

Fluorescent protein tags are the primary tool for labeling gene products and analyzing their dynamics using live-cell imaging. A quantitative comparison is made of fluorescent protein brightness and photostability in an in vivo animal model system, and tools and recommendations are given for optimal fluorescent protein selection.
Abstract
Fluorescent protein tags are fundamental tools used to visualize gene products and analyze their dynamics in vivo. Recent advances in genome editing have expedited the precise insertion of fluorescent protein tags into the genomes of diverse organisms. These advances expand the potential of in vivo imaging experiments and facilitate experimentation with new, bright, photostable fluorescent proteins. Most quantitative comparisons of the brightness and photostability of different fluorescent proteins have been made in vitro, removed from biological variables that govern their performance in cells or organisms. To address the gap, we quantitatively assessed fluorescent protein properties in vivo in an animal model system. We generated transgenic Caenorhabditis elegans strains expressing green, yellow, or red fluorescent proteins in embryos and imaged embryos expressing different fluorescent proteins under the same conditions for direct comparison. We found that mNeonGreen was not as bright in vivo as predicted based on in vitro data but is a better tag than GFP for specific kinds of experiments, and we report on optimal red fluorescent proteins. These results identify ideal fluorescent proteins for imaging in vivo in C. elegans embryos and suggest good candidate fluorescent proteins to test in other animal model systems for in vivo imaging experiments.
INTRODUCTION
For more than two decades, cell and developmental biologists have used genetically encoded fluorescent protein fusion tags to visualize proteins in living cells and organisms. Efforts to engineer and discover superior fluorescent proteins have resulted in variants with diverse emission wavelengths and photophysical properties (Tsien, 1998; Matz et al., 1999; Shaner et al., 2004, 2007, 2013; Shaner, 2014; Shcherbo et al., 2009). The color, brightness, and photostability of a fluorescent protein are critical parameters to consider for experiments in which proteins will be imaged in vivo (Shaner et al., 2005; Shaner, 2014; Davidson and Campbell, 2009). However, most brightness and photostability measurements are made with purified fluorescent proteins in vitro (Shaner et al., 2005; Cranfill et al., 2016). Although this approach provides information about the intrinsic optical properties of each fluorescent protein, it does not replicate many of the conditions of an in vivo biological system.
Historically, many methods used to express fluorescently tagged proteins resulted in nonphysiological levels of proteins of interest, limiting the interpretation of some experiments (Huang et al., 2000; Krestel et al., 2004; Doyon et al., 2011). However, genome engineering techniques based on the CRISPR/Cas9 system have recently made it possible to edit more precisely the genomes of diverse cell types and organisms (Doudna and Charpentier, 2014; Gilles and Averof, 2014; Harrison et al., 2014; Hsu et al., 2014; Peng et al., 2014) and routinely insert fluorescent protein tags into endogenous genomic loci in some organisms, as has long been standard in yeast (Dickinson et al., 2013, 2015; Auer et al., 2014; Bassett et al., 2014; Ma et al., 2014; Paix et al., 2014; Xue et al., 2014; Aida et al., 2015; Perry and Henry, 2015; Ratz et al., 2015). With this technological advance comes an increase in the need for information about the best fluorescent proteins to use for in vivo imaging studies. Fortunately, advances in genome editing techniques have also created an opportunity to close this gap in knowledge by facilitating the comparison of fluorescent proteins in vivo.
Our goal in this study was to make a systematic comparison of some of the brightest known fluorescent proteins that would answer the question: What fluorescent protein should one use in vivo for a given experiment? A previous systematic analysis of fluorescent proteins performed in Saccharomyces cerevisiae revealed clear information about which tags to use in vivo in yeast (Lee et al., 2013). Since that study, new fluorescent proteins have been characterized, including some reported to be brighter than green fluorescent protein (GFP; Shaner et al., 2013). Here we report direct comparisons of monomeric green (GFP, mNeonGreen [mNG]), yellow (mNG, monomeric yellow fluorescent protein for energy transfer [mYPet]), and red (TagRFP-T, mRuby2, mCherry, mKate2) fluorescent proteins in vivo in a multicellular animal model organism. We used CRISPR/Cas9-triggered homologous recombination in Caenorhabditis elegans to express the same transgene tagged with optimized versions of various fluorescent proteins from the same genomic locus. This allowed us to quantitatively compare the brightness and photostability of these fluorescent proteins in embryos imaged under typical experimental conditions. Because we made observations in vivo, encapsulated in our measurements are the variables that govern a given fluorescent protein’s performance, including intrinsic brightness, transcript or protein stability, and maturation rate, all of which contribute to practical use in live-imaging experiments.
Our findings provide quantitative data that are useful for choosing which fluorescent proteins to use for in vivo experiments in C. elegans. The results suggest a set of candidate fluorescent proteins for testing in other model systems, and, more generally, they demonstrate the value of testing fluorescent protein performance in vivo. We also give novel tools for the field, including constructs containing optimized fluorescent proteins and an Excel-based tool to assist investigators in choosing the best fluorescent proteins to use with their imaging resources.
RESULTS AND DISCUSSION
Predictions of fluorescent protein brightness
Before making in vivo measurements, we made quantitative predictions about which fluorescent proteins were expected to be brightest. We calculated the predicted brightness of each fluorescent protein by the product of the quantum yield and extinction coefficient as reported in the literature (Figure 1A; Yang et al., 1996; Shaner et al., 2004, 2008, 2013; Nguyen and Daugherty, 2005; Shcherbo et al., 2009; Lam et al., 2012; Lee et al., 2013). Because imaging conditions such as the excitation wavelength and emission filter sets used affect the observed brightness of a fluorescent protein, we sought to use these values to make more useful predictions of fluorescent protein brightness for directly comparing with our results.
FIGURE 1:

Predicted brightness of fluorescent proteins and embryo autofluorescence. (A) Reported brightness for fluorescent proteins at peak excitation wavelengths. (B) Predicted brightness of fluorescent protein comparisons performed in Figure 2. Excitation and emission wavelengths are at the top. (C) Embryo autofluorescence. Lines are averages of multiple embryos, and small points are individual embryos acquired using a spectral detector. Large points represent spinning-disk confocal autofluorescence data.
To facilitate the visual and quantitative evaluation of fluorescent protein spectra with the specific laser lines and filter sets that are used by us and others, we developed a simple and customizable Microsoft Excel-based tool that we call the Spectrum Viewer (Supplemental File S1). Using this tool, we calculated a predicted brightness for each fluorescent protein by integrating the portion of the fluorescent protein emission peak under our emission filter and multiplying by the quantum yield (Figure 1B). We then used the Spectrum Viewer to plot the normalized absorbance and emission spectra for the fluorescent proteins in our comparisons with the excitation wavelength and emission filter sets we used for imaging (Figure 2, A–D, third column). The Spectrum Viewer is available as Supplemental File S1.
FIGURE 2:

In vivo fluorescent protein brightness. (A–D) Left, embryos mounted side by side and imaged under the same conditions used for quantification. Center, quantification of each comparison. Each data point represents a single embryo. Black bars indicate the mean and 95% CIs. Right, excitation (top) and emission spectra (bottom) of compared fluorescent proteins. The illumination wavelength (Ex., blue line) and filter sets used for detection are indicated (Em., gray shading).
Measuring C. elegans embryo autofluorescence at different wavelengths
Because single-copy fluorescent transgenes sometimes produce weak fluorescence signal in vivo, we quantitatively assessed the endogenous autofluorescence levels of C. elegans embryos. We measured autofluorescence using two different techniques. In one case, we used a spectral detector to measure autofluorescence at various emission wavelengths. In the other, we used a spinning-disk confocal microscope with standard lasers and filter sets and an electron-multiplying charge-coupled device (EM-CCD) camera. The results of both experiments were consistent (Figure 1C) and are likely to be similar on other comparable imaging systems. We found autofluorescence to be most prominent under 488-nm excitation across a broad range of emission wavelengths (Figure 1C). Thus, when expressed at low levels, fluorescent proteins excited by 488-nm light, including GFP, will have significant background noise in C. elegans embryos. Embryos had considerably less autofluorescent background with 514-nm excitation (Figure 1C). This suggests that when imaging proteins expressed at low levels in embryos, 514-nm excitation and yellow fluorescent proteins such as mNeonGreen and mYPet may be superior to GFP and 488-nm illumination. We found autofluorescence to be lowest using 405- and 442-nm excitation, but we generally avoid live imaging in these wavelengths due to increased phototoxicity.
Generating single-copy transgene knock-ins
To directly compare fluorescent proteins in vivo, we used CRIPSR/Cas-9 to generate single-copy transgene knock-in strains expressing distinct fluorescent proteins. Constructs used to create these strains were identical except for the fluorescent protein sequences encoded in each case, and each transgene was inserted into the same locus in the C. elegans genome (Figure 2; see Materials and Methods). We confirmed the knock-ins by observation of the predicted fluorescence localization pattern at the plasma membrane, and we confirmed that knock-ins were single copy by PCR genotyping and sequencing (Supplemental Figure S1B).
In vivo fluorescent protein brightness
To assess the brightness of this set of fluorescent protein transgenes in vivo, we imaged staged C. elegans embryos, in some cases mounted side by side for direct comparisons, by spinning-disk confocal microscopy. We first compared GFP and mNG by quantifying the fluorescence from embryos illuminated with 488-nm excitation. Although mNG was predicted to be brighter than GFP based on in vitro data (Figure 1, A and B), we found that the GFP signal was nearly twice as bright as the mNG signal in vivo (Figure 2A). Mean values within each comparison are significantly different (p < 0.05) except where indicated with ns (not significantly different; determined by Student’s t test with Welch’s correction), and all significance values (p values) are reported in Supplemental Figure S2B.
With 514-nm illumination, mYPet was also brighter than mNG (Figure 2B). Although our calculations predicted that mYPet would be almost twice as bright as mNG (Figure 1B), we observed mYPet to be about four times as bright as mNG on average (Figure 2B). The data from the comparisons of mNG with GFP and mYpet suggest that mNG is not as bright in vivo as we predicted based on the published extinction coefficient and quantum yield (Shaner et al., 2013; Figures 1B and 2, A and B).
Next we examined the brightness of four red fluorescent proteins (TagRFP-T, mRuby2, mCherry, and mKate2). We performed experiments with two different emission filter sets, 561LP and 630/75BP, which are well matched to some or all of these red fluorescent proteins. The 561LP emission filter is optimal because it collects the majority of the peak emission for each fluorescent protein (Figure 2C). A bandpass filter, such as the 630/75BP, is less optimal (compare right column, Figure 2, C and D), although it might be useful for decreasing spectral overlap for two- or three-color imaging.
Using 561-nm illumination we measured the brightness of the four red fluorescent proteins. We found that TagRFP-T was the brightest using the 561LP filter set (Figure 2C). Using the 630/75BP filter set, the average fluorescence intensity of TagRFP-T was indistinguishable from that of mCherry (Figure 2D). These results are consistent with the orange-shifted emission spectrum of TagRFP-T and with our calculated predictions for these fluorescent proteins (Figures 1B and 2, C and D). mRuby2, which was predicted to be the brightest of the four red fluorescent proteins (Figure 1B), was the least bright regardless of the emission filter set we used (Figure 2, C and D). Taken together, these data reveal fluorescent protein brightness in vivo that did not always match predictions made using parameters measured in vitro.
Variation in fluorescent protein brightness between single-copy transgenes
Because we predicted that mNG would be ∼1.8 times brighter than GFP, we were surprised to find that GFP embryos were significantly brighter than mNG embryos (Figures 1B and 2A). Germline silencing in C. elegans can have heterogeneous effects on certain single-copy transgenes (Shirayama et al., 2012). Consequently fluorescent protein transgenes that are in every other way identical could be expressed at different levels, causing discrepancies between predicted and observed brightness. To ask whether differences in fluorescent protein abundance could account for the differences in fluorescence intensity we observed, we analyzed protein levels in each of our single-copy transgenic strains by Western blot (Supplemental Figure S1). We observed approximately twofold higher levels of mex-5-driven GFP::PH protein compared with mNG::PH protein (Supplemental Figure S1C; paired t test, p = 0.0408), which may be due to partial transgene silencing or posttranscriptional regulation of these transgenes.
To further investigate the discrepancy between our predictions and observations, we compared a second set of identical GFP and mNG single-copy transgene knock-in strains. These fluorescent proteins were fused to the C-terminus of a histone gene (his-58). As expected, the resulting fluorescence was brightest in nuclei (Figure 3A). To control for effects of cell cycle timing on histone protein abundance, we staged embryos to within 3 min of one another. We measured the fluorescence intensity in the nucleus of one embryonic cell (the EMS cell) in each embryo and found that the average fluorescence intensities of the GFP-histone–expressing embryos and the mNG-histone embryos were not significantly different (Figure 3A). Although in our initial comparison of membrane-localized transgenes we found that GFP-expressing embryos were significantly brighter than those expressing mNG (Figure 2A), both results suggest that in early C. elegans embryos, mNG is not as bright compared with GFP, as predicted (Figure 1B).
FIGURE 3:

Comparisons of GFP and mNeonGreen in single-copy transgenic strains and as knock-ins in endogenous genes. (A–C) Each data point represents a single embryo or animal; black bars represent the mean and 95% CIs. (A) Embryos expressing histone–fluorescent protein fusions. Fluorescence intensity of the EMS cell nucleus was measured (white arrowheads). (B) Young adult worms expressing membrane tag-fluorescent protein fusions in the pharynx (white arrowheads). The insert is a DIC image of the worms. (C) gex-3 knock-in, (D) rap-1 knock-in, and (E) nmy-2 knock-in and wild-type embryos were imaged using 488- and 514-nm illumination. Dashed lines outline embryos not visible under the given imaging conditions.
Because protein levels in the C. elegans germline and early embryo can be affected by silencing mechanisms (Shirayama et al., 2012), we compared GFP and mNG in a C. elegans tissue that has not been reported to exhibit the silencing. We replaced the germline promoter in our original GFP and mNG::PH repair template constructs with the myo-2 promoter, which drives expression in the pharynx (Okkema et al., 1993), and generated single-copy transgene knock-ins at the same genomic locus used for our initial comparison. We imaged staged worms and quantified GFP and mNG fluorescence and again found no significant difference between average GFP and mNG intensities (Figure 3B). These data are consistent with our findings in early embryos and are consistent with the possibility that factors outside of germline silencing may also play a role in determining the observed fluorescence from single-copy transgenes.
Comparing green fluorescent proteins as endogenous tags
We next set out to compare GFP and mNG inserted into existing genes at their endogenous loci. We picked three genomic loci for which N-terminal mNG knock-ins already exist—gex-3, rap-1, and nmy-2 (Dickinson et al., 2015)—and we generated identical GFP knock-ins at those loci by the same method used to create the original mNG strains. We imaged embryos from the paired strains side by side at the same developmental stage as in our previous comparisons (Figure 3, C–E). Using 488-nm illumination, we found no consensus in our comparisons: mNG::GEX-3 was brighter than GFP:GEX-3; mNG and GFP::RAP-1 were equally bright; and GFP::NMY-2 was brighter than mNG::NMY-2 (Figure 3, C–E).
Because background embryo autofluorescence is higher at 488-nm illumination (Figure 1C), we also imaged these embryos using 514-nm illumination. Background autofluorescence is most prevalent when fluorescent protein signal levels are low. Therefore we were most interested, in this comparison, in gex-3 knock-in embryos because we had observed that it has the lowest expression of the three genes we tagged (Figure 3, C–E). Although we could not quantitatively compare fluorescence intensity of embryos illuminated with 488- versus 514-nm wavelengths due to differences in image acquisition setup (e.g., laser power, filter sets), we observed that mNG::GEX-3 imaged with 514-nm illumination gave qualitatively the best results under these imaging conditions (Figure 3C). The wild-type embryos in each image show the level of autofluorescent background contributed under the given imaging conditions.
Photostability of fluorescent proteins in vivo
The brightness of a fluorescent protein, together with its photobleaching rate, determines how useful a fluorescent protein is for time-lapse imaging (Shaner et al., 2005; Shaner, 2014; Davidson and Campbell, 2009). To test the rate of photobleaching of the fluorescent proteins used in our initial comparison in Figure 2, we imaged embryos over time under continuous illumination (Figure 4, A–C). Fluorescence intensities were normalized to initial brightness measured for each embryo, and averages were plotted for each strain over time (Figure 4, A–C, left). Each photobleaching curve was fit to a one-phase exponential decay, and the half-life was calculated (Supplemental Figure S3B). To estimate a photon budget (Lee et al., 2013), or the amount of signal emitted by each fluorescent protein over time, we integrated the fluorescence intensity measured for each embryo up to 50% of its initial intensity (Figure 4, A–C, right).
FIGURE 4:
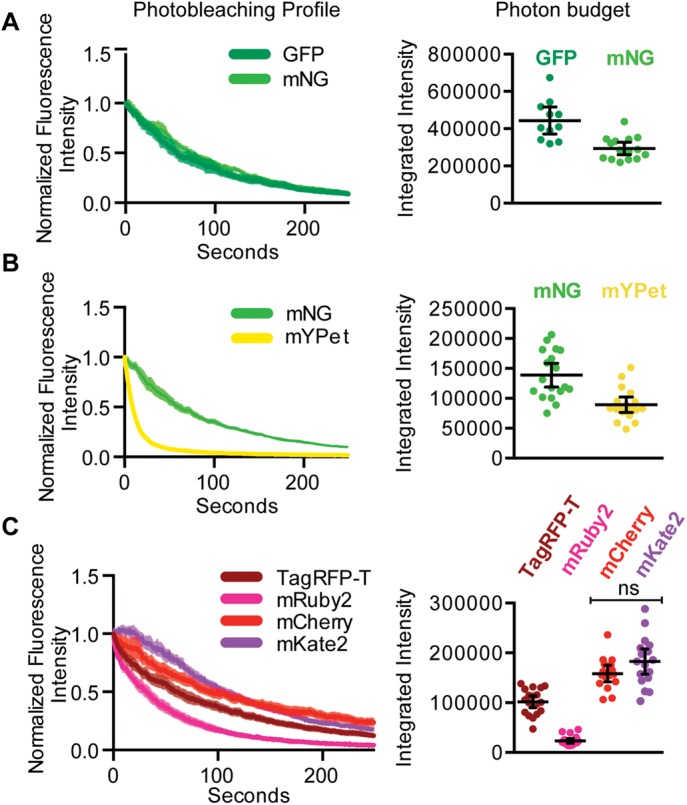
In vivo fluorescent protein photostability. (A–C) Fluorescence intensity was measured in embryos over time. Photobleaching profile and photon budget are compared for membrane-associated fluorescent protein fusions. Each data point represents a single embryo, and the black bars represent the mean and 95% CIs.
GFP and mNG displayed similar photobleaching half-lives, with mNG being slightly more photostable (Figure 4A and Supplemental Figure S3B). However, because the GFP embryos are brighter, on average, the integrated intensity, or photon budget, of the GFP embryos was slightly higher than that of mNG (Figure 4A). mYPet was observed to photobleach far faster than mNG, as expected (Supplemental Figure S3B; Shaner et al., 2013). Because mYPet is significantly brighter than mNG, its photon budget is only slightly less than that of mNG (Figure 4B). Of the red fluorescent proteins we tested, mKate2 had the slowest average photobleaching rate and about the same photon budget as mCherry (Figure 4C). The photobleaching profile of mKate2 suggests that it exhibits kindling (photoactivation) in the first few frames of illumination (Figure 3C and Supplemental Figure S3). Photoactivation was not reported in the initial characterization of mKate2 but had been observed for its precursor protein, mKate (Shcherbo et al., 2009). We conclude that mRuby2 and mYPet exhibited relatively poor photostability in vivo and that GFP, mNG, and mKate2 were the most photostable.
Summary and recommendations
Our results suggest specific recommendations for fluorescent proteins to use in in vivo experiments in C. elegans embryos, forming a baseline for comparisons in other in vivo systems. In general, we observed a lower-than-expected brightness for mNG. In some comparisons, GFP and mNG performed similarly (Figures 2A and 3), but, surprisingly, in some experiments, each was clearly brighter than the other. GFP was brighter than mNG in a germline transgene expressed at high levels and in an nmy-2 endogenous tag (Figures 2A and 3E). However, mNG was brighter than GFP in the more weakly expressed gex-3 endogenous tag (Figure 3C). These results suggest that GFP and mNG may each be ideal in different contexts and that testing may be required to identify the best green fluorescent protein for a specific experiment. mYPet was significantly brighter than mNG, but its high rate of photobleaching makes it an unattractive choice for long-term imaging (Figures 2B and 4B). The four red fluorescent proteins we tested were only compared under one set of conditions, which is a limitation of this study. However, TagRFP-T, mCherry, and mKate2 performed similarly in terms of brightness, and mKate2 had the superior photobleaching dynamics in vivo (Figures 2, C and D, and 4C).
Our measurements of autofluorescence in the early C. elegans embryo highlight the value of taking such measurements before designing in vivo imaging experiments. For C. elegans embryos, using 488-nm illumination gave higher background than imaging using 514-nm illumination (Figure 1C). Therefore, for genes with low expression levels, better signal-to-noise ratios may be achieved using a yellow fluorescent protein and exciting with 514-nm illumination rather than a green fluorescent protein and 488-nm illumination (Figure 3C). Because of the rapid photobleaching we observed for mYPet (Figure 4B), we would choose mNG to tag proteins expressed at lower levels in C. elegans embryos for long-term, live-cell imaging. Although these measurements are informative for considering which fluorescent proteins to use in vivo, because of variability in detector sensitivity and emission light scattering at different wavelengths, they may not reflect the actual autofluorescent properties of C. elegans embryos.
We observed several differences between our predictions and our in vivo measurements of fluorescent protein brightness, most notably for mNeonGreen and for mRuby2 (Figures 1B and 2). These results demonstrate the value of direct in vivo comparisons for selecting fluorescent proteins to use in vivo. Because we measured fluorescent protein performance head to head, in vivo, we expect that the differences in brightness we observed were due to the combination of intrinsic differences in fluorescent protein brightness and the cumulative effects of any regulatory mechanisms (at the mRNA or protein level) at play in the biological system that we used. Cases in which our quantitative expectations based on the intrinsic properties of fluorescent proteins were violated suggest that other regulatory mechanisms are indeed a factor in determining fluorescent protein performance.
Although identifying variables other than intrinsic brightness that might affect fluorescent protein brightness in vivo is outside the scope of this work, there are a variety of interesting possibilities to consider. One possibility is that coding sequence differences in fluorescent proteins result in differential silencing of the transgenes we compared. Germline silencing in C. elegans has been shown to have heterogeneous effects on certain single-copy transgenes (Shirayama et al., 2012). Consequently fluorescent protein transgenes that are in every other way identical could be expressed at different levels, causing discrepancies between predicted and observed brightness. Any effects of silencing on expression and observed brightness would likely differ in different model systems. Another possibility is that fluorescent proteins mature and decay at different rates in vivo than in vitro (Iizuka et al., 2011; Hebisch et al., 2013). Temperature could affect the performance of fluorescent proteins designed for expression in mammalian systems (37°C), as C. elegans is maintained and imaged near room temperature (20–25°C).
Identifying the cellular and organismal mechanisms underlying the context-specific performance of fluorescent proteins is important for understanding how universal findings in any one system may be. At present, it is unknown how applicable the specific results of this study are in model systems beyond C. elegans. The fluorescent proteins we found to be optimal were not the same as in a comprehensive in vivo comparison of fluorescent proteins in yeast (Lee et al., 2013), suggesting some potential for organism-specific rules for governing fluorescent protein performance in vivo. Future studies in diverse systems are needed to reveal whether there is a universally best set of fluorescent proteins. We used exclusively spinning-disk confocal microscopy for our comparisons. However, differences in illumination source and detectors used in different light microscopy techniques (e.g., wide field, total internal reflection fluorescence, light sheet) might change the observed performance of fluorescent proteins in live-imaging experiments.
This study gives information of practical value about which fluorescent proteins to use for in vivo experiments, as well as a tool for researchers to use to evaluate the spectra of different fluorescent proteins relative to their own imaging resources. The findings are especially applicable for experiments in C. elegans and suggest the value of performing similar experiments in other model systems, as well as offer good candidate fluorescent proteins to test.
MATERIALS AND METHODS
C. elegans strains and maintenance
All C. elegans strains used in this study are listed in Supplemental Figure S1 and were handled using standard techniques (Brenner, 1974). The strains were raised at 25°C in incubators in the dark and fed Escherichia coli OP50 except where otherwise indicated. The HT1593 (unc-119(ed3) III) strain, used as the parent to the LP306, LP274, LP402, LP193, LP307, LP308, LP401, LP403, and LP404 strains generated in this study, was raised at 15°C and fed E. coli HB101 before injection (Hochbaum et al., 2010; Dickinson et al., 2013).
Fluorescent protein selection
Because of their current widespread use, we chose to compare GFP and mCherry with newer green and red fluorescent proteins that are less commonly used but have been described as having superior brightness and/or photostability. We used a GFP variant, GFP S65C, commonly used in C. elegans, which we refer to as GFP (Green et al., 2008). S65C and S65T (eGFP) variants perform similarly (Heim and Tsien, 1996), and a previous in vivo study of fluorescent proteins in yeast reported that S65T outperformed certain green fluorescent protein variants (such as Clover and Emerald) in a direct comparison (Lee et al., 2013). mNG is a newer, monomeric green fluorescent protein (peak excitation, ∼506 nm) that is reported to be up to three times as bright and more photostable than eGFP in vitro (Shaner et al., 2013). We therefore compared mNG to GFP in our in vivo system. To assess the practical value of mNG’s yellow-shifted excitation spectrum (Shaner et al., 2013), we compared mNG with a yellow fluorescent protein, mYPet—the brightest reported yellow fluorescent protein (Nguyen and Daugherty, 2005). We chose three red fluorescent proteins to compare with mCherry: TagRFP-T, mKate2, and mRuby2. A direct comparison in yeast found that all three were brighter than mCherry in vivo (Lee et al., 2013). These red fluorescent proteins range in peak emission from 584 to 633 nm (Shaner et al., 2008; Shcherbo et al., 2009; Lam et al., 2012), making them useful in combination with different fluorescent proteins for two- or three-color imaging.
Fluorescent protein optimization and repair template construction
Single-copy transgenic knock-in strains (LP306, LP274, LP402, LP193, LP307, LP308, LP401, LP403, LP404) were generated using the method described in Dickinson et al. (2013). Fluorescent protein sequences were obtained from Heim and Tsien (1996), Shaner et al. (2004, 2008, 2013), Nguyen and Daugherty (2005), Shcherbo et al. (2009), and Lam et al. (2012). mNeonGreen was licensed from Allele Biotechnology. To increase the monomeric character of YPet, we introduced a well-characterized mutation to the original YPet sequence (A206K) to generate mYPet (Zacharias et al., 2002; Ohashi et al., 2007).
Repair template constructs were identical, except for the sequences of the fluorescent proteins tested. Each transgene construct consisted of a germline promoter sequence (Pmex-5) driving the expression of a fluorescent protein fused to the N-terminus of the same polypeptide: the pleckstrin homology domain from phospholipase C-δ1 (PH domain) and a 2× Flag epitope tag. The PH domain localizes to the plasma membrane by binding phosphatidylinositol 4,5-bisphosphate (Audhya et al., 2005). Because many of our source sequences are optimized for expression in mammalian systems, we sought to mitigate any effects that the presence of codons rarely used in C. elegans might have on translational efficiency. Therefore the nucleotide sequences of the fluorescent proteins and PH domain were optimized for expression in C. elegans using the C. elegans Codon Adapter (Codon Adaptation Index, ∼1; Redemann et al., 2011). Synthetic C. elegans introns were added to each fluorescent protein to facilitate expression of the transgenes (Fire et al., 1990). The fluorescent protein genes were synthesized in ∼500–base pair overlapping gBlock fragments (Integrated DNA Technologies), assembled using Gibson Assembly Master Mix (NEB, Ipswich, MA), PCR amplified, and cloned using the Zero Blunt TOPO PCR cloning kit (Invitrogen, Carlsbad, CA).
All repair template constructs were made using a derivative of the pCFJ150 vector backbone modified for Cas9-mediated homologous recombination (Frøkjær-Jensen et al., 2008; Dickinson et al., 2013). The mex-5 promoter, the C. elegans sequence-optimized mNeonGreen fluorescent protein and PH domain, and the tbb-2 3′ untranslated region (UTR) were added using Gibson Assembly (NEB) to create the vector pAP006. To generate repair templates with different fluorescent protein sequences, pAP006 was amplified into a linear fragment using the forward primer 5′ CACGGACTCCAAGACGAC (binds after the mex-5 promoter) and reverse primer 5′ TCTCTGTCTGAAACATTCAATTGATTATC (binds at the start of the C. elegans optimized PH domain). Fluorescent protein genes were amplified using gene-specific primers with minimum 30–base pair overlapping sequence to the parent vector fragment (forward, 5′ CGATAATCAATTGAATGTTTCAGACAGAGA + FP sequence; reverse, 5′ GCCGGCCACGGACTCCAAGACGACCCAGACCTCCAAG + FP sequence). The vector backbone fragment and fluorescent protein genes were assembled using Gibson Assembly (NEB). The repair templates for strains LP403 and LP404 were made using a similar strategy to exchange the mex-5 promoter for the myo-2 promoter sequence.
We deposited constructs containing the optimized fluorescent proteins in Addgene. Addgene detected an error in our original mKate2 plasmid that we used to generate the strain used in this study (LP307). The mutation causes a nonsynonymous change in the PH domain of this construct (A735T). Because the mutation was not in the fluorescent protein and the construct localizes to the plasma membrane, we predicted that the mutation would have no effect on observed fluorescence. The mutation was corrected, and the two strains were compared side by side; no difference in fluorescence intensity was detected (Supplemental Figure S3C).
Insertion and confirmation of transgene knock-ins
Single-copy transgenes were inserted into the C. elegans genome via Cas9-triggered homologous recombination, using the reagents and methods described in Dickinson et al. (2013). The transgenes were inserted near the ttTi5605 MosI insertion site on C. elegans chromosome II. This site has been used for both CRISPR/Cas-9 and Mos1 transposon–based transgene insertions and is known to permit the expression of transgenes in the germline (Frøkjær-Jensen et al., 2008; Dickinson et al., 2013). We used a guide RNA with the target sequence 5′-GATATCAGTCTGTTTCGTAA (Dickinson et al., 2013). Single-copy knock-ins were confirmed by rescue of the HT1593 uncoordinated phenotype, observation of the predicted fluorescence localization pattern at the plasma membrane, and PCR genotyping (Figure 2 and Supplemental Figure S1B). PCR genotyping was performed on genomic DNA extracted from putative knock-in animals, using primers outside the insertion site (5′-AGGCAGAATGTGAACAAGACTCG and 5′-ATCGGGAGGCGAACCTAACTG) as described in Dickinson et al. (2013). We further confirmed the integrity of the inserts by sequencing the promoter, coding regions, and 3′ UTRs of each strain. All seven transgenes resulted in minimal embryonic lethality at 25°C (Supplemental Figure S2A).
JA1699 was made with standard MosSCI methods using pJA449 (mtm-3 associated HOT core/his-58/mNeonGreen::tbb-2 3′ UTR), which was constructed using triple gateway into pCFJ150 using the mtm-3 promoter in pDONRP4P1R, pJA273 (his-58 coding in pDONR221), and pJA448 (C. elegans optimized mNeonGreen::tbb-2 3′ UTR in pDONR P2R-P3; Zeiser et al., 2011; Dickinson et al., 2013). The construction of strain JA1610 is described in Chen et al. (2014). LP431 (GFP::gex-3), LP574 (GFP::rap-1), and LP572 (GFP::nmy-2) were made using the strategy described for LP362 (mNG::gex-3) in Dickinson et al. (2015). PCR genotyping was performed to confirm knock-ins.
Predicted-brightness calculation
We calculated the predicted brightness of each fluorescent protein imaged with excitation and emission settings that match the settings we used for our comparisons. For each fluorescent protein at a given wavelength, we quantified the fraction of the total emission peak covered by the emission filter and multiplied by the brightness at a given illumination wavelength. To determine the fraction of the total emission peak, we took the sum of the normalized emission values over the range of the emission filter used for imaging (the area under the emission peak within the shaded region; Figure 2, third column) and divided by the sum of the total normalized emission values (the area under total emission peak; Figure 2, third column). To determine the brightness at a given excitation wavelength (Figure 2, blue line, third column), we took the product of the quantum yield (literature value) and the extinction coefficient times the fraction of excitation peak at the imaging wavelength (Yang et al., 1996; Shaner et al., 2004, 2008, 2013; Nguyen and Daugherty, 2005; Shcherbo et al., 2009; Lam et al., 2012; Lee et al., 2013).
Microscopy
Imaging embryos.
C. elegans embryos were dissected for imaging and mounted in egg buffer at the two- to three-cell stage on poly-l-lysine–coated coverslips with 2.5% agar pads. Embryos expressing different fluorescent proteins were initially imaged side by side, as shown in Figures 2 and 3 (three pairs/groups per comparison). To increase the number of embryos imaged for quantification, multiple embryos from the same strain were mounted in groups, and images were acquired using the same settings as the initial side-by-side comparisons. To minimize the effect of any unavoidable minor variation in imaging conditions, embryos from strains for a given comparison were imaged alternately using identical settings. HIS-58::GFP and mNG embryos were mounted at the three-cell stage—a short (∼3 min), identifiable stage between cell divisions. Fluorescence intensity was measured in the EMS cell nucleus. For the GFP and mNeonGreen endogenous knock-in strain comparisons, embryos from each strain plus an N2 wild-type embryo were imaged and compared in groups.
All embryos were imaged with a Nikon Eclipse Ti spinning-disk confocal microscope (CSU-X1 spinning-disk head; Yokogawa, Tokyo, Japan) using a Hamamatsu ImagEM X2 EM-CCD camera (C9100-13) and a 60×/1.4 numerical aperture (NA) Plan Apo oil immersion objective (Nikon, Tokyo, Japan). Samples were illuminated using solid-state lasers of wavelengths 488, 514, and 561 nm. The following emission filter sets were used for a given excitation wavelength: 488 nm, ET525/50 m (Chroma, Foothill Ranch, CA); 514 nm, ET545/40 m (Chroma); 561 nm, ET630/75 m (Chroma); and 561l p (Semrock, Rochester, NY).
Imaging whole worms.
Whole worms were mounted at the L4 developmental stage and immobilized using nanoparticles as previously described (Kim et al., 2013). Worms were imaged using a Nikon Eclipse Ti spinning-disk confocal microscope (Yokogawa CSU-X1 spinning-disk head) using a Hamamatsu ImagEM X2 EM-CCD camera (C9100-13; Hamamatsu City, Japan) and a 10×/0.30 NA Plan Fluor objective (Nikon) with 488-nm excitation and ET525/50x emission filter.
Image quantification.
For membrane-labeled strains, fluorescence intensity was quantified using MetaMorph software (Molecular Devices, Sunnyvale, CA) by taking the average of a 3-pixel-wide line scan perpendicular to the plasma membrane in the posteriormost embryonic cell (the P2 cell). For each time point, the maximum intensity from this line scan was recorded and average off-embryo background subtracted. GraphPad Prism software (GraphPad, La Jolla, CA) was used to plot the mean and 95% confidence intervals (CIs) for all initial brightness measurements and at each time point for bleaching measurements. To determine the half-life of a given fluorescent protein, the individual photobleaching traces were fit to a standard one-phase decay curve, the “half-life” for each curve was recorded, and the mean and 95% CIs were recorded for each fluorescent protein. The photon budget was determined by integrating the fluorescence intensity measured for each embryo until the intensity reached 50% of the initial intensity.
For histone fusion proteins and pharyngeal-labeled strains, the images were thresholded and segmented using ImageJ to define a region for measurement (either the nucleus or pharynx). For GFP and mNeonGreen knock-in strains, a region was drawn around each embryo. The average fluorescence intensity of the given region was calculated by measuring the average integrated intensity of the region and subtracting average off-embryo background for each image. Each embryo was displayed as an individual data point, and the mean and 95% CIs were plotted using GraphPad Prism software.
Unpaired, two-tailed t tests with Welch’s correction were used to compare means in all imaging experiments, and all statistical analyses were performed using GraphPad Prism software. All comparisons are significantly different (p < 0.05), unless otherwise indicated (“ns”). Statistics for individual experiments are given in Supplemental Figure S2B.
Quantifying autofluorescence in C. elegans embryos
We measured embryo autofluorescence in two separate experiments. For both, wild-type (N2) embryos were mounted in egg buffer on poly-l-lysine–coated coverslips with 2.5% agar pads. In one experiment, we used the same microscope, objective (60×), and camera described previously for imaging fluorescent embryos. We used a laser photodiode sensor (7Z02410 and filter 688657; OPHIR Photonics, Jerusalem, Israel) to adjust the settings so that laser power for each wavelength was 1 mW at the objective. We then imaged embryos under these conditions for each wavelength (445 nm, n = 13; 488 nm, n = 16; 514 nm, n = 16; and 561 nm, n = 14) with a common exposure time and the filter settings previously described. The emission filters used in this experiment range in the breadth of wavelengths they transmit and allow different amounts of light to pass through. In the second experiment, embryos were imaged using a Nikon A1R laser scanning confocal microscope. The excitation wavelengths used were 405, 442, 488, 515, and 561 nm. The illumination settings for each wavelength were set to a common wattage in the Nikon Elements software. Images of embryo autofluorescence were collected using a multispectral detector and emission fingerprinting for each of the given wavelengths.
For both experiments, image analysis was performed using ImageJ. Pixel intensity values were measured for three regions per embryo and averaged. Average off-embryo background was subtracted for each embryo, and the resulting fluorescence intensity was plotted at each detection wavelength. (To graph both experiments together, the results of the first experiment were scaled by multiplying the measured values [arbitrary units] by 500.) The x-axis value used for the first experiment was the center wavelength of the emission filter used for detection.
Western blotting
For quantifying protein levels, we picked L4-stage worms of each strain to three separate plates. After 12–14 h at 25C, gravid young adults were collected from each plate. Three lysates were generated for each strain at a concentration of 1 worm/μl (60 worms were picked into 45 μl of M9 Buffer, and 15 μl of 4× sample buffer was added). Samples were frozen in liquid nitrogen and sonicated in boiling water for 10 min twice. Lysates were separated on 12% NuPAGE Novex Bis-Tris Protein Gels (Invitrogen) and transferred to an Immobilon PVDF-FL membrane (EMD Millipore, Darmstadt, Germany) for immunoblotting. Fluorescent proteins expressed by transgenes were detected using a mouse anti-FLAG BioM2 (F9291; Sigma-Aldrich, St. Louis, MO) antibody at 1:1000 dilution, and a rabbit anti-HCP-3 (Monen et al., 2005) was used at 1:1000 dilution as a loading control. The following fluorescent secondary antibodies were used (1 μl/blot): Alexa Fluor 680 goat anti-mouse and Alexa Fluor 790 goat anti-rabbit (A31562 and A11369, respectively; Invitrogen). Three independent samples were collected and one blot from each biological replicate was performed. Blots were scanned using an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE), and fluorescence intensity was quantified using ImageJ. The ratio of transgene protein intensity (∼45-kDa band in the 680-nm channel) to loading control intensity (∼80-kDa upper band in the 790-nm channel) was measured for each lane on a given blot. These measurements were normalized by dividing the ratio measured for each lane by the total average ratio of all the lanes on a given blot. These normalized protein levels were plotted together with an average and 95% CIs using GraphPad Prism. Gel images were inverted and cropped slightly at the edges, and brightness and contrast were adjusted using ImageJ. The dashed line in Supplemental Figure S1C indicates where blank lanes were cropped.
Spectrum viewer
The fluorescence spectrum viewer (Supplemental File S1) was designed as a user-extensible collection of fluorescence spectra, dichroic filter spectra, and laser lines. Data were collected and digitized from a range of published fluorophore spectra using the WebPlotDigitizer software package (http://arohatgi.info/WebPlotDigitizer/). Digitized spectra were resampled at 1-nm wavelength increments, and excitation and emission spectra were each normalized to a maximum value of 1 relative fluorescence unit. Dichroic fluorescence filter data were similarly digitized from commercial plots. The spectrum viewer was implemented in Microsoft Excel using only worksheet range functions, avoiding the use of macrolanguage constructs. Up to four fluorophores, four fluorescent filters, and three laser lines may be selected and compared in an Excel chart through a simple graphical user interface. Possible spectral data listed in the user interface are populated from a DataList database worksheet, which in turn consists of spectrum names and accompanying worksheet ranges for stored spectral data. User selection of a spectrum to display populates a Current data worksheet via indirect references stored in the DataList database. The spectral chart is automatically updated to reflect changes in the Current data worksheet.
New fluorophore and fluorescent protein spectral data can be added to existing worksheets or as new worksheets. Indirect worksheet references must then be added to either the fluorophore or filter section of the DataList worksheet. The user interface is automatically repopulated with new choices. Simple, user-defined bandpass, short-pass, and long-pass filter sets can also be defined on the User Filters worksheet for comparison to fluorophore spectra.
Supplementary Material
Acknowledgments
We thank Kurt Thorn, Dave Matus, and Michael Werner for suggestions and comments on the manuscript. We also thank Terrence Wong for materials and plates. Strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). This work was supported by National Institutes of Health Grants T32 CA009156 (D.J.D. and A.M.P.) and F32 GM115151 (A.M.P.), a Howard Hughes postdoctoral fellowship from the Helen Hay Whitney Foundation (D.J.D.), a National Science Foundation graduate research fellowship (J.K.H.), and National Institutes of Health Grant R01 GM083071 and National Science Foundation Grant IOS 0917726 (B.G.).
Abbreviations used:
- CRISPR
clustered, regularly interspersed, short palindromic repeats
- GFP
green fluorescent protein
- mNG
monomeric NeonGreen
- mYPet
monomeric yellow fluorescent protein for energy transfer.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0063) on July 6, 2016.
REFERENCES
- Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T, Tanaka K. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 2015;16:87. doi: 10.1186/s13059-015-0653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audhya A, Hyndman F, McLeod IX, Maddox AS, Yates JR, III, Desai A, Oegema K. A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J Cell Biol. 2005;171:267–279. doi: 10.1083/jcb.200506124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014;24:142–153. doi: 10.1101/gr.161638.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, Liu JL. Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol Open. 2014;3:42–49. doi: 10.1242/bio.20137120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RAJ, Stempor P, Down TA, Zeiser E, Feuer SK, Ahringer J. Extreme HOT regions are CpG-dense promoters in C. elegans and humans. Genome Res. 2014;24:1138–1146. doi: 10.1101/gr.161992.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranfill PJ, Sell BR, Baird MA, Allen JR, Lavagnino Z, de Gruiter HM, Kremers G-J, Davidson MW, Ustione A, Piston DW. Quantitative assessment of fluorescent proteins. Nat Methods. 2016;13:557–562. doi: 10.1038/nmeth.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MW, Campbell RE. Engineered fluorescent proteins: innovations and applications. Nat Methods. 2009;6:713–717. doi: 10.1038/nmeth1009-713. [DOI] [PubMed] [Google Scholar]
- Dickinson DJ, Pani AM, Heppert JK, Higgins CD, Goldstein B. Streamlined genome engineering with a self-excising drug selection cassette. Genetics. 2015;200:1035–1049. doi: 10.1534/genetics.115.178335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10:1028–1034. doi: 10.1038/nmeth.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096–00. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, et al. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat Cell Biol. 2011;13:331–337. doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Wayne Davis M, Hopkins CE, Newman BJ, Thummel JM, Olesen S-P, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles AF, Averof M. Functional genetics for all: engineered nucleases, CRISPR and the gene editing revolution. EvoDevo. 2014;5:43. doi: 10.1186/2041-9139-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RA, Audhya A, Pozniakovsky A, Dammermann A, Pemble H, Monen J, Portier N, Hyman AA, Desai A, Oegema K. Expression and imaging of fluorescent proteins in the C. elegans gonad and early embryo. Methods Cell Biol. 2008;85:179–218. doi: 10.1016/S0091-679X(08)85009-1. [DOI] [PubMed] [Google Scholar]
- Harrison MM, Jenkins BV, O’Connor-Giles KM, Wildonger J. A CRISPR view of development. Genes Dev. 2014;28:1859–1872. doi: 10.1101/gad.248252.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebisch E, Knebel J, Landsberg J, Frey E, Leisner M. High variation of fluorescence protein maturation times in closely related Escherichia coli strains. PLoS One. 2013;8:e75991. doi: 10.1371/journal.pone.0075991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- Hochbaum D, Ferguson AA, Fisher AL. Generation of transgenic C. elegans by biolistic transformation. J Vis Exp. 2010;42: 2090. doi: 10.3791/2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W-Y, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med. 2000;6:482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- Iizuka R, Yamagishi-Shirasaki M, Funatsu T. Kinetic study of de novo chromophore maturation of fluorescent proteins. Anal Biochem. 2011;414:173–178. doi: 10.1016/j.ab.2011.03.036. [DOI] [PubMed] [Google Scholar]
- Kim E, Sun L, Gabel CV, Fang-Yen C. Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization. PLoS One. 2013;8:e53419. doi: 10.1371/journal.pone.0053419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krestel HE, Mihaljevic ALA, Hoffman DA, Schneider A. Neuronal co-expression of EGFP and β-galactosidase in mice causes neuropathology and premature death. Neurobiol Dis. 2004;17:310–318. doi: 10.1016/j.nbd.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Lam AJ, St-Pierre F, Gong Y, Marshall JD, Cranfill PJ, Baird MA, McKeown MR, Wiedenmann J, Davidson MW, Schnitzer MJ, et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat Methods. 2012;9:1005–1012. doi: 10.1038/nmeth.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lim WA, Thorn KS. Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One. 2013;8:e67902. doi: 10.1371/journal.pone.0067902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Ma J, Zhang X, Chen W, Yu L, Lu Y, Bai L, Shen B, Huang X, Zhang L. Generation of eGFP and Creknockin rats by CRISPR/Cas9. FEBS J. 2014;281:3779–3790. doi: 10.1111/febs.12935. [DOI] [PubMed] [Google Scholar]
- Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov S. Fluorescent proteins from nonbiouminescent Anthozoa species. Nat Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- Monen J, Maddox PS, Hyndman F, Oegema K, Desai A. Differential role of CENP-A in the segregation of holocentric C. elegans chromosomes during meiosis and mitosis. Nat Cell Biol. 2005;7:1248–1255. doi: 10.1038/ncb1331. [DOI] [PubMed] [Google Scholar]
- Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- Ohashi T, Galiacy SD, Briscoe G, Erickson HP. An experimental study of GFP-based FRET, with application to intrinsically unstructured proteins. Protein Sci. 2007;16:1429–1438. doi: 10.1110/ps.072845607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Sequence requirements for myosin gene expression and regulation in Ceanorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Wang Y, Smith HE, Lee C-Y, Calidas D, Lu T, Smith J, Schmidt H, Krause MW, Seydoux G. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 2014;198:1347–1356. doi: 10.1534/genetics.114.170423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Clark KJ, Campbell JM, Panetta MR, Guo Y, Ekker SC. Making designer mutants in model organisms. Development. 2014;141:4042–4054. doi: 10.1242/dev.102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry KJ, Henry JQ. CRISPR/Cas9-mediated genome modification in the mollusc, Crepidula fornicata. Genesis. 2015;53:237–244. doi: 10.1002/dvg.22843. [DOI] [PubMed] [Google Scholar]
- Ratz M, Testa I, Hell SW, Jakobs S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci Rep. 2015;5:9592. doi: 10.1038/srep09592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redemann S, Schloissnig S, Ernst S, Pozniakowsky A, Ayloo S, Hyman AA, Bringmann H. Codon adaptation–based control of protein expression in C. elegans. Nat Methods. 2011;8:250–252. doi: 10.1038/nmeth.1565. [DOI] [PubMed] [Google Scholar]
- Shaner NC. Fluorescent proteins for quantitative microscopy: important properties and practical evaluation. Methods Cell Biol. 2014;123:95–111. doi: 10.1016/B978-0-12-420138-5.00006-9. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods. 2013;10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J Cell Biol. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Shcherbo D, Murphy CS, Ermakova GV, Solovieva EA, Chepurnykh TV, Shcheglov AS, Verkhusha VV, Pletnev VZ, Hazelwood KL, Roche PM, et al. Far-red fluorescent tags for protein imaging in living tissues. Biochem J. 2009;418:567–574. doi: 10.1042/BJ20081949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee H-C, Gu W, Ishidate T, Conte D, Jr, Mello CC. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Xue Z, Ren M, Wu M, Dai J, Rong YS, Guanjun G. Efficient gene knock-out and knock-in with transgenic Cas9 in Drosophila. G3 (Bethesda) 2014;4:925–929. doi: 10.1534/g3.114.010496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T-T, Cheng L, Kain SR. Optimized codon usage and chromophore mutations provide enhanced sensitivity with the green fluorescent protein. Nucleic Acids Res. 1996;24:4592–4593. doi: 10.1093/nar/24.22.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- Zeiser E, Frøkjær-Jensen C, Jorgensen E, Ahringer J. MosSCI and Gateway compatible plasmid toolkit for constitutive and inducible expression of transgenes in the C. elegans germline. PLoS One. 2011;6:e20082. doi: 10.1371/journal.pone.0020082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.