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Expert review of proteomics. 2011;8:43. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Klinger AL, Kiselar J, Ilchenko S, Komatsu H, Chance MR, Axelsen PH. Biochemistry. 2014;53:7724. doi: 10.1021/bi5010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fändrich M, Meinhardt J, Grigorieff N. Prion. 2009;3:89. doi: 10.4161/pri.3.2.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xu G, Chance MR. Chem. Rev. 2007;107:3514. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- (23).Hambly DM, Gross ML. J. Am. Soc. Mass Spectrom. 2005;16:2057. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- (24).Kheterpal I, Lashuel HA, Hartley DM, Walz T, Lansbury PT, Wetzel R. Biochemistry. 2003;42:14092. doi: 10.1021/bi0357816. [DOI] [PubMed] [Google Scholar]
- (25).Xie B, Sharp JS. Analytical Chemistry. 2015;87:10719. doi: 10.1021/acs.analchem.5b02865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li Z, Moniz H, Wang S, Ramiah A, Zhang F, Moremen KW, Linhardt RJ, Sharp JS. The Journal of Biological Chemistry. 2015;290:10729. doi: 10.1074/jbc.M115.648410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang J, Ramachandran P, Kumar R, Gross ML. Journal of the American Society for Mass Spectrometry. 2013;24:450. doi: 10.1007/s13361-012-0555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Slysz GW, Percy AJ, Schriemer DC. Analytical Chemistry. 2008;80:7004. doi: 10.1021/ac800897q. [DOI] [PubMed] [Google Scholar]
- (29).Gau BC, Sharp JS, Rempel DL, Gross ML. Anal. Chem. 2009;81:6563. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Morris AM, Watzky MA, Agar JN, Finke RG. Biochemistry. 2008;47:2413. doi: 10.1021/bi701899y. [DOI] [PubMed] [Google Scholar]
- (31).Pallitto MM, Murphy RM. Biophys. J. 2001;81:1805. doi: 10.1016/S0006-3495(01)75831-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wolff M, Unuchek D, Zhang B, Gordeliy V, Willbold D, Nagel-Steger L. PLoS ONE. 2015;10:e0127865. doi: 10.1371/journal.pone.0127865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cruz L, Rao JS, Teplow DB, Urbanc B. The Journal of Physical Chemistry B. 2012;116:6311. doi: 10.1021/jp301619v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Besson C, Finney EE, Finke RG. Chemistry of Materials. 2005;17:4925. [Google Scholar]
- (35).Stravalaci M, Beeg M, Salmona M, Gobbi M. Biosens. Bioelectron. 2011;26:2772. doi: 10.1016/j.bios.2010.10.038. [DOI] [PubMed] [Google Scholar]
- (36).Crespo R, Rocha FA, Damas AM, Martins PM. Journal of Biological Chemistry. 2012;287:30585. doi: 10.1074/jbc.M112.375345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Eden K, Morris R, Gillam J, MacPhee CE, Allen RJ. Biophys. J. 2015;108:632. doi: 10.1016/j.bpj.2014.11.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Taouatas N, Heck AJR, Mohammed S. Journal of Proteome Research. 2010;9:4282. doi: 10.1021/pr100341e. [DOI] [PubMed] [Google Scholar]
- (39).Urbanc B, Betnel M, Cruz L, Bitan G, Teplow DB. J. Am. Chem. Soc. 2010;132:4266. doi: 10.1021/ja9096303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Klinger AL, Kiselar J, Ilchenko S, Komatsu H, Chance MR, Axelsen PH. Biochemistry. 2014;53:7724. doi: 10.1021/bi5010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vácha R, Linse S, Lund M. J. Am. Chem. Soc. 2014;136:11776. doi: 10.1021/ja505502e. [DOI] [PubMed] [Google Scholar]
- (42).Campioni S, Carret G, Jordens S, Nicoud L, Mezzenga R, Riek R. J. Am. Chem. Soc. 2014;136:2866. doi: 10.1021/ja412105t. [DOI] [PubMed] [Google Scholar]
- (43).Jones LM, B. Sperry J, A. Carroll J, Gross ML. Anal. Chem. 2011;83:7657. doi: 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.