

Abstract
Dengue virus (DENV), transmitted predominantly in tropical and subtropical regions by the mosquito Aedes aegypti, infects millions of people and leads to dengue fever and thousands of deaths each year. There are no direct-acting antivirals to combat DENV, and molecular and structural knowledge is required to develop such compounds. The dengue NS2B/NS3 protease is a promising target for direct-acting antivirals, as viral polyprotein cleavage during replication is required for the maturation of the viral particle. The NS2B/NS3 protease processes 8 of the 13 viral polyprotein cleavage sites to allow viral maturation. Although these sites share little sequence homology beyond the P1 and P2 positions, most are well conserved among the serotypes. How the other substrate residues, especially at the P′ side, affect substrate recognition remains unclear. We exploited the tight-binding general serine protease inhibitor aprotinin to investigate protease–substrate interactions at the molecular level. We engineered aprotinin’s binding loop with sequences mimicking the P′ side of DENV substrates. P′ residues significantly modulate substrate affinity to protease, with inhibition constants varying from nanomolar to sub-millimolar. Structural and dynamic analysis revealed the molecular basis of this modulation and allowed identifying optimal residues for each of the P′ positions. In addition, isothermal titration calorimetry showed binding to be solely entropy driven for all constructs. Potential flaviviral P′ side inhibitors could benefit from mimicking the optimal residues at P′ positions and incorporate hydrophobicity and rigidity to maintain entropic advantage for potency.
Keywords: substrate recognition, viral protease, dengue, protease inhibitor, peptide sequence
Graphical Abstract
Dengue virus (DENV), a member of the family Flaviviridae, is an enveloped virus with a positive single-strand RNA genome. There are four different serotypes of dengue virus (DENV 1–4), and each serotype shares 65–70% sequence homology of the viral genome.1,2 Dengue fever, which is caused by DENV, is a worldwide infectious disease endemic in more than 110 countries. Approximately 96 million of those infected develop disease symptoms, with about 20,000 annual deaths.3–5 The mosquito Aedes aegypti is the major vector of dengue virus, and due to the narrow temperature tolerance of Aedes, DENV is transmitted predominantly in tropical and subtropical regions. No antiviral drug to combat dengue infections is available, and the first-ever dengue vaccine (CYD-TDV vaccine, Dengvaxia) has been approved for use in Mexico, Brazil, El Salvador, and Philippines.6 DENV is in the same flaviviral family as the Zika virus, which is also transmitted by A. aegypti causing major outbreaks in tropical and subtropical regions, with no available treatment. A better understanding of DENV will contribute to the development of a targeted therapy against these global health threats.
The dengue RNA genome has one open reading frame, which encodes a single polyprotein including three structural proteins (C, prM, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5)7 (Figure 1a). The viral polyprotein gets processed at the lumen side of the rough endoplasmic reticulum membrane by the host cell peptidase, whereas dengue NS2B/NS3 protease cleaves the polyprotein at the cytoplasmic side.7 Dengue NS2B/NS3 protease is a serine protease of the chymotrypsin family with a classic Ser-His-Asp catalytic triad.8 The hydrophilic core of NS2B (cNS2B; amino acids 1394–1440) is required for the proper function of NS3 protease (NS3pro185; amino acids 1476–1660)9 and participates in substrate recognition.10 Dengue protease is responsible for processing 8 of the 13 polyprotein cleavage sites (C, NS2A, NS2A-NS2B, NS2B-NS3, NS3, NS3-NS4A, NS4A, NS4B-NS5) (Figure 1a).11 Polyprotein processing is required for the maturation of the viral particle.
Figure 1.
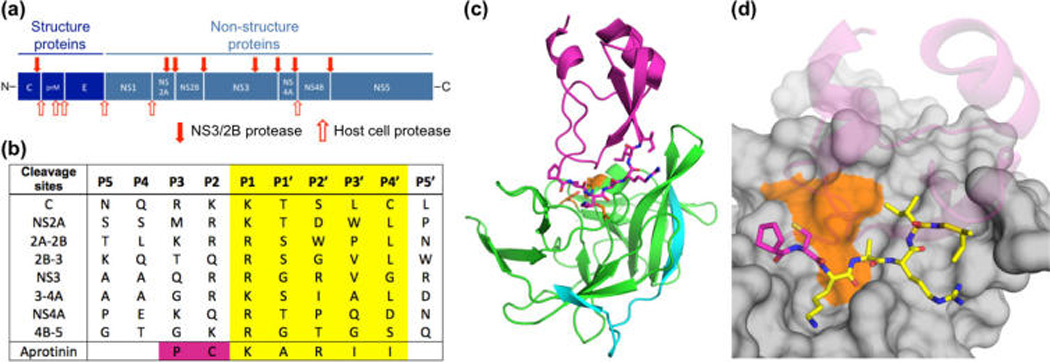
Design of aprotinin constructs mimicking dengue protease substrates. (a) Dengue virus polyprotein cleavage sites. (b) Polyprotein cleavage site sequences of DENV3 protease. (c) Aprotinin–DENV3 protease complex structure (3u1j). NS3 protease domain is in green, NS2B cyan, and aprotinin purple.10 (d) The binding loop of aprotinin is displayed as sticks and the residues screened with corresponding P1–P4′ substrate sequences are colored yellow.
The cleavage site sequences that DENV protease processes share little homology (Figure 1b). Two basic residues at P2 and P1 positions and a small polar residue at P1′ are preferred in general by flaviviral NS3 proteases.12 However, in DENV, some substrates have nonbasic residues at the P2 position, and the residues at P3–P5 and P′ positions are quite diverse (Figure 1b). Although diverse between different cleavage sites, the sites themselves are well conserved across all serotypes (and even in some cases with Zika), in particular, NS3, NS4A, and NS2B-NS3 (Table S1), implying that these sequences may be required for regulating the temporal processing of the polyprotein. However, how dengue protease recognizes these diverse substrates is not well understood.
Dengue protease does not share P′ substrate sequence preference with other serine proteases, but does so at P1 and P2 positions (furin RXRR, thrombin P1 R, trypsin P1 R). Thus, the peptidomimetic dengue protease inhibitors designed on the basis of the conserved P2 and P1 positions of substrate sequences alone may not be specific.13–15 Incorporating P′ moieties to current inhibitors could improve specificity. P′ residues at each position have been screened to investigate the favored amino acids using linear peptides.16,17 However, elucidation of the interdependence and key physical features of P′ recognition is still lacking. Linear peptides corresponding to either the P or P′ products have been used to investigate dengue protease product inhibition. However, prime side products have very low affinities (with no observed inhibition at concentrations of 1 mM) and hence do not allow biochemical characterization.18
In this study, we exploited the high-affinity binding and protease-bound structural availability of aprotinin to investigate P′ side substrate interactions with dengue protease (PDB: 3u1j).10 The binding loop of aprotinin shares close homology with the DENV NS3 cleavage site (Figure 1b). The homology makes this structure a useful template to investigate how different P′ native substrate residues may interact with the protease. We replaced the aprotinin binding loop with corresponding P1–P4′ substrate sequences of the eight DENV3 protease cleavage sites (C, NS2A, NS2A/B, NS2B/3, NS3, NS3/4A, NS4A, NS4B/5) (Figure 1b). To elucidate how P′ substrate sequence affects binding affinity, we measured the inhibition constant for each aprotinin construct in enzymatic assays. These were complemented with molecular dynamics simulations based on molecular models of the aprotinin–DENV3 complex. Isothermal titration calorimetry (ITC) revealed that binding is solely entropy driven, and enthalpy contributions are unfavorable. These constructs varied in binding affinity by 5 orders of magnitude, and their dynamic behavior implicates a complex interdependent recognition of the various cleavage sites. Thus, P′ side interactions can significantly affect ligand binding, and incorporating these interactions could help achieve additional specificity for dengue protease inhibition.
RESULTS
Design of Aprotinin Constructs Mimicking Dengue Protease Substrates
Dengue protease recognizes substrates with diverse sequences and favors two basic residues at P1 and P2 positions (Figure 1b), but little is known about how the protease recognizes the entire substrate sequences, which are well conserved between serotypes. To investigate the role of the prime side in conferring specificity to DENV protease, we used the serine protease inhibitor aprotinin, the binding loop of which shares high homology with the DENV NS3 cleavage site sequence (Figure 1b). Aprotinin has previously been shown to bind DENV2 protease with an inhibition constant (Ki) of 26 nM,19 and we measured a Ki of 3.7 nM to DENV3 protease (Figure 2). The crystal structure of the aprotinin–dengue protease complex (PDB: 3u1j)10 (Figure 1c) reveals how P′ positions bind at the active site, with extensive interactions at the P1 position and intermolecular interactions extending from the P3 to the P4′ position (Figure 1d),10 enabling engineering of constructs mimicking the P′ side of substrates.
Figure 2.
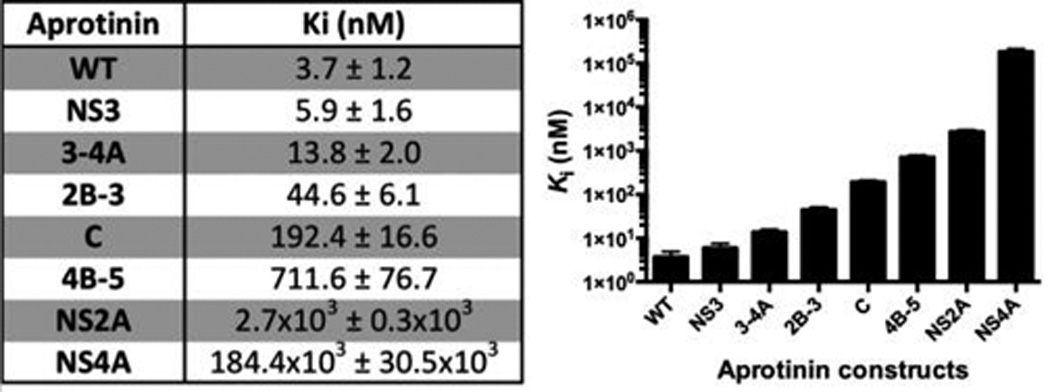
Inhibition constants (Ki) of aprotinin constructs and WT-AP DENV3 WT protease ordered from tightest to weakest binder.
In this study the binding loop (BL) residues in aprotinin (AP) were replaced with P1–P4′ substrate sequences of eight protease cleavage sites (C, NS2A, NS2A/2B, NS2B/3, NS3, NS3/4A, NS4A, NS4B/5; Figure 1). These cleavage site residues will be referred to as (cleavage site)BL-AP for clarity, and DENV protease residues will be distinguished using single-letter amino acid codes. Because the disulfide bond between Cys14 (P2 residue) and Cys38 is required for the proper folding of aprotinin,20 residues at the P2 and P3 positions were kept as the aprotinin sequence in all constructs. The eight engineered aprotinin constructs as well as the wild-type aprotinin were expressed and purified. The core domain of aprotinin forms the supporting scaffold for the binding loop and helps maintain the flat shape of the binding loop, which fits into cognate proteases’ active site and prevents deformation of the reactive site required for peptide bond cleavage.20 To test whether the constructs maintained the aprotinin fold, protein secondary structure was confirmed by circular dichroism spectroscopy (Figure S1). Seven of the eight engineered constructs, with the exception of NS2A-2B, maintained the aprotinin-like fold. The NS2A-2BBL-AP construct has a proline at the P2′ position, instead of an arginine in WT aprotinin, and replacement of this charged surface residue with proline may alter the protein expression and solubility. The remaining seven constructs were used for all further experiments, with wild-type aprotinin as a control.
Substrate Sequences Have Widely Varying Affinity to DENV3 Protease
To investigate how diverse P′ substrate sequences affect the binding to dengue protease, the inhibition constants of wild-type aprotinin and engineered constructs against DENV3 WT protease were determined using a FRET-based enzymatic assay. Whereas WT aprotinin has a Ki of 3.7 nM against DENV3 protease, the aprotinin constructs have a wide variety of Ki values ranging from 5.9 nM (NS3BL-AP) to 184 µM (NS4ABL-AP) (Figure 2). The 5 orders of magnitude difference in binding affinity strongly suggests that these sequences modulate the specificity of DENV3 protease. The sequences are distributed throughout the range of affinities with three sequences (NS3BL-AP; NS3-4ABL-AP; NS2B-3BL-AP) binding in the low to mid nanomolar range, three sequences (CBL-AP; NS4B-5ABL-AP; NS2ABL-AP) binding in the sub- to low micromolar range, and finally NS4ABL-AP in the sub-millimolar affinity. The tightest binding NS3BL-AP is very similar to the corresponding cleavage site in Zika (Table S1). Overall, different P′ residues significantly change the binding interactions between DENV3 protease and the substrate, over several orders of magnitude, and this variation in affinity likely provides some specificity for the regulation of the viral polyprotein processing.
Binding Interactions Are Solely Entropy Driven
The binding thermodynamics of the constructs to DENV3 protease were evaluated using ITC experiments. ITC measures the contributions of enthalpy and entropy to the binding free energy, which dictates the binding affinity. Reliable ITC data for CBL-AP binding to DENV3 protease, possibly due to the high protein concentration required causing disulfide bond formation and oligomerization, were not obtainable. Otherwise, as expected, the equilibrium dissociation constants (Kd) obtained from ITC experiments (Table S2) are in very good agreement with the inhibition constants (Ki) from enzymatic assays (Figure 2) (correlation coefficient = 0.999). The breakdown of the binding free energy to the contributions of enthalpy and entropy for each construct (Figure 3) indicates that the entropic contributions (−TΔS) are negative (favorable), whereas the enthalpy of binding is positive (unfavorable) for all constructs. Thus, binding is solely entropy driven and, with the exception of 4B-5BL-AP, the binding entropy strictly follows the trend of inhibition constants. Although still having an overall unfavorable binding enthalpy, the constructs of the natural DENV substrate sequences are more favorable than the WT-AP (Figure S2; Table S2) by ~2–4.5 kcal, suggesting that these natural substrate constructs are more complementary to the specific binding surface of DENV protease.
Figure 3.
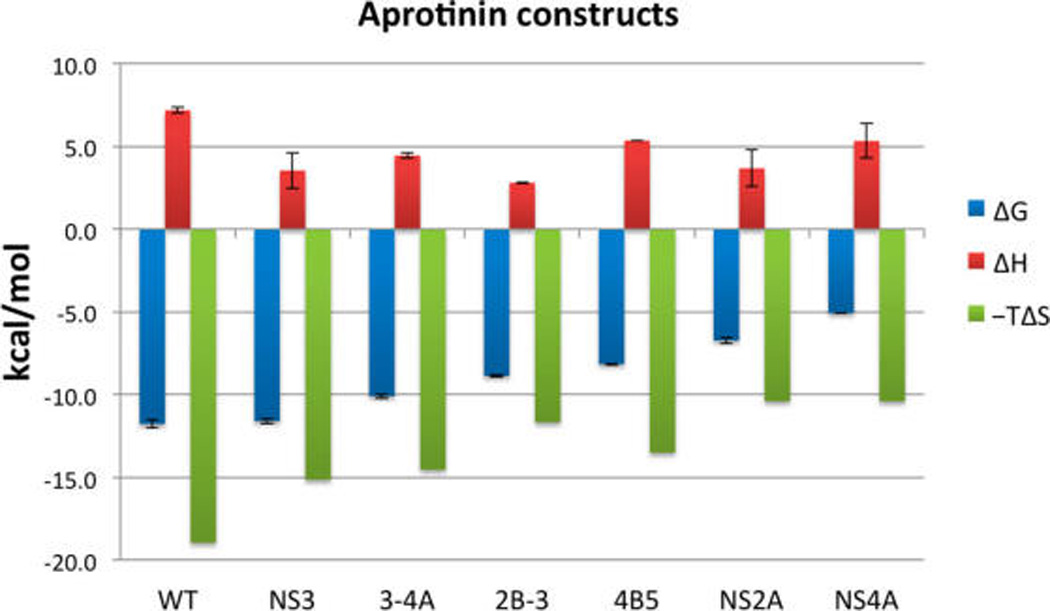
Thermodynamics measured by ITC of aprotinin constructs and WT-AP binding to DENV3 WT protease ordered from tightest to weakest binder. The binding of WT-AP and aprotinin constructs to DENV3 protease is entropy driven. Changes of Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (−TΔS) upon binding are displayed in blue, red, and green, respectively.
Binding Loop Fluctuations Correlate with Affinity
To investigate the molecular interactions between different substrate residues and DENV3 protease, extensive fully solvated MD simulations were performed starting from structural models generated on the basis of the complex crystal structure (PDB: 3ulj), where the binding loop was replaced by corresponding substrate sequences in silico.21 The binding loop of wild-type aprotinin was stable over the simulation, with well-overlapping snapshots and relatively low root-mean-square fluctuation (RMSF) values around ~1 Å (Figure 5a; Figure S4).
Figure 5.
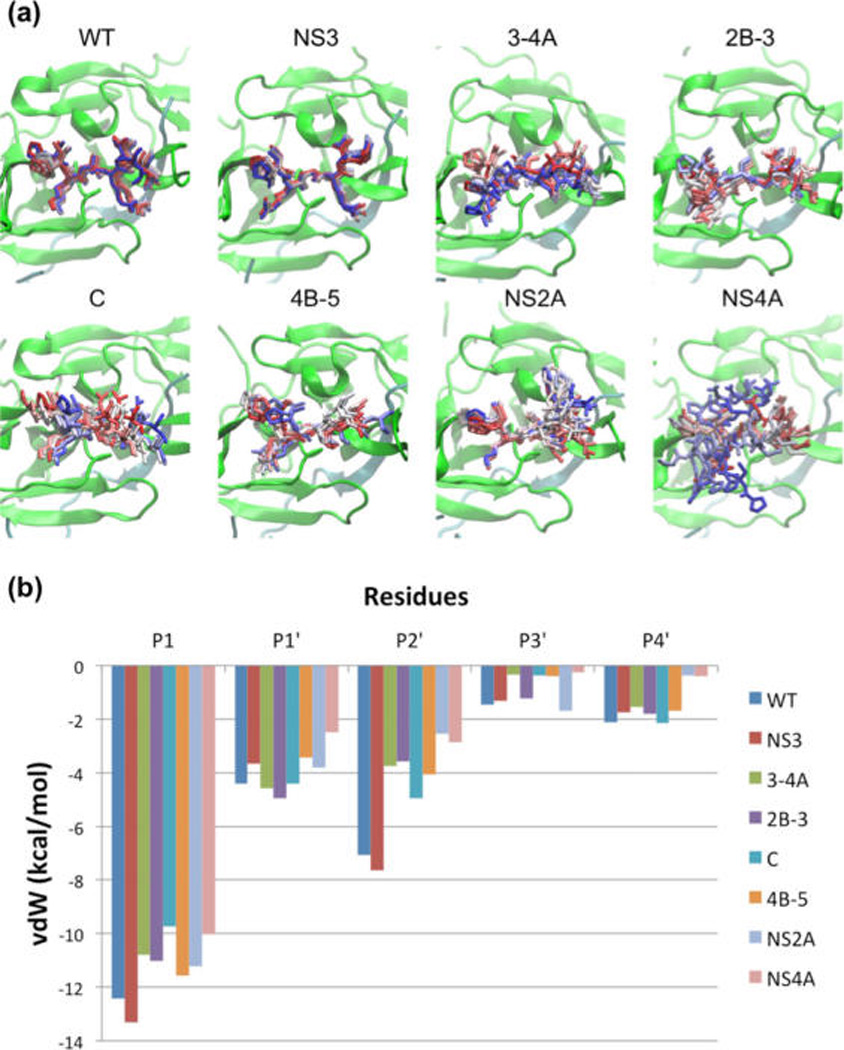
Molecular dynamics (MD) of binding loops bound to DENV protease active site. (a) Snapshots from MD simulation trajectories of WT-AP and aprotinin constructs bound to DENV3 protease. The protease is shown in cartoon representation and the aprotinin binding loops as sticks from start (in red) to end (in blue) of the trajectory (simulations were performed with full-length aprotinin, but only residues corresponding to native substrates are shown for clarity). (b) Binding loop vdW contacts during MD simulations of WT-AP and aprotinin constructs against DENV3 protease.
Aprotinin constructs with binding loops corresponding to the various substrate sequences had various levels of loop stability. The extent of the RMSF values corresponded closely with the affinities of the various substrate sequences, where the NS3BL-AP had the lowest RMSF of 1.2 Å and the tightest experimental binding affinity, and NS4ABL-AP the highest RMSF value of 4.3 and the weakest binding to DENV3 protease. In fact, the interactions between DENV3 protease and NS4ABL-AP were not strong enough to hold the binding loop in the starting pose, and by the end of the simulation only P1 Arg of NS4ABL-AP and D129 of the protease were still forming ionic interactions. Overall, fluctuations of residues corresponding to substrate sequences in each construct correlated well with the experimental Ki values.
Substrate Sequences with Higher Affinity Have Better Packing at the Protease Active Site
To investigate how packing of substrate residues at the protease active site contribute to protease binding, the average vdW contact energy was calculated over the simulation time and mapped onto the protease surface (Figure 4b). The H51 of the catalytic triad packs against Cys14 and Cys38 (outside the binding loop of aprotinin), and F31 of protease packs against Thr32 of aprotinin located outside the binding loop. To determine the interactions that determine substrate specificity, the overall vdW contact energy (P1–P4′) of residues in the binding loop of different aprotinin constructs was calculated. The two constructs with the lowest (most favorable) vdW interactions are the two tightest binders (Figure S3), WT-AP and NS3BL-AP (−27.5 and −27.7 kcal/mol, respectively). The NS3BL-AP substrate residues pack against protease residues G151, S135, T134, G133, P132, and V36, with vdW contact energies ranging from −1.3 (G151) to −4.0 (P132) kcal/mol. Other residues with significant amounts of contact energy include packing of Y161 (−4.0 kcal/mol) against P1 residue, P132 (−4.0 kcal/mol) against P1 and P2′ residues, V36 (−3.2 kcal/mol) against P3′ residue, and I30 (−3.5 kcal/mol) against P4′ residue. The binding loops of the other constructs (2B-3BL-AP, CBL-AP, 4B-5BL-AP, 3-4ABL-AP, and NS2ABL-AP) have similar level of vdW energies (−22.6 to −19.6 kcal/mol), and the weakest binder NS4ABL-AP has the highest (least favorable) contact energy (−16.0 kcal/mol). With the exception of 3-4ABL-AP, the overall contact energies of the various constructs reflect the differences observed in the experimental Ki values, where stronger binders have more contacts (lower energy) and hence pack better against protease active site residues.
Figure 4.
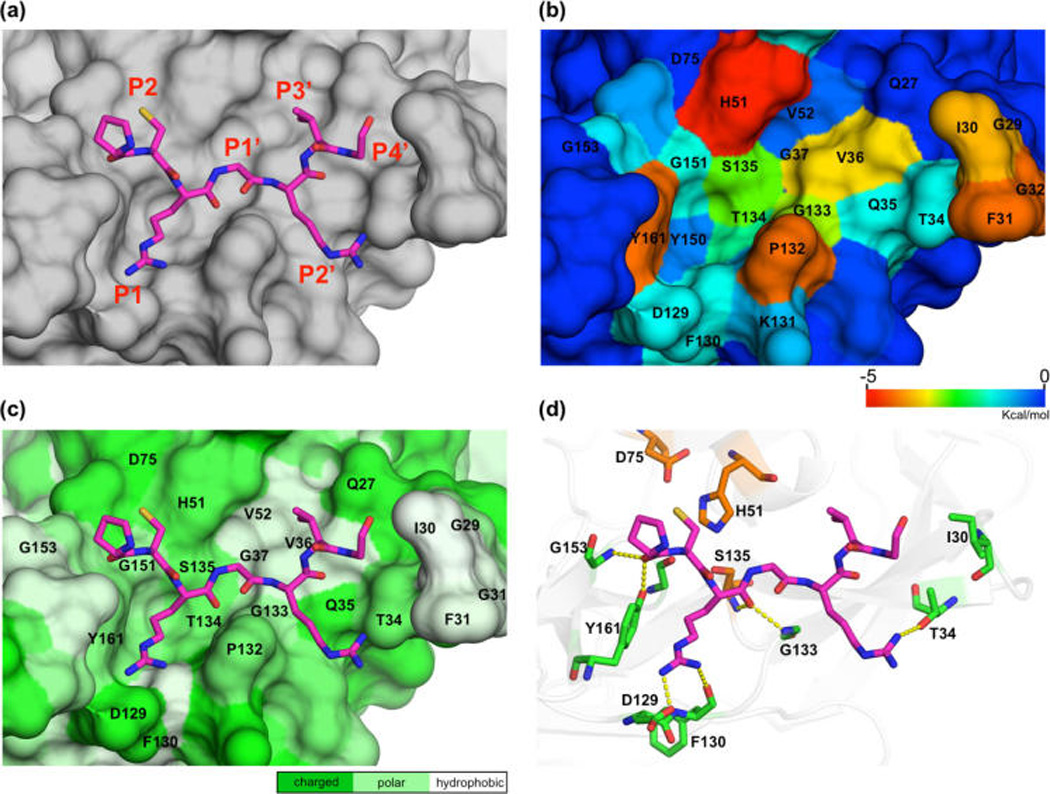
Interaction of substrate residues with DENV3 protease (NS3BL-AP binding to DENV3 protease is shown as an example). (a) NS3BL is displayed as sticks and DENV3 protease as surface. (b) DENV3 protease’s surface colored according to the extent of vdW contacts with NS3BL-AP. (c) DENV3 protease’s surface is colored on the basis of polarity. (d) Hydrogen bonds between NS3BL-AP and DENV3 protease are displayed as yellow dashes.
Tightest Binders Have More Hydrogen Bonds
To quantify the contribution of hydrogen bonds to the binding of aprotinin constructs to DENV3 protease, the percentage of time each hydrogen bond existed during the MD trajectories was calculated (Table 1). The most highly populated hydrogen bonds are formed with P1 and P2′. The binding loop of WT-AP had five hydrogen bonds (three strong and two weak ones) with DENV3 protease that existed >40% of the simulation time. NS3BL-AP had four hydrogen bonds (three strong and one weak) (Figure 4d; Table 1). All other constructs had three hydrogen bonds with DENV3 protease, except 4B-5BL-AP, which had only two hydrogen bonds. Thus, the number of hydrogen bonds overall correlates with binding affinity.
Table 1.
Hydrogen Bonds between Substrate Residues at the Binding Loop and DENV3 Protease for the Various Aprotinin Constructs and the Percentage of Time (Yellow, >40%; Red, >60%) the Hydrogen Bonds Existed during the MD Simulations
| (Aprotinin) | Protease | WT | NS3 | 3-4A | 2B-3 | C | 4B-5 | NS2A | NS4A |
|---|---|---|---|---|---|---|---|---|---|
| P3 (P13-main) | Y161-side | 54.7% | 21.7% | 13.4% | 40.2% | 23.1% | 33.3% | 33.3% | |
| P1 (K15-side) | D129-sides | 86.4% | 73.1% | 86.8% | 110.9% | 79.8% | 73.6% | 87.7% | 126.6% |
| P1 (K15-side) | F130-main | 71.4% | 45.9% | 73.5% | 37.9% | 63.7% | 37.2% | 62.8% | 40.2% |
| P1 (K15-main) | G133-main | 78.5% | 66.2% | 49.9% | 54.3% | 61.9% | 76.2% | 80.2% | 45.9% |
| P2′ (R17-side) | T34-side | 54.3% | 61.3% |
The ionic bond between the P1 side chain and protease D129 side chain is the strongest interaction and is conserved in all constructs (Figure 4d). This hydrogen bond existed >70% of the simulation time for all constructs (Table 1). P1 forms additional side chain and backbone hydrogen bonds with the backbone atoms of F130 and G133 in the protease, respectively. Overall, the P1 residue contributes the most hydrogen bonds compared to other residues in the binding loop, acting as an anchor to ensure binding.
The major difference in hydrogen bonds of different aprotinin constructs was at the P2′ position. There is one extra hydrogen bond between WT-AP or NS3BL-AP’s P2′ Arg side chain and T34 side chain of DENV3 protease (Figure 4d), which existed 54.3 and 61.3% of simulation time, respectively (Table 1). Despite this additional hydrogen bond, the overall experimental binding enthalpy, as described above, is still unfavorable, and the binding free energies are dominated by the entropic contributions. The extra hydrogen bond does stabilize the P2′ residue in both WT-AP and NS3BL-AP, resulting in more favorable vdW contacts compared to P2′ residues in other constructs (Figure 5b). Thus, Arg is more favorable than other amino acids in the P2′ position of substrate sequences.
Interactions at Individual Positions Reveal Amino Acids for Optimal Packing
The contribution of various residues to intermolecular interactions at individual positions of the binding loop was further examined (Figure 5b; Table S3). The P1 position has the best vdW contacts across all constructs, with the P1 arginine of NS3BL-AP both forming hydrogen bonds and having the lowest vdW contact energy (−13.3 kcal/mol), lower by 3.5 to 1.7 kcal/mol compared to the other constructs. However, in all constructs this side chain is either Arg or Lys, so the relative level of interaction is reflective of the interdependence of the rest of the binding site recognition.
At the P1′ position, serine in both the 2B-3BL-AP and 3-4ABL-AP constructs has slightly better contact energy compared to other P1′ residues. This serine can form a weak hydrogen bond with the backbone of protease H51 or V36 (12.1–32.6% of simulation time, respectively) and appears to be the most favored side chain at this position. NS3BL-AP and 4B-5BL-AP have a Gly at the P1′ position, resulting in contact energy below −4 kcal/mol. The worst contact energy at the P1′ position is of NS4ABL-AP’s Thr; however, this reflects the interdependent loss of contacts and high loop fluctuations mentioned above (also observed for P2′–P4′ positions) during the simulation.
At the P2′ position, vdW contact energy has major variations among substrates, with WT-AP’s and NS3BL-AP’s P2′ Arg having significantly lower contact energy than other constructs’ P2′ residues. In addition to being the largest side chain with the most contact surface, the extra hydrogen bond between P2′ Arg and T34 of the protease stabilizes this residue (Table 1). CBL-AP has the third most contacts, with the P2′ serine also forming a hydrogen bond with the Q35 backbone, but this hydrogen bond (24.0% of the simulation time) is not as strong as the one formed by Arg. Serine has a smaller contact surface than arginine, resulting in less favorable contact energy compared to WT-AP and NS3BL-AP. The P2′ Thr of 4B-5BL-AP can potentially form a weak hydrogen bond (3.4 Å away) with the Q35 side chain of the protease, but we did not observe this hydrogen bond in the simulation. The closest protease hydrogen bond donor to the P2′ Asp of NS2ABL-AP is the side chain of T34, which is too far away to form a hydrogen bond (5 Å). Therefore, these two residues, Thr and Asp at P2′, are less stable and contribute fewer vdW contacts compared to Arg. Hence, Arg followed by Ser is favored for vdW contacts and hydrogen bonds at the P2′ position.
At the P3′ position, NS2ABL-AP’s Trp has the lowest contact energy. Tryptophan has a much larger side chain compared to the P3′ residues in other constructs, and the extra surface area contributes to more vdW interactions. However, Trp is too big to optimally fit in the S3′ pocket and cannot maintain stable interactions with the protease throughout the simulation time (Figure 5a). The poor fit and stability of this Trp causes the neighboring P2′ and P4′ residues to drift away from the correlated binding pockets, resulting in the worst vdW contacts at these two positions among all of the constructs. The hydrophobic P3′ residues of WT-AP, 2B-3BL-AP, and NS3BL-AP (Ile, Val, and Val, respectively) have lower contact energy (−1.22 to −1.46 kcal/mol) compared to the remaining four constructs (−0.26 to −0.41 kcal/mol). Hence, P3′ side chains smaller than Trp better fill the hydrophobic S3′ pocket, by packing against V36 in the protease active site (Figure 4c).
Finally, for the P4′ position, aspartic acid in the NS4ABL-AP has the least vdW contacts with the protease as this residue cannot pack against protease’s hydrophobic loop (G29, I30, F31, G32), which defines the S4′ pocket. This unfavorable interaction caused the P4′ residue to drift away from the active site, and NS4ABL-AP was bound to DENV3 protease only through the interaction between P1 Arg and D129 of protease at the end of the simulation. Because of the hydrophobic nature of the S4′ surface, a hydrophobic P4′ residue would be favorable at this position.
Overall, the intermolecular contacts contributed by different aprotinin constructs correlate well with the Ki values (Figure 2; Table S3) and reveal which combination of residues might be optimal.
DISCUSSION
In this study, the binding interactions of P′ substrate residues to dengue protease were investigated by exploiting the high-affinity serine protease inhibitor aprotinin. Substrate affinity was strongly modulated by different P′ sequences with the inhibition constant varying over 5 orders of magnitude (Figure 2), which indicates protease’s affinity and processing may highly vary, potentially in both rate and temporal sequence, among various cleavage sites. The order of polyprotein processing can correlate with binding affinities to the cleavage sites and has been shown to be crucial for other viruses, such as the HIV-1 viral fitness/infectivity.22,23 However, to the best of our knowledge, the order of overall polyprotein processing during replication is not known for dengue virus. The biological function of the internal cleavage at NS3 (tightest binder) is not clear;24 however, processing of 3-4A and 2B-3 (second and third tightest binders, respectively) is required to release mature NS3 protein,25 which can then cleave the other cleavage sites. Hence, P′ sequences likely play a key role in regulating the specificity of polyprotein processing.
Previously, substrate peptides have been used to investigate residue preference at each position, with contradictory results. For example, acidic residues Glu and Asp were found to be favored at the P2′ position in the context of nKRR-X(P2′)XX (X, amino acid mixture except Cys and isostere norleucine instead of Met),16 but Glu was found to be the least favorable when screened within GLKR-G(P2′)AK.17 Using amino acid mixtures as a background can be misleading and mask the effect of specific residues at a given position, such as due to intramolecular interactions, whereas using a fixed background sequence may miss other key interdependencies. Linear substrate peptides corresponding to natural sequences spanning from P4 to P4′ have been used to investigate substrate cleavage.17 However, with diverse residues at the P positions (both basic and nonbasic P2 residues, distinct P3 and P4 residues), distinguishing the contributions of individual P′ sites was very challenging.
Therefore, instead of changing P′ substrate residues one by one or the whole cleavage site at once, our results reveal the effect of P1–P4′ positions as a contiguous sequence. Within the context of these viral substrate sequences, we have identified the favored amino acids at each substrate position: basic residues Arg and Lys at P1, Ser at P1′, Arg at P2′, and hydrophobic residues at P3′ and P4′ sites. Compared to previous peptide-based screening results,16 the main differences are at P2′ and P3′, which suggested acidic residues and serine to be favorable at P2′ and P3′, respectively. For the P2′ pocket, hydrogen bonds and vdW contacts are likely the dominant forces, and an acidic residue can form the same hydrogen bonds with the T34 residue that Arg interacts with (Figure 4d), so both an acidic residue or an arginine may be favorable at this position. With regard to the P3′ position, Ser may interact with Q27 through a hydrogen bond. However, we found hydrophobic substrate residues to pack well against V36 of the protease. Hydrophobic protease residues 30 and 31, which define the S4′ pocket, were proposed to interact with the membrane,26,27 and further studies are required to investigate the effect of hydrophobic interactions at this pocket. Thus, Arg/Lys-Ser-Arg-Ile/Val-Leu is the optimal sequence for P1–P4′ positions of the dengue protease substrates. None of the natural sequences contain this combination (Figure 1b), potentially suggesting an evolutionary advantage not to have the highest affinity combination for a particular cleavage site. This sequence may be a potent pan-flaviviral protease binder, based on the high level of homology among cleavage sites in the dengue viral serotypes and other flaviviruses such as the recently pandemic Zika virus. Comparison of the crystal structures of aprotinin bound to DENV protease (3u1j)10 or WNV protease (2ijo),28 shows the main difference is at the P2′ Arg. This Arg forms a hydrogen bond with the side chain of Thr34 with DENV protease and with the backbone carbonyl of Thr132 in complex with WNV protease. Residue 34 on the WNV protease is a tyrosine pointing away from the S2′ pocket. For the DENV protease, both the side chain of Thr34 and the backbone carbonyl group of Pro132 can form a hydrogen bond with P2′ Arg, potentially contributing to DENV protease’s preference for arginine at this position.
Thus, we suggest extending inhibitors to P′ sites to enhance both affinity and specificity against dengue protease. Previous peptidomimetic dengue protease inhibitors have been designed mainly on the basis of only P site substrate sequences13–15 and usually contain basic P2 and P1 residues. Because dengue protease has substrate sequence preferences (basic P1 or P1/P2 residues) similar to those of human serine proteases (thrombin, trypsin, and furin), further optimization of these inhibitors is required to increase specificity. In this study, we found that P′ amino acids can significantly affect the binding affinity. Dengue protease does not share P′ site sequence preference with other human serine protease; therefore, inhibitor design extending to or based on P′ sites could increase both affinity and specificity.
Designing potent DENV protease inhibitors will likely require using a combination of strategies. Inhibitors spanning P1–P1′ positions can mimic the transition state of the peptide cleavage reaction. Transition state mimicking compounds have proved successful in targeting HCV proteases,29 and enzymes have evolved to bind most strongly to the transition state, rather than substrate or product. Therefore, incorporating chemical moieties into inhibitors to mimic the transition state could increase inhibitor affinity. In fact, the peptidomimetic inhibitor Bz-Nle-Lys-Arg-Arg-B(OH)2, reported to bind DENV2 protease tightly (Ki value = 43 nM),15 uses a boronic acid as a warhead for the catalytic serine with the hydroxyl group mimicking the transition state. Because most P site inhibitors are highly charged and polar, designing inhibitors forming hydrophobic interactions with the prime site pocket (P3′ and P4′) can help adjust the hydrophobicity of these compounds, to potentially increase their cell permeability. As binding is solely entropy driven, relatively rigid inhibitors with nonrotatable bonds or macrocycles may leverage the entropically driven interactions. Even though the binding enthalpies of substrate-sequence-based aprotinin variants are better than that of the native aprotinin, implying the binding is more specific, the intermolecular interactions need to be further improved. This improvement will possibly require establishing additional van der Waals packing interactions or hydrogen bonds by introducing non-natural amino acid side chains during inhibitor design. Designing peptide-based inhibitors starting from the favored amino acids identified here and rigidifying the peptide by cyclization to maintain the binding loop structure is a feasible approach, with subsequent optimization to increase both affinity and specificity. Future studies will need to explore and efficiently combine a series of these potential strategies for the design of DENV, or other flaviviruses such as Zika, protease inhibitors to ensure both potency and specificity.
MATERIALS AND METHODS
Protease Gene Construction, Protein Expression, and Purification
Synthetic DENV3 protease gene (cDNA encoding NS2B (cNS2B; aa 1394–1440) and NS3 protease (NS3pro185; aa 1476–1660) with a G4SG4 linker between) was constructed using codon optimization for protein expression in Escherichia coli. The linker between NS2B and NS3, which may affect protein conformational flexibility as shown for serotype 2 with NMR,30 was kept identical as in the construct of the available crystal structure (3u1j)10 to enable a direct comparison. Protease gene was constructed between BamH1 and Xho1 sites of pGEX6p1 (GE Healthcare). The plasmid was transformed into BL21(DE3) for protein expression. We followed the published protein expression and purification protocols.16
Aprotinin Gene Construction, Protein Expression, and Purification
The cDNA encoding SUMO and aprotinin with His6 tag at the N terminus was constructed between Nde1 and Sac1 of pET28a. The plasmid was transformed into BL21-(DE3) cells for protein expression. We followed the published aprotinin expression and purification protocols.31
Aprotinin Construct Gene Construction
Mutagenesis primers were used to engineer substrate residues corresponding to each cleavage site onto the aprotinin binding loop, and polymerase chain reaction was performed to amplify the DNA. The transformation, expression, and purification protocols of aprotinin constructs were the same as for wild-type aprotinin.31
Enzyme Inhibition Assay
A FRET-based enzymatic cleavage assay was used to measure inhibition constants of aprotinin constructs against DENV3 WT protease. DENV3 NS2B/NS3 protease (100 nM) was incubated with various concentrations of aprotinin for 60 min in 50 mM Tris assay buffer (20% glycerol, 1 mM CHAPS, pH 8.5).32 Proteolysis reactions were initiated by adding 5 µM protease substrate (21st Century Biochemicals) and monitored using the EnVision plate reader (PerkinElmer) at excitation and emission wavelengths of 340 and 490 nm, respectively. The protease substrate used in the enzymatic reaction was fluorescence labeled and had the amino acid sequence of the 2A-2B cleavage site Ac-[D-EDANS]KRRSWP[K-DABCYL]-amide based on the substrate previously used in dengue protease inhibition constant measurements (Bz-Nle-Lys-Arg-Arg-AMC),16 but extended to prime sites to mimic natural cleavage reaction. The initial cleavage reaction velocities were determined using nonlinear fit to one-phase association of the whole progress curves.33 Apparent inhibition constants (Ki) were obtained by nonlinear regression fitting of initial velocity versus inhibitor concentration to the Morrison equation using Prism 6 (GraphPad Software). Data were collected in triplicate and processed globally to calculate the shared inhibition constant and standard deviation.
Molecular Dynamics Simulations
All molecular dynamics simulations were performed in triplicate following previously published protocols.34 Briefly, aprotinin constructs were modeled on DENV3 protease binding pocket based on the aprotinin and DENV3 WT protease complex structure (PDB: 3u1j). The modeled structures were prepared for simulations by keeping the crystallographic waters within 4.0 Å of any protease or aprotinin atom but removing the buffer salts from the coordinate file. By using the Protein Preparation Tool from Schrodinger,35 hydrogen atoms were added to the structures and the optimal protonation states for the ionizable side chains were determined. The hydrogen-bonding network of the initial structures was optimized, and the structures were minimized in vacuum using the Impact refinement module with the OPLS2005 force field.36 The prepared systems were solvated in a truncated octahedron solvent box with the TIP3P water model extending 10 Å beyond the protein in all directions, using the System Builder utility. The overall charge was neutralized by adding the necessary number of counterions (Na+ or Cl−). Desmond was used in all simulations with OPLS2005 force field. Each simulation was carried out at 300 K and 1 bar for 100 ns, and the coordinates and energies were recorded every 5 ps.
Structural Analysis
Hydrogen bonds were determined using VMD.37 A hydrogen bond was defined by a distance between the donor and acceptor of <3 Å and a donor–hydrogen–acceptor angle of >150°.
The vdW contacts between the protease and aprotinin binding loop were calculated using a simplified Lennard-Jones potential V(r) = 4ε [(σ/r)12 − (σ/r)6], with the well depth (ε) and hard sphere diameter (σ) for each protease–aprotinin atom pair. The V(r) for all protease–aprotinin atom pairs was computed within 6 Å, and when the distance between nonbonded pairs was less than e, V(r) was considered equal to −e. More details of the rationale for this modification to the original 6–12 Lennard-Jones potential were described before.38 The total ΣV(r) of the protease–aprotinin complex was computed using this simplified potential for each nonbonded pair.
Isothermal Titration Calorimetry
The ITC experiments were performed using Microcal ITC200 (Malvern) at 20 °C with the protease in the sample cell and aprotinin constructs in the syringe. The assay buffer was 20 mM Tris, pH 8.5. By fitting the data with nonlinear regression using Origin 7.0, the change in enthalpy (ΔH) and corresponding dissociation constant (Kd) were determined. The changes in Gibbs free energy (ΔG) and entropy (ΔS) were derived from these values.
Supplementary Material
Acknowledgments
Funding
This work was supported by NIH R01 AI085051.
We thank Sagar Kathuria for help with CD spectroscopy.
ABBREVIATIONS USED
- DENV
dengue virus
- P site
N terminus of polyprotein cleavage site
- P′ site
C terminus of polyprotein cleavage site
- BL
binding loop
- AP
aprotinin
- Kd
dissociation constant
- Ki
inhibition constant
- MD
molecular dynamics
- RMSF
root-mean-square fluctuation
- vdW
van der Waals
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Circular dichroism spectra of aprotinin variants (Figure S1); sequence alignments of dengue and Zika proteases’ polyprotein cleavage sites (Table S1); relative enthalpic contribution to the binding free energy of aprotinin constructs versus WT aprotinin (Figure S2); thermodynamic parameters from ITC experiments (Table S2); overall binding loop van der Waals (vdW) contacts from simulations (Figure S3 and Table S3); relative enthalpic contribution to the binding free energy of aprotinin constructs versus WT aprotinin to DENV3 protease (Figure S4); overall binding loop vdW contacts with DENV3 protease calculated from MD simulations of WT-AP and aprotinin constructs (Figure S5); aprotinin constructs’ binding loop fluctuations correlate with affinity (Figure S6) (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Rico-Hesse R. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology. 1990;174:479–493. doi: 10.1016/0042-6822(90)90102-w. [DOI] [PubMed] [Google Scholar]
- 2.Green S, Rothman A. Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr. Opin. Infect. Dis. 2006;19:429–436. doi: 10.1097/01.qco.0000244047.31135.fa. [DOI] [PubMed] [Google Scholar]
- 3.WHO. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control: New Edition. Geneva, Switzerland: 2009. [PubMed] [Google Scholar]
- 4.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monath TP. Dengue: the risk to developed and developing countries. Proc. Natl. Acad. Sci. U. S. A. 1994;91:2395–2400. doi: 10.1073/pnas.91.7.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simmons CP. A Candidate Dengue Vaccine Walks a Tightrope. N. Engl. J. Med. 2015;373:1263–1264. doi: 10.1056/NEJMe1509442. [DOI] [PubMed] [Google Scholar]
- 7.Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 8.Bera AK, Kuhn RJ, Smith JL. Functional characterization of cis and trans activity of the Flavivirus NS2B-NS3 protease. J. Biol. Chem. 2007;282:12883–12892. doi: 10.1074/jbc.M611318200. [DOI] [PubMed] [Google Scholar]
- 9.Yusof R, Clum S, Wetzel M, Murthy HM, Padmanabhan R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J. Biol. Chem. 2000;275:9963–9969. doi: 10.1074/jbc.275.14.9963. [DOI] [PubMed] [Google Scholar]
- 10.Noble CG, Seh CC, Chao AT, Shi PY. Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virol. 2012;86:438–446. doi: 10.1128/JVI.06225-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falgout B, Pethel M, Zhang YM, Lai CJ. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991;65:2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chambers TJ, Weir RC, Grakoui A, McCourt DW, Bazan JF, Fletterick RJ, Rice CM. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. U. S. A. 1990;87:8898–8902. doi: 10.1073/pnas.87.22.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nitsche C, Steuer C, Klein CD. Arylcyanoacrylamides as inhibitors of the Dengue and West Nile virus proteases. Bioorg. Med. Chem. 2011;19:7318–7337. doi: 10.1016/j.bmc.2011.10.061. [DOI] [PubMed] [Google Scholar]
- 14.Yin Z, Patel SJ, Wang WL, Chan WL, Ranga Rao KR, Wang G, Ngew X, Patel V, Beer D, Knox JE, Ma NL, Ehrhardt C, Lim SP, Vasudevan SG, Keller TH. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg. Med. Chem. Lett. 2006;16:40–43. doi: 10.1016/j.bmcl.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 15.Yin Z, Patel SJ, Wang WL, Wang G, Chan WL, Rao KR, Alam J, Jeyaraj DA, Ngew X, Patel V, Beer D, Lim SP, Vasudevan SG, Keller TH. Peptide inhibitors of Dengue virus NS3 protease. Part 1: Warhead. Bioorg. Med. Chem. Lett. 2006;16:36–39. doi: 10.1016/j.bmcl.2005.09.062. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Lim SP, Beer D, Patel V, Wen D, Tumanut C, Tully DC, Williams JA, Jiricek J, Priestle JP, Harris JL, Vasudevan SG. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 2005;280:28766–28774. doi: 10.1074/jbc.M500588200. [DOI] [PubMed] [Google Scholar]
- 17.Shiryaev SA, Kozlov IA, Ratnikov BI, Smith JW, Lebl M, Strongin AY. Cleavage preference distinguishes the two-component NS2B-NS3 serine proteinases of Dengue and West Nile viruses. Biochem. J. 2007;401:743–752. doi: 10.1042/BJ20061136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chanprapaph S, Saparpakorn P, Sangma C, Niyomrattanakit P, Hannongbua S, Angsuthanasombat C, Katzenmeier G. Competitive inhibition of the dengue virus NS3 serine protease by synthetic peptides representing polyprotein cleavage sites. Biochem. Biophys. Res. Commun. 2005;330:1237–1246. doi: 10.1016/j.bbrc.2005.03.107. [DOI] [PubMed] [Google Scholar]
- 19.Mueller NH, Yon C, Ganesh VK, Padmanabhan R. Characterization of the West Nile virus protease substrate specificity and inhibitors. Int. J. Biochem. Cell Biol. 2007;39:606–614. doi: 10.1016/j.biocel.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 20.Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- 21.Schrodinger LLC. Release 2015-3: Maestro, version 10.3. New York: 2015. [Google Scholar]
- 22.Pettit SC, Sheng N, Tritch R, Erickson-Viitanen S, Swanstrom R. The regulation of sequential processing of HIV-1 Gag by the viral protease. Adv. Exp. Med. Biol. 1998;436:15–25. doi: 10.1007/978-1-4615-5373-1_2. [DOI] [PubMed] [Google Scholar]
- 23.Pettit SC, Henderson GJ, Schiffer CA, Swanstrom R. Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J. Virol. 2002;76:10226–10233. doi: 10.1128/JVI.76.20.10226-10233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arias CF, Preugschat F, Strauss JH. Dengue 2 virus NS2B and NS3 form a stable complex that can cleave NS3 within the helicase domain. Virology. 1993;193:888–899. doi: 10.1006/viro.1993.1198. [DOI] [PubMed] [Google Scholar]
- 25.Zhang L, Padmanabhan R. Role of protein conformation in the processing of dengue virus type 2 nonstructural polyprotein precursor. Gene. 1993;129:197–205. doi: 10.1016/0378-1119(93)90269-9. [DOI] [PubMed] [Google Scholar]
- 26.Assenberg R, Mastrangelo E, Walter TS, Verma A, Milani M, Owens RJ, Stuart DI, Grimes JM, Mancini EJ. Crystal structure of a novel conformational state of the flavivirus NS3 protein: implications for polyprotein processing and viral replication. J. Virol. 2009;83:12895–12906. doi: 10.1128/JVI.00942-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chappell KJ, Stoermer MJ, Fairlie DP, Young PR. West Nile Virus NS2B/NS3 protease as an antiviral target. Curr. Med. Chem. 2008;15:2771–2784. doi: 10.2174/092986708786242804. [DOI] [PubMed] [Google Scholar]
- 28.Aleshin AE, Shiryaev SA, Strongin AY, Liddington RC. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 2007;16:795–806. doi: 10.1110/ps.072753207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, Hart A, Hesk D, Ingravallo P, Jiang C, Kong R, Lu J, Pichardo J, Prongay A, Skelton A, Tong X, Venkatraman S, Xia E, Girijavallabhan V, Njoroge FG. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother. 2006;50:1013–1020. doi: 10.1128/AAC.50.3.1013-1020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YM, Gayen S, Kang C, Joy J, Huang Q, Chen AS, Wee JL, Ang MJ, Lim HA, Hung AW, Li R, Noble CG, Lee le T, Yip A, Wang QY, Chia CS, Hill J, Shi PY, Keller TH. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013;288:12891–12900. doi: 10.1074/jbc.M112.442723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun Z, Lu W, Jiang A, Chen J, Tang F, Liu JN. Expression, purification and characterization of aprotinin and a human analogue of aprotinin. Protein Expression Purif. 2009;65:238–243. doi: 10.1016/j.pep.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J. Biol. Chem. 2001;276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- 33.Salykin A, Kuzmic P, Kyrylenko O, Musilova J, Glatz Z, Dvorak P, Kyrylenko S. Nonlinear regression models for determination of nicotinamide adenine dinucleotide content in human embryonic stem cells. Stem Cell Rev. 2013;9:786–793. doi: 10.1007/s12015-013-9454-3. [DOI] [PubMed] [Google Scholar]
- 34.Ozen A, Lin KH, Kurt Yilmaz N, Schiffer CA. Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc. Natl. Acad. Sci. U. S. A. 2014;111:15993–15998. doi: 10.1073/pnas.1414063111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 36.Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 37.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics. 1996;14(33–38):27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 38.Ozen A, Haliloglu T, Schiffer CA. Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J. Mol. Biol. 2011;410:726–744. doi: 10.1016/j.jmb.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.