

Abstract
The human urinary proteome provides an assessment of kidney injury with specific biomarkers for different kidney injury phenotypes. In an effort to fully map and decipher changes in the urine proteome and peptidome after kidney transplantation, renal allograft biopsy matched urine samples were collected from 396 kidney transplant recipients. Centralized and blinded histology data from paired graft biopsies was used to classify urine samples into diagnostic categories of acute rejection, chronic allograft nephropathy, BK virus nephritis, and stable graft. A total of 245 urine samples were analyzed by liquid chromatography–mass spectrometry using isobaric Tags for Relative and Absolute Quantitation (iTRAQ) reagents. From a group of over 900 proteins identified in transplant injury, a set of 131 peptides were assessed by selected reaction monitoring for their significance in accurately segregating organ injury causation and pathology in an independent cohort of 151 urine samples. Ultimately, a minimal set of 35 proteins were identified for their ability to segregate the 3 major transplant injury clinical groups, - comprising the final panel of 11 urinary peptides for acute rejection (93% AUC), 12 urinary peptides for chronic allograft nephropathy (99% AUC), and 12 urinary peptides for BK virus nephritis (83% AUC). Thus, urinary proteome discovery and targeted validation can identify urine protein panels for rapid and non-invasive differentiation of different causes of kidney transplant injury, without the requirement of an invasive biopsy.
Keywords: kidney transplantation injury, acute rejection, urine proteomics, protein biomarkers, non-invasive biomarkers
INTRODUCTION
Kidney transplantation is the optimal choice of treatment for end-stage kidney disease1. Despite improved short-term outcomes2, long-term outcomes and graft survival rates remain suboptimal2, 3. Recent studies by our group and others have demonstrated that sub-clinical inflammation followed by tissue injury is an ongoing process in the transplanted kidney, and is a primary cause of graft loss4–7. This injury cannot be identified by the currently used clinical biomarkers-serum creatinine and increased urine protein load, or proteinuria. Both markers reflect non-specific and late organ injury, and cannot distinguish between causes of graft dysfunction that may require diametrically opposed approaches to therapy, such as immunosuppression intensification for graft rejection and immunosuppression reduction for BK viral nephritis. Both entities, if untreated, result in chronic tubulointerstitial loss and fibrosis2, 8.
In this study, we have continued our efforts9–13 to fully map and decipher changes in the urine proteome and peptidome after kidney transplantation, and to understand the perturbations in specific proteomic panels in the urine during biopsy-confirmed injury to the organ, defined by acute rejection (AR)2, BK viral nephritis (BKVN)14, and chronic allograft nephropathy (CAN)15 versus stable renal allograft (STA)15. In addition, we have evaluated the biological processes that drive these specific injuries, and assessed variances in perturbations of transcriptional and translational programs in graft and urine samples from the same patient.
RESULTS
Transplant injury-specific proteins segregate transplant injuries by proteomics using either iTRAQ-based or label-free LC-MS in 264 unique urine samples analyzed by different methodologies and in independent sample sets
Application of a 2D-LC-MS/MS strategy using iTRAQ reagents on 108 urine samples pooled into 6 pools/phenotype (5 independent phenotype specific samples for AR, CAN and STA and 3 independent urine samples for BKVN (due to limited available cases)), identified a total of 6379 unique peptides (false discovery rate, FDR <0.1%), spanning 958 unique human proteins (Supplemental Table S1A)13. Principal component analysis (PCA) of this data (Figure 1A) demonstrated that urine proteins generally segregate samples with injuries from stable grafts and also clustered sample groups into different phenotypes. Within this cluster of proteins, fibrinogen β and fibrinogen γ have been previously confirmed by independent ELISA validation to be significantly (p<0.05) elevated in AR over the other injury phenotypes13. This approach allowed for interrogation of the dynamic range of protein abundance measurements in transplant dysfunction categories prior to proceeding with larger numbers of individual samples to be analyzed by label-free LC-MS and SRM.
Figure 1.

The label-free LC-MS datasets from 137 individual samples identified 26,462 peptides mapped to 2,291 proteins. Each peptide was evaluated to determine if there was adequate data for an Analysis of Variance or a qualitative G-test16. Outlier and contaminant data were filtered using the log2 robust Mahalnobis distance17. Thus, at the end of filtering and outlier discovery there were 133 samples associated with 16,218 human peptides from 1,574 proteins. PCA demonstrated that these proteins cluster to separate injuries from stable grafts, and also show scatter differences based on different injury phenotypes (Figure 1B).
Urine proteins uncover biological mechanisms of graft injury
A significant number (n=811) of the total proteins (n=1719) identified were impacted during kidney graft injury (Supplemental Table S1B and S1C). Major changes were noted for major molecular processes in transplant injury: the immune response (n=179 proteins; p=4.01E-23, response to external stimulus (n=224, p=5.68E-24), and extracellular matrix organization (n=118, p=2.05E-51) (Supplemental Table S1B). A total of 517 urine proteins were significantly dysregulated in AR; 228 were increased and 289 were decreased in AR compared to other clinical categories (p<0.05) (Supplemental Table S1C), and were uniquely enriched for regulation of cell adhesion (n=51, p=2.98E-17), wound healing (n=48, p=2.45E-12), regulation of body fluid levels (n=47,1.68E-11) (Supplemental Table S1D). A set of 186 proteins were specific to CAN (p<0.05; 99 increased and 87 decreased) when compared to AR, STA, and BKVN urine samples; these proteins were uniquely enriched for biological processes involving negative regulation of protein metabolic process (n=19, p=1.25E-03), innate immune response (n=20, p=2.61E-3), multi-organism process (n=33, 6.19E-03) etc (Supplemental Table S1D). A set of 108 proteins specific to BKVN, showed overlapping biological processes with both AR and CAN, with unique enrichment of protein refolding (n=5, p=1.54E-04), regulation of response to stress (n=19, p=1.15E-03), xenobiotic catabolic process (n=3, p=1.73E-02) etc. (Supplemental Table S1D). Significant, and somewhat BKVN-specific urinary protein alterations were seen in lactotransferrin (TRFL), SUMO2 (SUMO2), granulins (GRN), haptoglobin related protein (HPTR), peptidase inhibitor 16 (PI16), alpha-1-antitrypsin (A1AT) and fibulin-1 (FBLN1).
Evaluation of transcriptomic data from matching urine and graft biopsies within patients shows specific overlapping and unique molecular processes in transplant injury
Out of 811 proteins that were identified as significantly changed in the urine protein dataset (Supplemental Table S1C), 26% (n=153) were also dysregulated at the mRNA level in the matching biopsies. There was a relatively high level of agreement in the tissue genes and urine proteins from the same patient, showing dysregulation in each specific injury group: 50% in AR, 67% in CAN, and 42% in BKVN. These overlapping pathways, at both the gene level in tissue and the protein level in the effluent from the same tissue, were significantly involved in tissue development (n=29, p=1.14E-07), extracellular matrix organization (n=16, p=2.85E-07), and cell adhesion (n=36, p= 4.75E-07). Enrichment analysis for cellular and biological processes showed a significant enrichment of proteins involved in the immune response in AR (p<0.001) (Figure 2A), and enrichment of both solute/ion transporters and extra-cellular matrix reorganization in CAN (p<0.001) (Figure 2B/C). We then analyzed proteins mapping to immune cell infiltrate specific transcriptional profiles from matching kidney biopsies [5], profiled on microarrays with AR, CAN and STA phenotypes; similar patterns of immune subset cell specific enrichment was seen at the kidney mRNA and the urine protein level in the same patient groups. There was an overlap of similar patterns of immune cell infiltrates in both acute and chronic rejection, with strongest enrichment for B cell, dendritic cell and granulocyte activation in both AR and CAN urine samples (Figure 2D/E). There was additional enrichment for monocyte and T cell specific proteins only in AR urine, which was not significant in CAN urine.
Figure 2.

Independent SRM Validation to Identify Minimal Biomarker Protein Fingerprints for Graft Injury
Of 296 peptides, mapping to 100 proteins selected for SRM validation based on the criteria provided in the methods section, SRM assays for 131 peptides mapping to 78 proteins were optimized. SRM validation for different injury sub-types was done across 151 samples that were randomly split for AR, CAN and STA into two independent sample groups: Validation Set 1 (22 AR, 27 STA, 31CAN; n=80) and Validation Set 2 (20 AR, 20 STA, 15 CAN, and 16 BKVN, n=71). Due to the smaller number of BKVN samples, these were all included in a single sample group to retain analysis power. As seen in Figure 3A, there were overall differences in Validation Set 1 samples, seen across the majority of peptides sampled (n=100), segregating transplant injury overall from stable samples (filter of >2 fold change and p <0.01 for transplant injury (Supplemental Table S1E). From the subset of perturbed proteins in transplant injury, using AltAnalyze (www.altanalyze.org), a smaller subset of 35 peptides from 33 proteins was identified (Table 2) to accurately differentiate the different types of transplant injuries (AR, CAN and BKVN) from STA samples in Validation Set 2 (Figure 3B). 11 peptides were specific to AR, 12 peptides were specific to CAN, and 12 peptides were specific to BKVN. Receiver operator curve (ROC) analysis for each injury subtype across AR-specific, BKVN-specific, and CAN-specific panels provided AUCs of 93.9% (93.3%–94.5%) for AR (Figure 3C1), 83.2% (82.2%–84.2%) for BKVN (Figure 3C2), and 99.5% (99.5%–99.5%) for CAN (Figure 3C3). We compared surveillance samples versus ‘for cause’ samples for their overall sample characteristics. Since all of the AR and BKVN samples were for cause, we used CAN samples for this analysis. There were 26 survillance biopsies from 46 CAN biopsies. ROC analysis resulted in almost the same AUCs for surveillance (AUC=99.4%) and for cause (AUC=99.5%) compared to an overall AUC of 99.5 %. We performed analysis that added clinical parameters at various levels. Addition of clinical parameters did not contribute to a better discrimination of injury from no-injury, in terms of AUC.
Figure 3.

Table 2.
Transplant injury specific peptide-panels for AR, BKVN, and CAN discovered by LC-MS and validated by SRM
| AR-specific | Peptide ID | logFC | adj.P.Val |
|---|---|---|---|
| 1 | COMP_HUMAN_ELQETNAALQDVR | 3.0 | 3.84E-12 |
| 2 | EGF_HUMAN_IESSSLQGLGR | 2.9 | 3.84E-12 |
| 3 | PGCB_HUMAN_ALHPEEDPEGR | 2.9 | 1.67E-10 |
| 4 | ZA2G_Human_EIPAWVPFDPAAQITK | 2.7 | 3.84E-12 |
| 5 | FIBA_Human_NSLFEYQK | 2.6 | 9.80E-13 |
| 6 | H2B1B_HUMAN_LLLPGELAK | 2.6 | 1.68E-12 |
| 7 | K2C75_HUMAN_NLDLDSIIAEVK | 2.4 | 9.80E-13 |
| 8 | K1C15_HUMAN_ALEEANADLEVK | 2.1 | 3.84E-12 |
| 9 | DAG1_HUMAN_VTIPTDLIASSGDIIK | 2.0 | 1.02E-11 |
| 10 | HV305_HUMAN_EVQLVESGGGLVQPGGSLR | 1.9 | 1.83E-12 |
| 11 | HPT_Human_VGYVSGWGR | 1.8 | 2.22E-13 |
| BK-specific | Peptide ID | logFC | adj.P.Val |
| 1 | HV303_HUMAN_AEDTAVYYCAK | 2.6 | 6.84E-08 |
| 2 | LMAN2_HUMAN_NCIDITGVR | 2.1 | 1.36E-06 |
| 3 | ENOA_HUMAN_GNPTVEVDLFTSK | 2.1 | 3.04E-07 |
| 4 | HV102_HUMAN_DTSTSTVYMELSSLR | 2.0 | 2.06E-05 |
| 5 | CFAB_Human_EAGIPEFYDYDVALIK | 2.0 | 2.29E-04 |
| 6 | APOH_HUMAN_FICPLTGLWPINTLK | 1.8 | 1.39E-05 |
| 7 | GRP78_Human_ITPSYVAFTPEGER | 1.7 | 5.97E-04 |
| 8 | DEF1_HUMAN_IPACIAGER | 1.5 | 2.84E-03 |
| 9 | CATG_Human_NVNPVALPR | 1.4 | 2.84E-03 |
| 10 | TRFL_HUMAN_NLLFNDNTECLAR | 1.3 | 2.84E-03 |
| 11 | CPXM1_Human_LLPQTWLQGGAPCLR | 1.2 | 5.68E-03 |
| 12 | ANXA2_HUMAN_GLGTDEDSLIEIICSR | 1.2 | 5.68E-03 |
| CAN-specific | Peptide ID | logFC | adj.P.Val |
| 1 | HV305_HUMAN_NTLYLNMNSLR | 6.3 | 4.99E-27 |
| 2 | RL18_HUMAN_ILTFDQLALDSPK | 4.8 | 1.03E-25 |
| 3 | F151A_HUMAN_AVGPSLDLLR | 2.7 | 2.41E-13 |
| 4 | TGFR2_HUMAN_LTAQCVAER | 2.6 | 4.21E-10 |
| 5 | LYAM1_HUMAN_AEIEYLEK | 2.5 | 1.61E-12 |
| 6 | PLGB_Human_AFQYHSK | 2.5 | 7.26E-13 |
| 7 | F151A_HUMAN_TYTQAMVEK | 2.4 | 4.50E-10 |
| 8 | K2C8_HUMAN_LSELEAALQR | 2.4 | 5.26E-13 |
| 9 | K1C19_HUMAN_ILGATIENSR | 2.3 | 1.38E-10 |
| 10 | IBP7_HUMAN_GTCEQGPSIVTPPK | 2.3 | 7.50E-11 |
| 11 | DSRAD_Human_YLNTNPVGGLLEYAR | 2.2 | 4.83E-10 |
| 12 | LV102_HUMAN_WYQQLPGTAPK | 2.2 | 3.94E-10 |
DISCUSSION
Understanding disease mechanisms and identifying clinically relevant and robust biomarkers for diagnostics and monitoring are critically important in organ transplantation11. Urine is used to monitor the status of the kidney9, 13, 18. Studying almost 400 unique, biopsy-paired urine samples, we report our success in identifying and validating urine proteins as biomarkers of specific categories of kidney transplant injury. Our study emphasizes (focuses on) injury phenotypes that require differing approaches for clinical patient management and are currently indistinguishable by any other available non-invasive assays. Previous studies5, 13 have shown that AR, CAN, and BKVN have many overlapping immune signatures that herald changes in allo- and innate immunity. Despite common injury cascades at the gene4, 19 and protein13 levels, AR is managed by immunosuppression intensification of T, BK, NK, and macrophage cell-dependent alloimmunity, whereas BKVN requires drastic immunosuppression reduction to allow for recovery of innate anti-viral immunity. Biomarkers specific to each phenotype, which are robust, stable, and easily accessible by urine testing, have immense clinical value in providing critical insight for the advancement of transplant therapy towards customization and personalization. This will also aid in mitigating transplant injury and guide optimal immunosuppression dosing.
Biomarker discovery and validation is inherently an arduous task because of issues including sample selection data analysis, patient heterogeneity, and other physiological confounders11, 18, 20. Here, we use cutting edge technological assays, controlled study design, and customized bioinformatics to identify panels of proteins that may be used as fingerprints of AR, BKVN, and CAN injuries. The utilization of independent sample populations with different methodologies, inclusive of SRM, allowed for elimination of false positives and recruitment of the most associative proteins within each transplant injury cohort. Our approach to reduce injury-specific proteins to minimal sets allowed for the generation of a combined set of 33 proteins that encompasses differentiation of three specific groups of transplant injuries, and can be rapidly profiled as a non-invasive assay in the out-patient setting, either as SRM or as ELISA-based multiplex, low-cost assays.
The inclusion of matched transcriptional profiling of biopsies allowed for an analysis of overlapping, functionally-relevant biological pathways in the graft and urinary ultrafiltrate. As seen in our previous proteomic studies 4, 13, 21, we found the immune-mediated and complement pathways most relevant for acute rejection injury, as well as disruption of tubular solute and ion transporters, extracellular matrix re-organization in the chronically injured graft, and innate immune responses and kinases disrupted in the SV40 infected graft. Interestingly, individual components of these pathways were variably regulated at the gene and protein level, which highlights the importance of the post-translational modifiers, activators, and regulators that close the gap between gene transcription and final protein function. Enrichment-based computational analysis of cell-specific expression data suggests a key role for B cells, granulocytes, and dendritic cells in alloimmune injury in the graft, with a more specific role for T cells in the acute phase of the rejection response. The absence of a T cell enrichment protein response in chronic injury may relate to the fact that most immunosuppressive drugs are T cell-centric, and thus result in satisfactory long-term suppression of the T cell response. The more recent and increasing clinical application of agents such as Rituximab, which selectively depletes immature B cells 22, Boretizumib that selectively targets late B cells and plasma cells 23, and Belatacept that possibly targets memory B cells 24, may result in the evolution of “escape” mechanisms seen in chronic graft injury. An earlier urine proteomic biomarker study published by our group used 9 a MALDI-TOF based “peptidomic” approach of analyzing protein digestion patterns from the activation of various proteases in urine during acute rejection. Though this previous study only focused on examining very small peptides <10 kDa after selective filtration, which is different from our study that now interrogates a different proteomic fraction of urine that is >10 kDa. There is a significant overlap of proteins (87%) identified in this study and another recent publication by our group, which uses the same approach on a smaller sample set that only contains AR and STA sample analysis13. More than two thirds of the proteins and peptides in this latter paper overlap with the current dataset in acute rejection. In this current study, the analysis of additional categories of graft dysfunction allows for the discovery of additional markers that now more fully map major categories of graft dysfunction.
CONCLUSIONS
In conclusion, years of carefully archiving urine samples collected with standardized protocols 10, 12, 25, matched with graft biopsies, and read by centralized histology 26, as well as a “back to basics” approach of whole proteome discovery using unbiased methods, resulted in biologically relevant information on graft injury. The bird’s eye view picture confirms our basic understanding of immune injury mechanisms, but a more granular view now provides details on critical players in these pathways that deserve closer attention. The limitations of the study mainly reside in the high cost and restricted availability of mass spectromery instrumentation. Given the experiment costs, we used a pooling strategy approach for the iTRAQ discovery to reduce the number of experiments, while retaining a larger sample analyte cohort. This could have potentially eliminated underlying disease heterogeneity of individual samples and “smoothened” out some biologically important signals that could track with disease severity in each classification class. Though this pooling approach can reduce the statisitcal power for discovery and limit extrapolation of data, the inclusion of individual samples, without pooling, in the validation cohort, allowed for very stringent analysis of a more restricted and smaller set of biomarkers. In addition, the deliverable of an SRM based panel to quantitatively measure the level of peptides to direct physicians to make decisions on transplant injury, impose the necessity of an available mass spectrometer in a clinical lab or hospital testing site, though these machines are now becoming increasingly availabile at lower costs. We propose to work on developing ELISA based assays for these targets to make the evaluation of this urine protein panel more cost effective. The strength of this study is access to large sample numbers, through the support of multicenter clinical trials, which allowed for the use of independent validation sets of samples with variable demographics for fine tuning a 33-protein biomarker panel in urine that provides an identification card for each transplant injury subset by a simple urine analysis27. The development of this highly selective urine protein panel for “at event” injury diagnosis, sets the stage for developing a urine biomarker panel to predict the onset of specific injuries, when histological injury is still in its most incipient stage and not detected by other clinical parameter change in function, such as a drift in the serum creatinine, proteinuria, or change in ultrasound-based Doppler resistive indices 28. Application of a protein panel-based, serial, and out-patient urine analysis approach carries the promise of predictive precision medicine for the renal transplant recipient.
BRIEF METHODS
Patient population and study samples
The study samples were selected from a biorepository of 2016 banked urine samples of which 770 were biopsy matched. A total of 396 unique and appropriate urine samples (sample selection flowchart in Figure 4A) were evaluated from kidney transplant patients with matched biopsies. They were read by a central pathologist and scored by the Banff and Chronic Allograft Damage Index (CADI)29–32 as acute cellular or humoral rejection with clinical graft dysfunction and tubulitis and/or vasculitis on histology (AR; n=112) 2, stable with no histological or clinical graft injury (STA; n=117), chronic allograft nephropathy with clinical graft dysfunction and chronic tubule-interstitial injury on histology (CAN; n=116) 15, and BK viral nephritis with SV40 staining on histology, with/without clinical graft dysfunction (BKVN; n=51). “Graft injury” in this study was defined as a greater than 20% increase in serum creatinine from its previous steady-state baseline value and an associated biopsy that was pathological. Acute rejection (AR) was defined at minimum, as per Banff Schema, a tubulitis score ≥ 1 accompanied with an interstitial inflammation score ≥1. Chronic allograft nephropathy (CAN) was defined at minimum as a tubular atrophy score ≥1 accompanied by an interstitial fibrosis score ≥1. BKVN was defined as positivity of polyomavirus PCR in peripheral blood, together with a positive SV40 stain in the concomitant renal allograft biopsy. Normal (STA) allografts were defined by an absence of significant injury pathology as defined by Banff schema. All samples were collected from pediatric and young adult recipients transplanted between 2000–2011 at Lucile Packard Children’s Hospital of Stanford University, under IRB approved protocols. The study was also approved by The Human Research Protection Program (HRPP) of the University of California, San Francisco to allow analysis of biobanked samples. The overall study design is summarized in Figure 4B.
Figure 4.
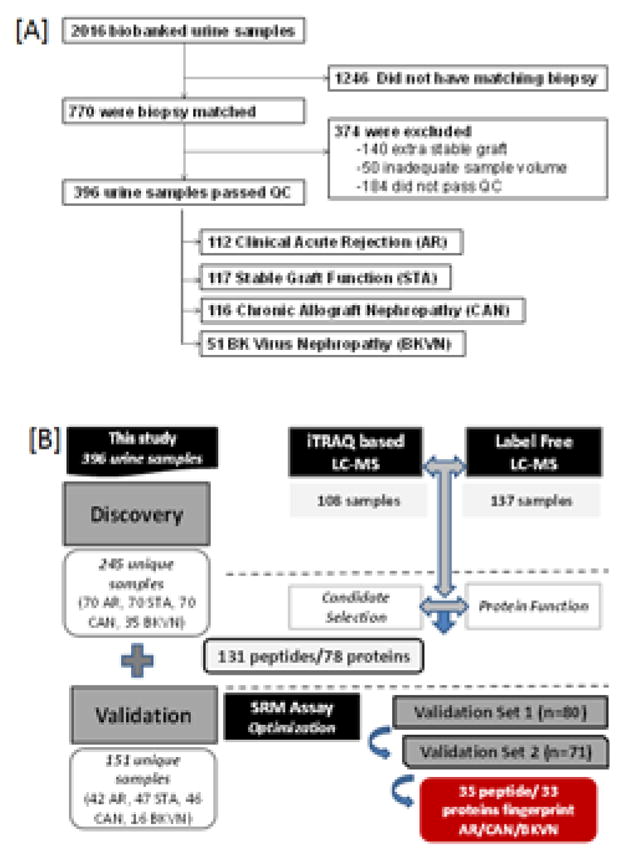
Urine collection, processing, and storage
We optimized our urine collection, processing, and storage protocols for proteomic studies from multiple clinical sites in our large biobank of urine samples33,11, 13. Second morning void midstream urine samples were collected and centrifuged at room temperature. All the urine samples in this study were collected prior to the biopsies. The detailed method is available in Supplemental Methods.
Discovery of urine proteomic repertoires by quantitative iTRAQ proteomics with a novel pooled sample approach and independent validation of phenotype specific proteins by shotgun proteomics using LC-MS/MS
To identify the repertoire and abundance range of proteins that were perturbed after transplantation of an HLA mismatched kidney, as well as their further alterations during immunological injury from innate (viral) or alloimmunity, we used a novel pooling approach for each distinct injury phenotype, with multiple samples/pool to control for pool heterogeneity 13. We prepared 6 unique pools for each phenotype and each pool contained 5 unique samples of the same injury phenotype/pool, with a sum total of 30 unique samples/clinical phenotypes except for BKVN, which contained 3 samples per pool. The pool diagnoses were similar to our target groups of interest: AR, BKVN, STA and CAN. Detailed mass spectrometric method is available in Supplemental Methods. For liquid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS analysis), we used a Thermo Fisher Scientific LTQ Orbitrap Velos MS outfitted with a custom electrospray ionization (ESI) interface assembled in-house. We used 40 AR, 40 STA, 40 CAN, and 17 urine samples for LC-MS/MS validation. The LC-MS/MS analyses for individual samples were performed on the same LC-MS platform (Orbitrap Velos MS) with similar parameters as the iTRAQ samples, except that top 10 collision-induced dissociation was used at a normalized collision energy of 40%. The raw data has been deposited into the MassIVE repository, with accession MSV000079262. It was also shared with ProteomeXchange, and assigned accession PXD002761.
Peptide selection, SRM assay configuration, and customized LC-SRM assays
Peptide selection for SRM is summarized in Supplemental Figure S1 provided in Supplemental Methods. Briefly, we used data generated from this study, as well as the data from the pooled study with iTRAQ reagents, to initiate the peptide selection. The detailed peptide selection method is provided in Supplemental Methods. Based on the selection criteria, crude synthetic peptides labeled with either C-terminal heavy lysine ([13C6, 15N2]-lysine) or C-terminal heavy arginine ([13C6, 15N4]-arginine) were purchased from Thermo Fisher Scientific (San Jose, CA). All cysteines were modified by carbamidomethylation (CAM). The details of the SRM assay is provided in Supplemental Methods.
Independent validation of most informative phenotype specific peptides by customized LC-SRM assays
Data analyses
Peptides from iTRAQ-based assay were identified based on tandem MS/MS spectra using the Sequest search algorithm against a human protein database (UniprotKB, released 2010-05). Details of peptide identification criteria for all LC-MS-based assays is available in the Supplemental Methods.
Data analysis for LC-SRM data
The data was analyzed with Skyline software 34. The best transition, highest in intensity and lowest in noises, was selected and used for quantification. The ratio of endogenous to heavy isotope-labeled internal standard was normalized with protein load and urine creatinine to achieve relative abundance of the peptide in the urine. All data analysis was performed under R 3.1.2 (http://www.r-project.org/). Data were log(2) transformed and quantile normalized. The batch effect was adjusted by empirical Bayes methods 35. Clustering heatmaps were drawn by computing correlation similarity metric and average linkage distance 36. Confounder analysis was done to eliminate any association between injury phenotypes and demographic parameters. Recipient age was significantly associated with patients’ phenotypes with a p-value < 0.001. We evaluated correlation of potential biomarker (log-transformed) and recipients’ age, however there was no significant association found (p=0.187–0.403). Based on this, we did not control for recipient age for the three potential biomarkers. There were significant associations between three potential biomarker proteins and initial infiltration amount at 95% confidence level. The correlations between biomarkers and mononuclear cell interstitial inflammation (i-score) were positive (pearson correlation range = 0.33–0.41). After this analysis, a panel of minimal peptides was selected by shrinking all peptides through minimizing the usual sum of squared errors of peptides, and using penalized maximum likelihood for linear regression models, with an algorithm called LASSO and its extension elasticnet 37, 38. The coefficients of selected peptides were derived from the linear regression model. Discrimination accuracy was calculated by cross validation, in which each group was randomly sampled 1000 times. Each time, 90% of random samples were used as a training data set and the remaining 10% as a test set. The prediction results from this approach, performed 1000 times, were pooled together and then bootstrapped 100 times on the pooled results to generate receiver operating characteristic (ROC) curves and the area under the curve (AUC).
Enrichment of functional protein groups in transplant injuries overlapping in between urine protein and gene expression of kidney biopsy by microarrays
Proteins were functionally characterized into groups for those central to the immune response (n=30), regulating solute/ion transport in the renal tubule (n=5), and critical for matrix and tissue remodeling (n=10). These proteins were further evaluated for their alterations in each functional category, across the clinical transplant phenotypes by hypergeometric enrichment analysis (double sided p value<0.05).
Enrichment analyses of within patient, matched biopsy mRNA/urine protein measurements
Human biopsy microarray data on a subset of the same patients, profiled by urine proteomics, was downloaded from GEO (GSE25902; 4), and immune cell enriched gene lists were examined, as provided in our previous publication 4, for B cell proliferation, T cell proliferation, NK cell activation, granulocyte migration, dendritic cell migration, mast cell activation, and macrophage activation. Enrichment significance of each immune cell type (e.g. B cell and T cell) was examined by using the geometric means of significant genes for each phenotype by Fisher’s exact test, using a significance threshold of p<0.05.
Intra-patient variances in tissue mRNA versus urine protein levels in transplant injury
Gene expression data for AR, CAN, and BKVN specific genes from kidney biopsy data (GEO GSE75693), were used to map 811 proteins that were identified as significantly altered in injury phenotypes AR, CAN, and BKVN, utilizing microarray data from matching biopsies from the same patient at the same time-point. The functional enrichment analysis was performed by using STRING database (http://string-db.org)39. The p value were false discovery rate (fdr) adjusted.
Supplementary Material
Supplemental Table S1A: 958 unique human proteins identified by iTRAQ based LC-MS method
Supplemental Table S1B: List of 1719 urine proteins identified by two methods combined
Supplemental Table S1C: List of proteins significantly increased in AR, CAN and BKVN urine phenotypes
Supplemental Table S1D: List of biological and functional pathways associated in different transplant injury
Supplemental Table S1E: List of 296 peptides screened for SRM assay
Supplemental Table S1F: List of 100 peptides that segregated transplant injury from stable graft
Supplemental Table S1G: List of urine samples that were included in this study with matching microarray data on GEO database. Both IDs refer to the database IDs.
Table 1.
Demographic information for 396 kidney transplant patients in the study
| 1A: 108 patients enrolled in the iTRAQ based LC-MS pooled sample analysis | ||||
|---|---|---|---|---|
| Phenotype | AR | STA | CAN | BKV |
| Number of Patients | 30 | 30 | 30 | 18 |
| Steroid-free/Steroid-based (%Steroid Free) | 57 | 50 | 50 | 33 |
| Recipient Gender (%male) | 53.3 | 50.0 | 60.0 | 66.6 |
| Recipient Age* | 13.8 ± 4.7 | 15.9 ± 4.2 | 12.1 ± 5.6 | 15.7 ± 3.4 |
| Donor Age* | 24.8 ± 11.5 | 30.6 ± 9.7 | 30.3 ± 9.3 | 29.1 ± 10.0 |
| post-txp (mo) | 24.4± 29.4 | 26.6± 37.1 | 13.6± 17.0 | 8.6± 5.6 |
| 1B: 137 patients enrolled in the label-free LC-MS individual sample analysis | ||||
|---|---|---|---|---|
| Phenotype | AR | STA | CAN | BKV |
| Number of Patients | 40 | 40 | 40 | 17 |
| Steroid-free/Steroid-based | 58% | 50% | 55% | 45% |
| Recipient Gender (%male) | 55.8 | 56.9 | 77.1 | 70.6 |
| Recipient Age* | 15.7± 4.9 | 16.5±4.4 | 11.4± 6.0 | 16.7 ± 3.5 |
| Donor Age* | 28.2 ± 11.7 | 30.7 ± 10.5 | 30.2 ± 8.6 | 28.3 ± 9.0 |
| post-txp (mo) | 72.9±77.5 | 24.1± 28.6 | 24.9± 27.3 | 9.8± 8.2 |
| 1C: 151 patients enrolled in the label-free LC-SRM analysis | ||||
|---|---|---|---|---|
| Phenotype | AR | STA | CAN | BKV |
| Number of Patients | 42 | 47 | 46 | 16 |
| Steroid-free/Steroid-based | 50% | 45% | 57% | 25% |
| Recipient Gender (%male) | 65 | 55 | 76 | 69 |
| Recipient Age* | 16.4± 3.9 | 16.4±4.6 | 11.7± 6.0 | 17.1 ± 3.1 |
| Donor Age* | 27.1 ± 11.0 | 30.7 ± 10.5 | 30.0 ± 8.8 | 28.1 ± 9.3 |
| post-txp (mo) | 82.7±82.7 | 24.2± 29.8 | 24.9± 27.9 | 10.1± 8.4 |
Recipient/Donor Age: mean ± stdev
Acknowledgments
Sources of support:
The authors acknowledge the funding support from NIDDK R01DK083447 (to M.M.S. and D.G.C.), DP2OD006668 (to W.J.Q.), and P41GM103493 (to R.D.S.). The experimental work described herein was performed in the Environmental Molecular Sciences Laboratory (EMSL), a U.S. Department of Energy (DOE) national scientific user facility located at PNNL in Richland, Washington and in the Sarwal Lab at University of California San Francisco (UCSF). We thank Kaelyn Caspillo for her help with manuscript preparation.
Footnotes
Disclosure: The authors declare no competing interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abecassis M, Bartlett ST, Collins AJ, et al. Kidney transplantation as primary therapy for end-stage renal disease: a National Kidney Foundation/Kidney Disease Outcomes Quality Initiative (NKF/KDOQITM) conference. Clinical journal of the American Society of Nephrology : CJASN. 2008;3:471–480. doi: 10.2215/CJN.05021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nankivell BJ, Alexander SI. Rejection of the kidney allograft. The New England journal of medicine. 2010;363:1451–1462. doi: 10.1056/NEJMra0902927. [DOI] [PubMed] [Google Scholar]
- 3.de Fijter JW. Rejection and function and chronic allograft dysfunction. Kidney international Supplement. 2010:S38–41. doi: 10.1038/ki.2010.421. [DOI] [PubMed] [Google Scholar]
- 4.Naesens M, Khatri P, Li L, et al. Progressive histological damage in renal allografts is associated with expression of innate and adaptive immunity genes. Kidney international. 2011;80:1364–1376. doi: 10.1038/ki.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarwal M, Chua MS, Kambham N, et al. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. The New England journal of medicine. 2003;349:125–138. doi: 10.1056/NEJMoa035588. [DOI] [PubMed] [Google Scholar]
- 6.Sigdel TK, Li L, Tran TQ, et al. Non-HLA antibodies to immunogenic epitopes predict the evolution of chronic renal allograft injury. Journal of the American Society of Nephrology : JASN. 2012;23:750–763. doi: 10.1681/ASN.2011060596. [DOI] [PubMed] [Google Scholar]
- 7.Stegall MD, Dean PG, Gloor J. Mechanisms of alloantibody production in sensitized renal allograft recipients. Am J Transplant. 2009;9:998–1005. doi: 10.1111/j.1600-6143.2009.02612.x. [DOI] [PubMed] [Google Scholar]
- 8.Trydzenskaya H, Juerchott K, Lachmann N, et al. The genetic predisposition of natural killer cell to BK virus-associated nephropathy in renal transplant patients. Kidney international. 2013;84:359–365. doi: 10.1038/ki.2013.59. [DOI] [PubMed] [Google Scholar]
- 9.Ling XB, Sigdel TK, Lau K, et al. Integrative urinary peptidomics in renal transplantation identifies biomarkers for acute rejection. Journal of the American Society of Nephrology : JASN. 2010;21:646–653. doi: 10.1681/ASN.2009080876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigdel TK, Kaushal A, Gritsenko M, et al. Shotgun proteomics identifies proteins specific for acute renal transplant rejection. Proteomics Clin Appl. 2010;4:32–47. doi: 10.1002/prca.200900124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sigdel TK, Sarwal MM. The proteogenomic path towards biomarker discovery. Pediatric transplantation. 2008;12:737–747. doi: 10.1111/j.1399-3046.2008.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sigdel TK, Nicora CD, Hsieh SC, et al. Optimization for peptide sample preparation for urine peptidomics. Clinical proteomics. 2014;11:7. doi: 10.1186/1559-0275-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigdel TK, Salomonis N, Nicora CD, et al. The identification of novel potential injury mechanisms and candidate biomarkers in renal allograft rejection by quantitative proteomics. Molecular & cellular proteomics : MCP. 2014;13:621–631. doi: 10.1074/mcp.M113.030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohl DL, Brennan DC. BK virus nephropathy and kidney transplantation. Clinical journal of the American Society of Nephrology : CJASN. 2007;2(Suppl 1):S36–46. doi: 10.2215/CJN.00920207. [DOI] [PubMed] [Google Scholar]
- 15.Fletcher JT, Nankivell BJ, Alexander SI. Chronic allograft nephropathy. Pediatric nephrology. 2009;24:1465–1471. doi: 10.1007/s00467-008-0869-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webb-Robertson BJ, Cannon WR, Oehmen CS, et al. A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics. 2010;26:1677–1683. doi: 10.1093/bioinformatics/btq251. [DOI] [PubMed] [Google Scholar]
- 17.Matzke MM, Waters KM, Metz TO, et al. Improved quality control processing of peptide-centric LC-MS proteomics data. Bioinformatics. 2011;27:2866–2872. doi: 10.1093/bioinformatics/btr479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigdel TK, Gao X, Sarwal MM. Protein and peptide biomarkers in organ transplantation. Biomarkers in medicine. 2012;6:259–271. doi: 10.2217/bmm.12.29. [DOI] [PubMed] [Google Scholar]
- 19.Roedder S, Sigdel T, Salomonis N, et al. The kSORT assay to detect renal transplant patients at high risk for acute rejection: results of the multicenter AART study. PLoS medicine. 2014;11:e1001759. doi: 10.1371/journal.pmed.1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarwal MM, Sigdel TK, Salomon DR. Functional proteogenomics--embracing complexity. Seminars in immunology. 2011;23:235–251. doi: 10.1016/j.smim.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Sigdel TK, Kaushal A, Gritsenko M, et al. Shotgun proteomics identifies proteins specific for acute renal transplant rejection. Proteomics Clinical applications. 2010;4:32–47. doi: 10.1002/prca.200900124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2006;6:859–866. doi: 10.1111/j.1600-6143.2006.01288.x. [DOI] [PubMed] [Google Scholar]
- 23.Neubert K, Meister S, Moser K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nature medicine. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 24.Melvin G, Sandhiya S, Subraja K. Belatacept: A worthy alternative to cyclosporine? Journal of pharmacology & pharmacotherapeutics. 2012;3:90–92. doi: 10.4103/0976-500X.92499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sigdel TK, Lau K, Schilling J, et al. Optimizing protein recovery for urinary proteomics, a tool to monitor renal transplantation. Clin Transplant. 2008;22:617–623. doi: 10.1111/j.1399-0012.2008.00833.x. [DOI] [PubMed] [Google Scholar]
- 26.Furness PN, Philpott CM, Chorbadjian MT, et al. Protocol biopsy of the stable renal transplant: a multicenter study of methods and complication rates. Transplantation. 2003;76:969–973. doi: 10.1097/01.TP.0000082542.99416.11. [DOI] [PubMed] [Google Scholar]
- 27.Sarwal MM, Ettenger RB, Dharnidharka V, et al. Complete steroid avoidance is effective and safe in children with renal transplants: a multicenter randomized trial with three-year follow-up. Am J Transplant. 2012;12:2719–2729. doi: 10.1111/j.1600-6143.2012.04145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gholami S, Sarwal MM, Naesens M, et al. Standardizing resistive indices in healthy pediatric transplant recipients of adult-sized kidneys. Pediatric transplantation. 2010;14:126–131. doi: 10.1111/j.1399-3046.2009.01180.x. [DOI] [PubMed] [Google Scholar]
- 29.Racusen LC. The Banff schema and differential diagnosis of allograft dysfunction. Transplant Proc. 2004;36:753–754. doi: 10.1016/j.transproceed.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 30.Racusen LC, Solez K, Colvin RB, et al. The Banff 97 working classification of renal allograft pathology. Kidney international. 1999;55:713–723. doi: 10.1046/j.1523-1755.1999.00299.x. [DOI] [PubMed] [Google Scholar]
- 31.Solez K, Colvin RB, Racusen LC, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8:753–760. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 32.Solez K, Colvin RB, Racusen LC, et al. Banff ‘05 Meeting Report: differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’) Am J Transplant. 2007;7:518–526. doi: 10.1111/j.1600-6143.2006.01688.x. [DOI] [PubMed] [Google Scholar]
- 33.Sigdel TK, Lau K, Schilling J, et al. Optimizing protein recovery for urinary proteomics, a tool to monitor renal transplantation. Clin Transplant. 2008;22:617–623. doi: 10.1111/j.1399-0012.2008.00833.x. [DOI] [PubMed] [Google Scholar]
- 34.MacLean BDT, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:3. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 36.Eisen MB, Spellman PT, Brown PO, et al. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. Journal of statistical software. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 38.Tibshirani R. Regression shrinkage and selection via the lasso. J Royal Statist Soc B. 1996;58:267–288. [Google Scholar]
- 39.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1A: 958 unique human proteins identified by iTRAQ based LC-MS method
Supplemental Table S1B: List of 1719 urine proteins identified by two methods combined
Supplemental Table S1C: List of proteins significantly increased in AR, CAN and BKVN urine phenotypes
Supplemental Table S1D: List of biological and functional pathways associated in different transplant injury
Supplemental Table S1E: List of 296 peptides screened for SRM assay
Supplemental Table S1F: List of 100 peptides that segregated transplant injury from stable graft
Supplemental Table S1G: List of urine samples that were included in this study with matching microarray data on GEO database. Both IDs refer to the database IDs.