

Quantitative knowledge of the recruitment of spindle assembly checkpoint (SAC) proteins by the kinetochore is essential to understanding the mechanisms that regulate protein recruitment and hence the strength of the SAC. Here this recruitment is quantified, and novel mechanisms are identified that strongly modulate SAC protein recruitment by the kinetochore.
Abstract
Recruitment of spindle assembly checkpoint (SAC) proteins by an unattached kinetochore leads to SAC activation. This recruitment is licensed by the Mps1 kinase, which phosphorylates the kinetochore protein Spc105 at one or more of its six MELT repeats. Spc105 then recruits the Bub3-Bub1 and Mad1-Mad2 complexes, which produce the inhibitory signal that arrests cell division. The strength of this signal depends, in part, on the number of Bub3-Bub1 and Mad1-Mad2 molecules that Spc105 recruits. Therefore regulation of this recruitment will influence SAC signaling. To understand this regulation, we established the physiological binding curves that describe the binding of Bub3-Bub1 and Mad1-Mad2 to the budding yeast kinetochore. We find that the binding of both follows the mass action law. Mps1 likely phosphorylates all six MELT repeats of Spc105. However, two mechanisms prevent Spc105 from recruiting six Bub3-Bub1 molecules: low Bub1 abundance and hindrance in the binding of more than one Bub3-Bub1 molecule to the same Spc105. Surprisingly, the kinetochore recruits two Mad1-Mad2 heterotetramers for every Bub3-Bub1 molecule. Finally, at least three MELT repeats per Spc105 are needed for accurate chromosome segregation. These data reveal that kinetochore-intrinsic and -extrinsic mechanisms influence the physiological operation of SAC signaling, potentially to maximize chromosome segregation accuracy.
INTRODUCTION
Proteins involved in spindle assembly checkpoint (SAC) signaling recognize and bind to unattached kinetochores in a dividing cell. This allows the SAC proteins to interact with one another and generate the mitotic checkpoint complex to delay cell division (Musacchio, 2015). Duration of the delay generated in this manner depends on the number of SAC proteins recruited by the kinetochore (Collin et al., 2013; Dick and Gerlich, 2013; Heinrich et al., 2013). In fact, to strengthen SAC signaling, unattached kinetochores can expand their physical capacity and recruit larger quantities of SAC proteins (Wynne and Funabiki, 2015). Therefore, to understand how the eukaryotic cell regulates the strength of the SAC, we must first define the intrinsic capacity of the kinetochore to bind SAC proteins, that is, its signaling capacity, and then identify any physiological factors that regulate the use of this capacity.
The biochemical interactions that recruit SAC proteins to the kinetochore are well understood (Figure 1A). An unattached kinetochore permits the Mps1 kinase to phosphorylate the kinetochore protein Spc105/KNL-1 to initiate the SAC signaling cascade (London et al., 2012; Aravamudhan et al., 2015). Phosphorylation of Spc105 occurs at one or more of the six sites in its phosphodomain, which are commonly referred to as the MELT repeats because of their Met-Glu-Leu-Thr consensus sequence. Each phosphorylated MELT repeat, which is referred to as MELpT here, can bind one molecule of the Bub3-Bub1 complex (Primorac et al., 2013). The kinetochore-bound Bub1 is also phosphorylated by Mps1, which enables it to bind the heterotetrameric Mad1-Mad2 complex (Chen et al., 1999; London and Biggins, 2014; Moyle et al., 2014). The kinetochore-localized Mad1-Mad2 then catalyzes a conformational transition in Mad2 and paves the way for the assembly of the mitotic checkpoint complex.
FIGURE 1:

Steady-state binding of SAC proteins to the kinetochore scales inversely with the number of signaling kinetochores. (A) Schematic of the biochemical reactions that recruit SAC proteins to an unattached kinetochore and generate the mitotic checkpoint complex. Cdc20 and Mad3/BubR1 are not shown. (B) Cartoon, one unattached sister kinetochore pair created using a galactose-repressible centromere as described previously (Tanaka et al., 2010). In these cells, the 15 bioriented kinetochores appear as two puncta ∼800 nm apart, whereas the unattached kinetochore pair appears as a diffraction-limited focus situated away from the bioriented kinetochores (asterisk in the micrograph; scale bar, ∼0.53 μm). This unattached sister kinetochore pair recruits Bub1 and Mad1. Middle, expected numbers of kinetochores: 15 in the puncta corresponding to the bioriented kinetochores and 2 in the focus corresponding to the unattached sister kinetochore pair, confirmed by quantifying the fluorescence of mCherry-labeled Ndc80 complex subunits. Right, quantification of Bub1 and Mad1 molecules recruited per kinetochore (mean ± 95% confidence interval). (C) Cartoon, spindle depolymerization by the microtubule poison nocodazole creates two or more kinetochore clusters in mitotic yeast cells. Kinetochores in larger cluster, which is situated proximal to the spindle pole, do not recruit any SAC proteins. Kinetochores in the smaller cluster recruit SAC proteins (micrographs). (D) Forced localization of Mps1 to the kinetochore using rapamycin-induced dimerization induces SAC signaling from all 32 kinetochores (Aravamudhan et al., 2015). Scale bar, ∼0.96 μm (C, D). (E) Number of Mad1 dimers and Bub1 molecules recruited as a function of the number of signaling kinetochores in the cell. Solid lines display model fit for a bimolecular reaction limited by the abundance of soluble proteins (mean ± SD). (F) Scaling between the number of Bub1 molecules and the number of Mad1 dimers recruited by signaling kinetochores in each of the three foregoing experiments (mean ± SEM).
These details indicate that the nominal signaling capacity of a kinetochore is determined by the number of MELT repeats per Spc105 and the number of Spc105 molecules per kinetochore. If these two numbers are known, then the nominal value of the maximal signaling capacity can be calculated. However, it is reasonable to expect that this maximal signaling capacity may not be operational in vivo because of a number of physiological factors: the phosphoregulation of Spc105, cellular abundance of SAC proteins, variations in the affinity of MELT repeats for the Bub3-Bub1 complex, and so on (Primorac et al., 2013; Krenn et al., 2014; Zhang et al., 2014; Overlack et al., 2015; Vleugel et al., 2015). To understand whether and how these factors regulate SAC protein recruitment, it is necessary to quantify the recruitment of SAC proteins by unattached kinetochores under physiological conditions. However, very few quantitative data are available that define how physiological factors regulate the in vivo operation of kinetochore-based SAC signaling (Howell et al., 2004; Vleugel et al., 2015).
To define quantitatively the physiological operation of the kinetochore-based SAC signaling reactions, we systematically studied the biochemistry underlying Bub3-Bub1 and Mad1-Mad2 recruitment by the kinetochore in the budding yeast Saccharomyces cerevisiae. Budding yeast offers unparalleled experimental advantages for this study. The cellular concentrations of the reactants—Spc105, Bub3, Bub1, and Mad1—are all known (Ghaemmaghami et al., 2003; Kulak et al., 2014; Wang et al., 2015). The steady-state copy numbers of the reaction products—the number of SAC proteins bound to the kinetochore—can be accurately measured using fluorescence microscopy (Joglekar et al., 2006; Aravamudhan et al., 2013). Finally, the measurements reported here will demonstrate that budding yeast kinetochores recruit SAC proteins via defined biochemical interactions; they do not expand or form a corona containing SAC proteins like metazoan kinetochores (Cooke et al., 1997; Hoffman et al., 2001; Wynne and Funabiki, 2015). Therefore quantitation of SAC protein recruitment directly reveals the operation of the underlying biochemical interactions. We exploited these advantages to identify novel mechanisms that strongly modulate the recruitment of SAC proteins by the kinetochore and thus potentially control the strength of SAC signaling.
RESULTS
The nominal value of the maximum SAC protein binding capacity of one yeast kinetochore can be easily calculated. The yeast kinetochore contains five Spc105 molecules (Joglekar et al., 2006; Aravamudhan et al., 2013), each Spc105 molecule contains six MELT repeats (London et al., 2012), and each phosphorylated MELT repeat, that is, MELpT, can bind one Bub3-Bub1 (Primorac et al., 2013). Therefore the yeast kinetochore is equipped to bind 30 Bub3-Bub1 molecules and at least an equal number of Mad1-Mad2 heterotetramers.
To study how this binding capacity is used in vivo, we quantified the recruitment of Bub1 and Mad1 in yeast cells containing exactly one pair of unattached sister kinetochores. To generate exactly one pair of unattached kinetochores, we conditionally inactivated and then reactivated the centromere on chromosome III using the method developed by the Tanaka lab (Tanaka et al., 2010). Briefly, we inserted the strong galactose-inducible promoter pGal1 upstream from the point centromere on chromosome III so that the activity of this centromere is controlled by pGal1. In glucose-containing media, pGal1 is strongly repressed. This allows the downstream centromere to function normally. However, when in galactose-containing media, pGal1 is strongly induced, and the centromere is inactivated (Hill and Bloom, 1987). This carbon source–dependent inactivation of one centromere was exploited in G1-synchronized cells that transition to and then arrest in metaphase. The inactivated centromere was reactivated after all other chromosomes established stable bipolar attachments (see Materials and Methods). On reactivation, this centromere rapidly assembles kinetochore components and recruits SAC proteins, enabling the quantitation of SAC protein recruitment by a single pair of unattached kinetochores (Krefman et al., 2015).
To quantify the Bub1 and Mad1 molecules recruited, we labeled the respective proteins at their C-terminus with a variant of the green fluorescent protein, GFP(S65T; hereafter referred to as GFP). We verified that the fluorescent tag did not compromise protein function (Supplemental Figure S1A). We found that when the cell contained exactly one unattached sister kinetochore pair, each kinetochore bound 9.1 ± 3.4 Bub1 molecules on average, using only 33% of its maximal capacity for binding Bub3-Bub1 (Figure 1B; also see Materials and Methods for a detailed description of the methodology used to deduce protein numbers from the measured fluorescence signal). We expected that the number of Bub3 molecules recruited would be the same as that of Bub1 because Bub1 binds to the MELpT motifs as part of the Bub3-Bub1 complex (Supplemental Figure S1, C and D; London et al., 2012). Next we quantified the number of Mad1-GFP recruited by the unattached sister kinetochore pair. Because Mad1 is recruited by Bub1, we expected that the number of Mad1-Mad2 heterotetramers would also be similar to the number of Bub3-Bub1 molecules (London and Biggins, 2014). However, each kinetochore bound 37.4 ± 8 Mad1-GFP molecules, or, equivalently, 18.7 ± 5.7 Mad1-Mad2 heterotetramers. Thus the number of Mad1-Mad2 heterotetramers is twice the number of Bub3-Bub1 molecules recruited by the kinetochores (Figure 1B).
Because the number of unattached kinetochores is close to its lowest possible value in the foregoing experiment, the availability of SAC proteins was not expected to limit their recruitment (protein abundance values are tabulated in Supplemental Table S1). However, the foregoing data show that the two unattached kinetochores recruit a surprisingly low copy number of SAC proteins under a condition that should favor maximal recruitment. The low recruitment is likely sufficient to delay the cell cycle (Spencer and Hieter, 1992; Kerscher et al., 2003). Note that we quantified Mad1 in a strain lacking the gene NUP60, in which Mad1 cannot localize to the nuclear envelope (Scott et al., 2005; Yuen et al., 2007). Consequently, the availability of Mad1 increases, enabling the signaling kinetochores to recruit more Mad1 molecules than the number in wild-type cells (Supplemental Figure S1E). Although this will likely influence the cell biological outcome of the strength of SAC signaling, it is unlikely to affect kinetochore-intrinsic regulation of Mad1 recruitment, which is the focus of this study.
We next investigated whether and how the use of the signaling capacity of the kinetochore changes when a dividing cell contains more signaling kinetochores. We used two methods to generate well-defined numbers of signaling kinetochores. First, we treated yeast cells with nocodazole to depolymerize spindle microtubules (see Materials and Methods). This treatment creates a cluster of 8–10 ± 2 unattached and SAC-active kinetochores identified as a fluorescent puncta situated distal to the collapsed spindle pole bodies (Figure 1C; quantification of kinetochore counts in Supplemental Figure S1C; Gillett et al., 2004). To create even more SAC-active kinetochores, we forced Mps1-Frb to dimerize with Fkbp12 domains fused with a kinetochore subunit (Aravamudhan et al., 2015). This method forces approximately one or two Mps1 molecules to localize to each kinetochore. As a result, all 32 kinetochores become SAC active even after they are stably attached to spindle microtubules (Figure 1D; also see Supplemental Figure S1F).
Quantification of the number of Bub1-GFP and Mad1-GFP molecules recruited by the kinetochores under these conditions revealed that as the number of signaling kinetochores in a cell increased, the cumulative number of SAC proteins recruited by these kinetochores reached saturation (Figure 1E). The average numbers of Bub1 and Mad1 molecules per kinetochore decreased correspondingly (Supplemental Figure S2A). Of interest, the kinetochores continued to recruit twice as many Mad1 dimers as Bub1 molecules (Figure 1E). This strict stoichiometry between the two proteins suggests that the recruitment of the excess Mad1 dimers is not due to adventitious binding; it results from a specific interaction (Figure 1F). Either each Bub3-Bub1 molecule binds two Mad1-Mad2 heterotetramers or the kinetochore contains an additional, but Bub3-Bub1 dependent, Mad1-binding site (Scott et al., 2005).
These data further confirm that the yeast kinetochore uses a small fraction of its maximal signaling capacity during mitosis. The rapid saturation of the cumulative number of Bub3-Bub1 molecules recruited with increasing number of signaling kinetochores suggests that the abundance of SAC proteins, a kinetochore-extrinsic factor, plays a significant role in limiting their recruitment by the kinetochore. At the same time, the surprisingly modest recruitment of SAC proteins even when there is only one unattached chromosome in the cell indicates that factors intrinsic to the kinetochore also modulate recruitment. To identify and characterize these kinetochore-intrinsic and -extrinsic factors, we studied the operation of biochemical reactions that recruit Bub3-Bub1 and Mad1-Mad2 to unattached kinetochores individually. The phosphorylation of MELT repeats in Spc105 is the first step in the SAC signaling cascade (Figure 1A). However, it is not possible to quantify the phosphoregulation of Spc105 using live-cell fluorescence microscopy. The binding of Bub3-Bub1 to Spc105, which comes next, can be directly quantified. Therefore we first established the physiological binding curve for the Bub3-Bub1-Spc105 interaction, treating Spc105 as the substrate. This quantification indirectly informs on the phosphorylation state of Spc105 because Bub3-Bub1 binds exclusively to MELpT in a 1:1 stoichiometry.
To study the Bub3-Bub1-Spc105 interaction, we systematically reduced Spc105 copy number per kinetochore and then quantified how this affected Bub1 recruitment. To reduce Spc105 copy number per kinetochore, we first replaced SPC105 with SPC105-GFP and then controlled its expression with pGalL, a weak, galactose-inducible promoter (Janke et al., 2004). In this strain, the steady-state concentration, and hence the copy number of Spc105-GFP per kinetochore, depends on the concentration of galactose in the growth medium (Figure 2A). We found that even after a significant reduction in Spc105 copy number, kinetochore biorientation appeared to be unaffected (Supplemental Figure S2B). Reduced Spc105 copy number may decrease the recruitment of Mps1 to the kinetochore by affecting the Mps1-recruitment site in the kinetochore: the Ndc80 complex (Kemmler et al., 2009; Aravamudhan et al., 2015; Hiruma et al., 2015; Ji et al., 2015). However, we found that even a fourfold reduction in the Spc105 copy number did not significantly reduce the number of Ndc80 complexes per kinetochore (Figure 2B). Furthermore, kinetochore-bound Mps1 is substoichiometric to Ndc80 in budding yeast (Aravamudhan et al., 2015). Therefore we assumed that the activity of Mps1 kinase in SAC signaling was not significantly affected in the following experiment.
FIGURE 2:

Spc105 molecules within one kinetochore recruit Bub3-Bub1 independently. (A) Top, representative micrographs of cells expressing different levels of Spc105-GFP (from the galactose-responsive pGalL promoter; galactose concentration in the medium is noted at the top of each micrograph, scale bar, ∼0.96 μm). Bottom, change in the number of Spc105 molecules per kinetochore in metaphase with the concentration of galactose in growth medium (mean ± SD). Solid line represents saturation binding fit to the data. (B) Top, representative micrographs display the Ndc80-mCherry fluorescence from kinetochore clusters containing different amounts of Spc105-GFP (scale bar, ∼0.96 μm). Scatter plot, each black circle represents the binned average number of Ndc80-mCherry molecules plotted against the average number of Spc105-GFP molecules per kinetochore in metaphase. Solid line represents linear regression of the data. (C) Change in Bub1 recruitment due to reduced numbers of Spc105 molecules per kinetochore. Gray circles represent the number of Bub1 molecules recruited by the cluster of unattached kinetochores containing the indicated number of Spc105 molecules in nocodazole-treated cells. Pale green squares represent averages calculated from binned data (mean ± SD). Black squares indicate change in the number of Bub1 molecules recruited when the number of signaling kinetochores is changed in wild-type cells (from Figure 1E). The y-axis on the right-hand side displays the concentration of the kinetochore-bound Bub1 molecules (nM) assuming that the yeast nucleus is 2 μm in diameter. Solid curve represents the model fit to the entire data set as a bimolecular reaction with the ligand depletion model (Bmax is the maximal binding and KD is the nanomolar dissociation constant predicted by the fit). (D) Schematic showing that the cumulative number of Spc105 molecules that participate in SAC signaling, and not their spatial distribution, determines the steady-state number of Bub3-Bub1 molecules recruited.
To study the effect of lowered copy number of Spc105 on Bub1 recruitment, we grew yeast cells expressing Spc105-GFP from pGalL in medium containing predefined galactose concentration for ∼20 h and then treated them with nocodazole. We used green fluorescent protein (GFP) fluorescence to calculate the number of Spc105 molecules that recruited Bub1 in each cell and used mCherry fluorescence to deduce the corresponding number of Bub1-mCherry molecules. These measurements revealed that the cumulative number of Bub1 molecules recruited by unattached kinetochores decreased proportionately with the decrease in the cumulative number of Spc105 molecules within those kinetochores (Figure 2C). The number of Bub1 molecules per Spc105 increased modestly (Supplemental Figure S2C), likely because of an increase in the concentration of Bub3-Bub1 relative to that of Spc105. Mad1 recruitment was not significantly reduced, probably because of the combined effect of the relatively modest reduction in the total number of kinetochore-bound Bub1 and the 2:1 Mad1-dimer:Bub1 stoichiometry (Supplemental Figure S2D). The proportionate decrease in the number of Bub1 molecules with the decrease in the number of Spc105 molecules is noteworthy. This trend suggests that the clustering of Spc105 molecules in the kinetochore, which could lead to cooperative binding, does not detectably enhance the steady-state recruitment of Bub1 and Mad1. It is informative to compare the number of Bub1 molecules per Spc105 measured in this experiment with the same number measured in the experiment in Figure 1E, in which the number of Spc105 molecules per kinetochore was unchanged but the number of signaling kinetochores was varied. This comparison reveals that Bub1 recruitment depends on the total number of Spc105 molecules involved in signaling but not on their distribution. Whether five Spc105 molecules are located in one kinetochore or distributed in three different kinetochores, they recruit the same total number of Bub3-Bub1 molecules (Figure 2D). In other words, Spc105 molecules in each kinetochore appear to recruit Bub3-Bub1 independently.
The independent recruitment of the Bub3-Bub1 complex by the five Spc105 molecules within each kinetochore suggests that the Bub3-Bub1-Spc105 interaction can be modeled as a bimolecular reaction that follows the mass action law and that its operation is limited by intracellular concentration of the Bub3-Bub1 complex (Figures 1E and 2C). To fit these values with the foregoing model, we converted protein copy numbers into concentrations by assuming that the yeast nucleus is a sphere of 2 μm diameter (see the Fitting equilibrium binding models section in Materials and Methods). This exercise yielded a satisfactory fit and established the physiological binding curve for the Bub3-Bub1-Spc105 interaction. This curve estimates the apparent dissociation constant for the Bub3-Bub1-Spc105 complex and the maximal steady-state concentration of this complex that can form at the physiological concentration of Bub3-Bub1. As discussed later, both parameters are essential for predicting the effects of experimental perturbations on Bub3-Bub1 recruitment.
We next assessed the steady-state phosphoregulation of Spc105 by Mps1 and the PP1 phosphatase Glc7. The number of MELpT in an unattached kinetochore is determined by the balance of the opposing activities of Mps1 kinase, which phosphorylates MELT repeats to enable Bub3-Bub1 recruitment, and Glc7 phosphatase, which dephosphorylates MELpT to suppress Bub3-Bub1 recruitment (Pinsky et al., 2009; London et al., 2012; Zhang et al., 2014). Therefore we reasoned that Glc7 inhibition should enhance Bub3-Bub1 recruitment up to the limit established by the Spc105-Bub3-Bub1 binding curve (Figure 2C). To test this, we inhibited Glc7 phosphatase activity using calyculin A, a cell-permeable inhibitor of PP1 phosphatases (Hoon et al., 2008). To assess the potency of calyculin A, we assayed the phosphorylation of the Snf1 kinase, a well-characterized target of Glc7. Glc7 maintains Snf1 predominantly in the dephosphorylated state. Therefore the inhibition of Glc7 by calyculin A increases the abundance of phosphorylated Snf1 (Orlova et al., 2008). Using a phosphospecific antibody against Snf1, we confirmed that calyculin A treatment significantly reduced the cellular activity of Glc7 (Supplemental Figure S2E). In spite of the reduction in Glc7 activity, Bub1 recruitment by unattached kinetochores did not increase (Figure 3A). Therefore Glc7 likely has little effect on the steady-state number of MELpT in the unattached yeast kinetochore, even though its abundance exceeds that of Mps1 by approximately two orders of magnitude (Supplemental Figure S2F and Supplemental Table S1).
FIGURE 3:

The abundance of Bub1 limits the steady-state recruitment of Bub3-Bub1 and Mad1-Mad2 to the yeast kinetochore. (A) Bub1 recruitment by unattached kinetochores in nocodazole-treated cells in the absence or presence of the PP1 phosphatase inhibitor calyculin A (Caly). (B) Estimation of the number of Bub1 molecules per cell using quantitative immunoblotting. Top, immunoblot of Bub1-GFP expressed from indicated promoters using monoclonal anti-GFP antibodies (cells grown in medium containing 1.5% galactose). *Unknown protein recognized by the anti-GFP antibody that comigrates with Bub1-GFP. To quantify the Bub1-GFP band intensity, we subtracted the intensity of this nonspecific band from that of the Bub1-GFP band. The abundance of Ndc80-GFP was similarly quantified using a strain that expressed Ndc80-GFP from the endogenous NDC80 promoter. Bottom: To calculate the average number of molecules per cell, we assumed that there are 1160 molecules of Ndc80 per cell as reported by Ghaemmaghami et al. (2003). The ratio of Bub1-GFP to Ndc80-GFP band intensities was multiplied by this number to obtain the average number of Bub1 molecules per cell. Black circles represent the numbers deduced from each experimental repeat. The horizontal lines indicate the average value. (C) The average number of Bub1-GFP molecules per unattached kinetochore as a function of the degree of Bub1-GFP overexpression in nocodazole-treated cells (mean ± 95% confidence intervals). Data from experiments involving the weaker pGalL promoter were fitted with a one-site specific binding model. The dissociation constant obtained from this fit was then used to constrain the fit for the data obtained with the pGAL1 promoter (also see Supplemental Figure S3A). (D) Recruitment of Bub1 by the unattached kinetochores requires phosphorylatable MELT repeats. Scatter plot displays the average number of Bub1-GFP molecules per Spc105 molecule in cells treated with nocodazole (mean ± 95% confidence intervals) when Bub1-GFP is overexpressed. The allele of Spc105 is indicated at the bottom. (E) The cumulative number of Bub3-mCherry molecules recruited by the cluster of unattached kinetochores in nocodazole-treated cells is tightly correlated with the number of Bub1-GFP molecules recruited by the same cluster (blue boxes indicate means calculated from binned data; error bars represent SD). Blue line displays linear regression constrained to pass through the origin. Gray lines indicate the Bub3 and Bub1 numbers measured in nocodazole-treated wild-type cells. (F) Change in the recruitment of Mad1-mCherry by unattached kinetochore clusters as a function of the number of Bub1-GFP molecules recruited by the respective clusters in nocodazole-treated cells that also overexpress Bub1-GFP (green circles indicate means calculated from pooled data; error bars represent SD). The curve represents one-site saturation binding fit to the data. Gray lines demarcate the average Mad1 and Bub1 numbers measured in nocodazole-treated wild-type cells.
The data so far show that each Spc105 recruits at most two Bub3-Bub1 complexes despite possessing the capacity to recruit six (Supplemental Figure S2C). Furthermore, this recruitment is not suppressed by Glc7 (Figure 3A). Following published observations, we reasoned that one or more of the following factors might modulate Bub3-Bub1 recruitment: 1) the cellular abundance of Bub3-Bub1 is low, 2) Mps1 phosphorylates only two or three of the six MELT repeats on average, 3) Spc105 is fully phosphorylated, but some of the MELpT are ineffective in recruiting Bub3-Bub1 (Primorac et al., 2013; Vleugel et al., 2013), and 4) steric hindrance disfavors the binding of more than two Bub3-Bub1 complexes to the same Spc105 molecule.
We first assessed the role of the cellular abundance of the Bub3-Bub1 complex in limiting Bub3-Bub1 recruitment. The intracellular concentration of this complex is determined by the concentrations of Bub3 and Bub1 and the dissociation constant of the Bub3-Bub1 complex (Larsen et al., 2007). Because the abundance of Bub1 is much lower than that of Bub3 (Supplemental Table S1), we reasoned that Bub1 overexpression should increase Bub3-Bub1 concentration. We used two different galactose-inducible promoters, pGalL and pGal1, to achieve different degrees of Bub1-GFP overexpression (Figure 3B). We grew these cells in predetermined galactose concentrations for ∼20 h, treated them with nocodazole to create unattached kinetochores, and then quantified Bub1-GFP recruited by the unattached kinetochores. We found that moderate overexpression of Bub1-GFP significantly increased its recruitment: on average, each Spc105 recruited ∼2 Bub1-GFP molecules (Figure 3C). When Bub1-GFP was strongly overexpressed, >>25-fold higher than the endogenous concentration (Figure 3B, bottom), each Spc105 recruited 5.6 ± 0.2 Bub1-GFP molecules on average (28 ± 0.8 Bub1-GFP molecules per kinetochore; Figure 3C). Thus, when Bub1 is highly abundant in the cell, nearly every MELT repeat in each Spc105 molecule binds one Bub3-Bub1 complex.
It is possible that experimental artifacts such as the weak dimerization of GFP contributed to increased kinetochore recruitment of Bub1-GFP upon its overexpression. To test this, we examined whether the observed localization of Bub1 required the MELpT sites. We overexpressed Bub1-GFP in a strain that expresses Spc105-5A(172T), an allele of Spc105 in which five of the six MELT motifs are nonphosphorylatable and hence incapable of binding Bub3-Bub1. Despite significant Bub1 overexpression, each Spc105-5A molecule recruited only one Bub1 molecule (Figure 3D). Thus the observed Bub1-GFP recruitment is mediated by MELT repeats. Any contribution from GFP dimerization is undetectable. This is consistent with the extremely low affinity of GFP dimers (dissociation constant, ∼110 μM; Zacharias et al., 2002), which is two to three orders of magnitude smaller than the affinity of the MELpT for Bub3-Bub1 (dissociation constant, ∼ 0.2–1 μM; Primorac et al., 2013).
We also examined whether the kinetochore contains a Bub1-binding site other than the MELT repeat. Structure of the ternary complex consisting of the Bub3-binding domain of Bub1, Bub3, and a peptide containing one phosphorylated MELT motif indicates that Bub1 depends on Bub3 for binding to MELpT but also enhances the affinity of the Bub3-MELpT interaction (Primorac et al., 2013). Therefore, if MELpT is the only binding site mediating the observed recruitment, then any increase in Bub1 binding should be accompanied by a corresponding increase in Bub3 recruitment. Consistent with this prediction, the number of kinetochore-bound Bub3 molecules maintained a 0.8:1 stoichiometry with kinetochore-bound Bub1 in cells that overexpressed Bub1 (Figure 3E). This experiment demonstrates that Bub1 overexpression is sufficient to increase the steady-state binding of the Bub3-Bub1 complex to unattached yeast kinetochores. Furthermore, Spc105 molecules in unattached yeast kinetochores are capable of recruiting up to six Bub3-Bub1 molecules, which shows that Spc105 is maximally phosphorylated under these conditions. Furthermore, these experiments show that each of the six MELT repeats in Spc105 is capable of binding one Bub3-Bub1 complex.
We used the enhanced recruitment of Bub3-Bub1 upon Bub1 overexpression to further confirm that Glc7 does not suppress Bub3-Bub1 recruitment by unattached kinetochores. We reasoned that the increased abundance of the Bub3-Bub1 under Bub1 overexpression conditions would more effectively reveal even a small increase in the number of MELpT motifs due to Glc7 inhibition. We inhibited Glc7 with calyculin A as before in cells that moderately overexpressed Bub1 and then quantified the recruitment of Bub1-GFP by unattached kinetochores. Bub1 recruitment did not increase in the presence of calyculin A compared with the levels measured under Bub1 overexpression conditions without the inhibitor (Supplemental Figure S3A). This result confirms that Glc7 activity does not influence the phosphorylation state of Spc105 in nocodazole-treated cells and further supports our finding that Spc105 is maximally phosphorylated in unattached kinetochores. Thus Bub1 abundance in the cell is the primary factor that limits Bub3-Bub1recruitment by Spc105.
We next examined whether the low-abundance Bub1 also suppresses the recruitment of Mad1-Mad2 by the kinetochores (Supplemental Table S1). If this is the case, then Bub1 overexpression should be sufficient to increase the recruitment of Mad1 to unattached kinetochores. Consistent with this expectation, we found that unattached kinetochores recruited significantly more Mad1 dimers in cells that overexpressed Bub1 (Figure 3F). At the highest Bub1 expression levels, the number of kinetochore-bound Mad1 dimers was threefold higher than for the wild type. The stoichiometry between Mad1 dimers and Bub1 decreased gradually from 2:1 at lower Bub1 concentrations to 1:1 at the highest concentration (Supplemental Figure S3B). The saturation in Mad1 level may be due to either incomplete phosphorylation of Bub1 by Mps1 or limited availability of soluble Mad1. As noted before, the nup60Δ strains used for these measurements increases the Mad1 recruitment relative to wild-type levels. Thus the availability of Mad1 also plays a significant role in dampening Mad1-Mad2 recruitment by unattached kinetochores in wild-type cells. In conclusion, although the yeast kinetochore is equipped and primed to recruit a large number of SAC proteins, vegetatively growing yeast cells use only a small fraction of this signaling capacity by expressing Bub1 at low levels and limiting the availability of free Mad1.
Our studies so far investigated the recruitment of Bub3-Bub1 mediated by Spc105 molecules that contain six MELT repeats within their phosphodomains (defined here as residues 1–329). It is possible that the spatial proximity of these repeats enhances Bub3-Bub1 recruitment. To test this, we measured the recruitment of Bub3-Bub1 by two Spc105 alleles containing only one phosphorylatable MELT motif, Spc105-5A(172T) or Spc105-5A(313T) (the phosphorylatable threonine is indicated by the number in parentheses; also see Figure 4A, bottom). We found that both alleles recruited Bub1 highly effectively such that every Spc105-5A molecule recruited one Bub1 molecule (1 ± 0.6 and 0.9 ± 0.5 for every spc105-5A(172T) or spc105-5A(313T); respectively; Figure 4A). Mad1 recruitment reflected the expected stoichiometry of 2:1 between Mad1 dimers and Bub1 molecules in both cases (Figure 4B).
FIGURE 4:

Binding of more than one Bub3-Bub1 molecule to an Spc105 phosphodomain is strongly disfavored. (A) Comparison of the number of Bub1-GFP molecules recruited per kinetochore in nocodazole-treated cells expressing wild-type Spc105 with those expressing Spc105-5A(172T) or Spc105-5A(313T) (mean ± 95% confidence intervals). Differences in mean values are not statistically significant. The sequences of the MELT repeats are noted at the bottom for comparison. (B) Comparison of the recruitment of Mad1-GFP under the same conditions as in A. Differences in mean values are not statistically significant. (C) Binding curves for MELpT and Bub3-Bub1 suggested by wild-type Spc105 (pale green) and the Spc105-5A alleles (dashed gray). Data from Figure 2C were replotted by converting the total number of Spc105 molecules in the cluster of unattached kinetochores into the number of MELpT repeats. The dashed curve displays the binding of Bub1 predicted by the affinity of spc105-5A molecules and the Bmax from Figure 2C. The open circles highlight the expected recruitment of Bub3-Bub1 as a function of the number of MELT repeats predicted by the two models. (D) Top, the two experimental schemes used to double the total number of MELT repeats in the kinetochore. Left, each Spc105 molecule contains two phosphorylatable MELT repeats. Right, in a strain expressing Spc105-5A(172T), rapamycin-induced dimerization of GFP-Spc105120:329-5A(313T)-Frb with Ndc80-2xFkbp12 is used to create kinetochores that contain twice as many phosphodomains as a wild-type cell. Because each phosphodomain contains one MELT motif, the number of MELT motifs per kinetochore is also doubled. Bottom, comparison of Bub1 recruitment by the indicated Spc105 alleles (mean ± 95% confidence intervals).
The recruitment of approximately one Bub1 by each Spc105-5A molecule is surprisingly large compared with the wild-type Spc105 containing six MELT repeats. This discrepancy is best understood by considering Bub1 recruitment by the Spc105-5A alleles in the context of the physiological binding curve between Spc105 and Bub3-Bub1. Because the Spc105-5A allele contains only one MELT motif, it is informative to first convert the x-axis of the binding curve from the concentration of Spc105 to that of MELpT repeats. Noting that 1) Bub3-Bub1 recruitment by Spc105 is additive (Figure 2C), 2) every MELT repeat is phosphorylated (Figure 3, A–D, and Supplemental Figure S3A), and 3) every MELT repeat can bind one Bub3-Bub1 complex (Figures 3C and 4A), we see that the concentration of Spc105 can be converted into that of MELpT (pale green squares in Figure 4C). This curve predicts the number of Bub3-Bub1 molecules that a given number of MELpT should recruit under physiological conditions. When the numbers of Bub3-Bub1 molecules recruited by the two spc105-5A alleles are graphed on the same plot (red squares in Figure 4C), it becomes clear that they are significantly higher than expected.
To understand quantitatively the implications of this finding, we examined the dissociation constant and maximal recruitment predicted by the MELpT-Bub3-Bub1 binding curve. The bimolecular interaction with ligand depletion model fitted to these data predicts an average dissociation constant KD ≈ 70 nM for the MELpT-Bub3-Bub1 complex (Figure 4C, pale green line). This value is approximately threefold to fourfold lower than the value measured in vitro, which was obtained from the binding of a partial fragment of Bub1 in complex with Bub3 to a peptide containing a phosphorylated MELT motif (Primorac et al., 2013). The fit also predicts the maximal recruitment of Bub3-Bub1 under physiological conditions, Bmax ≈ 29 nM. This value must be less than or equal to the endogenous Bub3-Bub1 concentration. Available data (Ghaemmaghami et al., 2003; Larsen et al., 2007) and our measurement of the number of Bub1 molecules in the nucleus validate this expectation (Supplemental Figure S3C). In contrast to the dissociation constant deduced from Bub1 recruitment by the wild-type Spc105, data for Bub1 recruitment by the two Spc105-5A alleles (red squares in Figure 4C) suggest a significantly lower dissociation constant: ∼1 nM. Thus the two binding curves predict distinctly different interaction affinities even though they pertain to the same biochemical.
To understand how the physiological recruitment of Bub3-Bub1 transitions from one binding affinity to the other, we created an allele of Spc105—Spc105-4A(172T, 313T)—that contains two phosphorylatable MELT motifs. The two binding affinities discussed earlier make distinct predictions regarding how Bub3-Bub1 recruitment should increase as a function of the number of MELpT. If the MELpT repeats in Spc105-4A behave like the MELpT in Spc105-5A, then there will be ∼76% increase in the number of Bub3-Bub1 molecules recruited by unattached kinetochores in nocodazole-treated cells (gray open circle in Figure 4C). On the other hand, if Spc105-4A behaves like the wild-type Spc105, then there will not be a significant increase in Bub3-Bub1 recruitment under the same conditions (pale green open circle in Figure 4C). To test this, we quantified Bub1 recruitment by unattached kinetochore clusters by subjecting Spc105-4A–expressing cells to nocodazole (Figure 4D). Despite containing two MELT repeats, spc105-4A(172T, 313T) recruited the same number of Bub1 molecules in nocodazole-treated cells as spc105-5A(172T) (or ∼0.8 ± 0.4 Bub1 per Spc105). Thus spc105-4A(172T, 313T) behaves like the wild-type protein rather than the spc105-5A alleles. These observations may be explained by invoking steric hindrance: the binding of the first Bub3-Bub1 molecule to the Spc105 phosphodomain may disfavor the binding of the second molecule, even though additional binding sites are available within the phosphodomain.
To validate this behavior further, we sought to circumvent the apparent steric hindrance by creating kinetochores that contain twice as many phosphodomains each containing one MELT motif. Because each phosphodomain contains only one MELT motif, each should now be capable of recruiting one Bub3-Bub1 complex. To create kinetochores with twice the number of Spc105 phosphodomains, we used rapamycin-induced dimerization to conditionally anchor Spc105 phosphodomains each containing one phosphorylatable MELT motif to the kinetochore in cells expressing Spc105-5A (Figure 4D, top). In the absence of rapamycin, unattached kinetochores in nocodazole-treated cells recruited the expected number of Bub1 molecules (Figure 4D). However, when we anchored an additional phosphodomain to these kinetochores by adding rapamycin, they recruited ∼33% more Bub1 molecules (Figure 4D). Although the observed increase is less than the expected 76% increase, it shows that kinetochores containing twice as many phosphodomains with one MELT motif recruit significantly more Bub3-Bub1 than those containing the wild-type number of phosphodomains with two or more MELT repeats. Therefore we conclude that the presence of multiple MELT repeats in the same phosphodomain suppresses Bub3-Bub1 recruitment at physiological concentration of Bub3-Bub1.
Our observations bring into question why Spc105 contains six MELT repeats. The cumulative recruitment of Bub3-Bub1and Mad1-Mad2 by unattached kinetochores in nocodazole-treated cells is the same whether the phosphodomain of Spc105 contains one MELT motif or six. The number of signaling kinetochores is a key parameter here. Nocodazole treatment creates a relatively large number of unattached kinetochores that all contribute to SAC signaling. Even if individual kinetochores are partially defective in signaling, the cumulative signal generated by a large number of such kinetochores might be sufficient to arrest the cell cycle. Can a small number of kinetochores that are partially defective in signaling also arrest cell division? To answer this question, we assessed the growth of strains expressing different alleles of Spc105-5A on media containing low doses of the microtubule-destabilizing drug benomyl (Figure 5). Because of the decreased stability of spindle microtubules, the kinetochores are expected to require more time to form stable, bipolar attachments. Therefore a dividing cell is likely to contain a small number of unattached kinetochores, and these kinetochores must activate and sustain the SAC robustly to arrest cell division. Consistent with this expectation, the spc105-6A strain, which is SAC-null, cannot grow on media containing benomyl. We found that the three Spc105-5A alleles that we tested also did not grow in medium containing low doses of benomyl (Figure 5A). This is surprising because when we generated unattached kinetochores in the same strains using nocodazole treatment, the kinetochores recruited the same cumulative number of SAC proteins as wild-type strains (Figure 4, A and B). We next sought to determine the minimum number of MELT repeats that confers benomyl resistance to vegetatively growing yeast cells. We found that the cited Spc105-4A allele fared marginally better than the Spc105-5A alleles, but it did not grow as well as the wild-type strain. In contrast, an allele of Spc105 containing three phosphorylatable MELT repeats grew just as well as wild-type cells, showing that a minimum of three MELT repeats are likely to be essential for growth in medium containing low doses of benomyl (Figure 5B).
FIGURE 5:
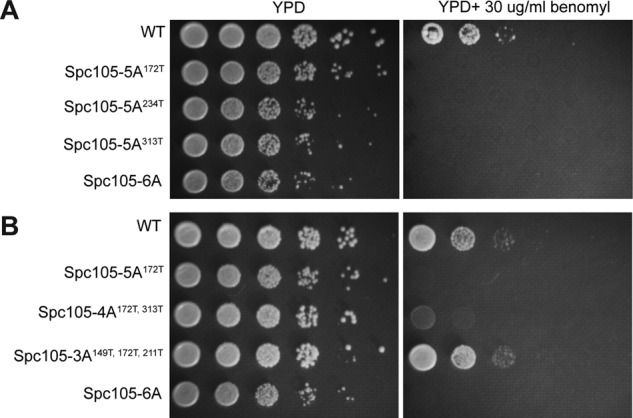
Benomyl sensitivity of cells expressing Spc105 alleles containing reduced numbers of MELT repeats. (A) Benomyl sensitivity of strains expressing three different Spc105 alleles, each containing only one phosphorylatable MELT motif. (B) Spc105 alleles containing two MELT repeats show a marginal improvement in benomyl resistance, whereas the Spc105 allele containing three MELT repeats imparts a wild-type level of benomyl resistance.
DISCUSSION
The systematic analysis of the SAC protein recruitment by unattached kinetochores presented here defines the steady-state operation of the respective biochemical reactions. It uncovers the kinetochore-intrinsic and -extrinsic mechanisms that modulate protein recruitment in budding yeast. Note that there are additional key quantities that we did not measure: the kinetics of SAC protein recruitment and the steady-state rate of turnover of the kinetochore-bound molecules. Because of the low abundance of SAC proteins, accurate measurement of these parameters, especially protein turnover, will require specialized analysis methods. We also did not quantify the physiological outcomes of the experimental perturbations: the mitotic delay induced and chromosome segregation accuracy. These outcomes depend on the operation of the kinetochore-based reactions reported here, as well as on the concentrations and diffusivity of additional proteins—Mad2, Mad3/BubR1, and Cdc20—that make up the mitotic checkpoint complex. Further experimentation is necessary to obtain these data and complete the quantitative understanding of SAC signaling in budding yeast. Nonetheless, our work provides novel insights into the regulation of the kinetochore-based reactions of the SAC.
Our data define the biochemical properties of key kinetochore-based signaling reactions. They show that Mps1 likely phosphorylates Spc105 completely in unattached yeast kinetochores and primes it to recruit the maximum number of Bub3-Bub1 molecules. We also find that each Spc105 is capable of binding six Bub3-Bub1 molecules. This is in contrast to the human KNL-1, in which a large fraction of MELT repeats appear to be degenerate because of their very weak affinity for Bub3-Bub1 (Vleugel et al., 2015). We find that Glc7 does not appreciably affect Bub3-Bub1 recruitment in unattached kinetochores. However, it is also possible that Bub3-Bub1 binding to the MELpT itself hinders Glc7 from dephosphorylating MELpT. Under physiological conditions, Spc105 recruits Bub3-Bub1 modestly in spite of its capacity to bind six Bub3-Bub1 molecules, for two reasons. First, the binding of multiple Bub3-Bub1 molecules to the same Spc105 phosphodomain is disfavored. The mechanistic basis of this effect is unknown. We speculate that the reduced binding results from conformational changes in the unstructured domain of Spc105 brought about by the binding of large protein complexes. This effect may serve a biological function by ensuring that the limited supply of Bub3-Bub1 molecules is equally distributed among all Spc105 molecules. Further studies are needed to elucidate the mechanistic basis of this apparent negative cooperativity and test whether it is functionally significant. In addition to this effect, the low abundance of the Bub3-Bub1 complex also limits its own recruitment and that of Mad1-Mad2. This modulation is likely necessary to ensure that the generation of the wait-anaphase signal does not scale linearly with the number of unattached kinetochores (Collin et al., 2013; Krefman et al., 2015). Given that one or only a few unattached kinetochores can delay cell division, concurrent signaling from all unattached kinetochores at maximal capacity is unnecessary and may likely lead to the accumulation of excess inhibitory signal that unnecessarily delays cell division even after attachments are made (Doncic et al., 2005; Sear and Howard, 2006). Finally, we also show that the yeast kinetochore is capable of recruiting two Mad1-Mad2 heterotetramers for every Bub3-Bub1. The molecular mechanism underlying this observation is unknown. However, doubling the Mad1-Mad2 recruitment can be expected to double the steady-state signaling from the kinetochore and thus make the signal more robust.
The difference in the effectiveness of SAC signaling in strains expressing Spc105-5A as measured by two different assays is notable. In nocodazole-treated cells containing a relatively large number of unattached kinetochores that activate the SAC, Spc105-5A alleles containing only one phosphorylatable MELT motif appear to be capable of recruiting SAC proteins and arresting cell division. However, for cells to proliferate in medium containing low doses of benomyl, at least three MELT repeats per Spc105 are essential. Under this condition, the number of unattached kinetochores in the cell is likely to be small. With only a small number of SAC active kinetochores, Spc105 molecules with three or more MELT repeats are necessary to recruit SAC proteins rapidly and in sufficient steady-state numbers that the SAC is robustly activated. Note that we examined the benomyl sensitivity of only one allele each of Spc105-4A (of the 15 possible alleles) and Spc105-3A (of the 20 possible alleles). Because each MELT motif has a unique position, as well as Bub3-Bub1 binding affinity, each Spc105-4A or Spc105-3A allele may differ in its ability to promote cell growth under this condition. It is also noteworthy that whereas a minimum of three MELT repeats are necessary for survival on benomyl-containing medium, the recruitment of two Bub3-Bub1 molecules per Spc105 when the cell contains only two unattached kinetochores suggests that two MELT motifs should be sufficient (Figure 1B). To address this potential discrepancy, it will be necessary to undertake a detailed analysis of the influence of individual characteristics of each of the six MELT motifs, as well as of the sequence of chromosome attachment to the spindle, in cells subjected to low doses of benomyl.
Because the biochemical reactions of SAC signaling studied here are highly conserved, similar mechanisms likely operate in the human kinetochore. It has been shown that human KNL-1 containing only one MELT motif flanked by additional Bub1 binding sites (known as the KI motifs) arrest cell division when the cells are treated with nocodazole (Krenn et al., 2014). However, our data indicate that it will be necessary to test whether this KNL-1 allele arrests cell division when the dividing cell contains only one or a few unattached kinetochores (Vleugel et al., 2013). Aberrant expression of SAC genes is very common in cancer cells (Kops et al., 2005). Our data suggest that underexpression may reduce the effectiveness of SAC signaling specifically in situations in which only one or a few unattached kinetochores are required to stall cell division. A systematic study of the regulation of SAC protein recruitment by the human kinetochore will illuminate such mechanistic connections between SAC protein concentrations and high chromosomal instability commonly found in cancer cells.
MATERIALS AND METHODS
Plasmid and strain construction
Strains and plasmids used in this study are listed in Supplemental Tables S2 and S3, respectively. Plasmids encoding mutations in Spc105 phosphodomain (pAJ335, pAJ407, pAJ554) were constructed from pSB1878 through quick-change mutagenesis (London et al., 2012). pAJ548 containing the anchorable minimal phosphodomain of Spc105 was constructed from pAJ349 using quick change mutagenesis (Aravamudhan et al., 2015). GFP(S65T) and mCherry tagged to proteins were used to localize kinetochores and quantify either the number of kinetochores or the copy numbers of SAC proteins. The tags were introduced at the endogenous locus through homologous recombination of PCR-amplified fragments (Bahler et al., 1998). A seven–amino acid linker (sequence RIPGLIN) separates the fluorescent protein from the C-terminus of the tagged protein. To drive gene expression using a galactose promoter, either the pGal1 or the pGalL promoter was inserted upstream from the start codon of the open reading frame of the selected gene via homologous recombination (Janke et al., 2004). Rapamycin-induced dimerization of Fkbp12 and Frb was used to conditionally localize Mps1-Frb at the C-terminus of either Ndc80-2xFkbp21 or Mtw1-2xFkbp12 as described previously (Haruki et al., 2008; Aravamudhan et al., 2015).
Strains expressing Spc105 alleles were constructed by plasmid shuffling. Briefly, the wild-type SPC105 was deleted in a strain supplemented with the wild-type SPC105 gene expressed from a centromeric plasmid (yCP50) containing the auxotrophic marker URA3. Plasmids encoding Spc105 alleles (pAJ335:spc105-5A(172T) or pAJ407: spc105-5A(313T)) were linearized with BstEII and integrated at the Leu-2 locus. Strains encoding only the Spc105 mutant alleles were generated by counterselecting for yCP50 on 5-FOA.
All strains were grown at 32°C in the yeast extract and peptone (YP) supplemented with 2% glucose (YPD) or galactose and raffinose in different ratios (adding up to 2%) or synthetic medium supplemented with essential amino acids and 2% glucose as needed. Yeast cells were imaged at room temperature for <30 min after sample preparation.
Microscope setup and fluorescence intensity measurement
Fluorescence microscopy was performed on a Nikon Ti-E inverted microscope with a 1.4 numerical aperture, 100× oil-immersion objective. Experiments involving one pair of unattached kinetochores were conducted with the 1.5× Optovar lens. Ten-plane image stacks were acquired using a piezostage to focus at different planes along the depth of the cell, with 200-nm separation between the planes.
Total fluorescence of kinetochore clusters (16 kinetochores in metaphase and ∼10 in nocodazole) was quantified by integrating intensities over a 6 × 6–pixel region centered on the maximum-intensity pixel. The median intensity of pixels immediately surrounding the 6 × 6–pixel area was used to correct for background fluorescence. Analysis was performed using a Matlab program as described previously (Aravamudhan et al., 2014). Analysis of single-kinetochores was performed manually using ImageJ. In this case, the integrated intensity of a 6 × 6 nuclear region adjacent to the kinetochore cluster was used in background subtraction.
Deduction of the number of protein molecules from fluorescence intensity measurements
Copy number of Spc105 per kinetochore.
In metaphase, each budding yeast kinetochore contains an invariable copy number of molecules of core kinetochore subunits: approximately eight copies of Ndc80 and Spc25 molecules and five molecules of Spc105-GFP (Joglekar et al., 2006; Aravamudhan et al., 2015). However, we discovered that the fluorescent protein tag may reduce the number of Spc105-GFP molecules per kinetochore. The kinetochore incorporates five copies of Spc105 when GFP is at or proximal to Spc105 C-terminus and eight copies of Spc105 when GFP is at or proximal to Spc105 N-terminus (Supplemental Figure S1B). The maximal number is the same as the number of Ndc80 complex molecules (Aravamudhan et al., 2013). However, to express GFP-Spc105, the fusion gene was integrated at an ectopic locus, which may result in protein overexpression. Therefore, in this study, we assume that the yeast kinetochore contains five Spc105 molecules. Note that even if this number turns out to be higher, it will only affect the stoichiometry between Spc105 and SAC proteins. The absolute number of SAC proteins will remain unchanged.
Calculation of the brightness of GFP and mCherry under the imaging conditions.
To deduce the number of SAC proteins from the measured fluorescence signal, we first calculated the brightness of GFP and mCherry molecules. The brightness values were obtained from the known number of Ndc80 complex molecules in the yeast kinetochore and the measured fluorescence signal of metaphase kinetochore clusters in a strain expressing Ndc80-GFP and Spc25-mCherry (AJY939). AJY939 was grown under same imaging conditions as the experimental strain, and the calibration data were acquired throughout the duration of this study. This calibration accounted for changes in the microscope setup over time.
Rationale behind the use of GFP and mCherry tags as reporters of protein copy number.
Intensities of GFP-tagged SAC proteins recruited by the kinetochore were reproducible between multiple strains with different genotypes (e.g., Figure 4, A and B). Thus the expression levels of SAC proteins are not strain specific, and GFP signal intensity is a robust reporter of protein numbers. Therefore, whenever possible, we used the GFP tag for quantification of SAC proteins. The use of mCherry as a reporter of protein numbers can be problematic because the maturation efficiency of mCherry varies significantly from strain to strain and produces variable mCherry fluorescence, even though the copy number of the protein tagged with mCherry itself is not expected to change (Joglekar et al., 2013). Therefore we mostly used mCherry to either visualize a cellular structure or estimate the number of kinetochores by tagging a kinetochore subunit with mCherry. In the latter case, the known number of kinetochores in the metaphase cluster enables us to obtain a strain-specific calibration for the mCherry signal corresponding to a single kinetochore. This calibration was used to deduce the number of unattached kinetochores in nocodazole-treated cells (Supplemental Figure S1C).
In a subset of the experiments, we examined how the number of an mCherry-tagged SAC protein recruited by a cluster of unattached kinetochores varies as a function of another GFP-tagged SAC protein recruited by the same cluster in nocodazole-treated cells (Figure 3, E and F). In these cases, we reasoned that the number of an SAC protein recruited by a fixed number of unattached kinetochores would be the same irrespective of the fluorescent protein tag on that protein. This reasoning implicitly assumes that the expression level of the SAC protein is insensitive to the fluorescent protein tag. Therefore the measured GFP and mCherry fluorescence for a given SAC protein (measured in two different strains) should correspond to the same number of molecules.
Identifying unattached kinetochores in strains in which additional Spc105 phosphodomains were anchored to the kinetochore
In this experiment (Figure 4D), Bub1 was mCherry labeled, but the kinetochore did not contain any fluorescently labeled subunits. Therefore we could not directly determine the number of unattached kinetochores in the absence of rapamycin. However, after the cells were treated with rapamycin, spc105120:329-5A-Frb-GFP molecules dimerized with the Fkbp12-tagged Ndc80 complex subunit. We assumed that the Bub1-recruiting clusters in cells treated with nocodazole contained the same number of unattached kinetochores in the presence and absence of rapamycin. In the latter case, we identified the unattached kinetochore cluster as the one that recruited high levels of Bub1.
Generating a single pair of unattached kinetochores
To generate experimentally a single pair of unattached kinetochores, we used the centromere reactivation system described by Tanaka et al. (2005). Briefly, we constructed strains containing a galactose-inducible promoter proximal to the CEN3 locus and a methionine-repressible promoter controlling CDC20 expression. The strains were grown to mid log phase in synthetic medium containing 2% raffinose and lacking methionine and then synchronized in α-factor for 2 h. The G1-synchronized cells were then transferred for 2 h to YP supplemented with 2% raffinose, 200 mM methionine to repress CDC20 and arrest the cells in metaphase, and 2% galactose to activate CEN3 and prevent kinetochore assembly. The cells thus synchronized in metaphase were transferred quickly to 2% glucose to activate the CEN3, which then assembles a kinetochore. We imaged SAC proteins recruited by these kinetochores within the next 15 min. A majority of the kinetochores achieved bipolar attachments by the end of the experiment.
Nocodazole treatment to depolymerize the mitotic spindle
Log-phase cultures were synchronized in G1 by treatment with α-factor for 2 h and then released into nocodazole (15 μg/ml) for 2 h to depolymerize the spindle and generate unattached kinetochores. We and others previously reported that this treatment generates two classes of kinetochore clusters in the cell: one or two smaller clusters consisting of ∼10 kinetochores and a larger kinetochore cluster that localizes proximal to the spindle pole bodies (Gillett et al., 2004; Aravamudhan et al., 2015). Only the smaller cluster contains completely unattached kinetochores that recruit Bub1 and Mad1. Kinetochores in the larger cluster recruit very little Bub1 and undetectable levels of Mad1, potentially because these kinetochores retain residual microtubule attachment. The number of SAC proteins localizing to kinetochores reported in this work corresponds to the number of molecules recruited by the smaller, SAC-active kinetochore cluster.
Anchoring Mps1 and Bub1 to the kinetochore using rapamycin-induced dimerization
To conditionally force Mps1 to localize to the kinetochores, we used previously described methodology involving the rapamycin-induced dimerization of Fkbp12 and Frb domains (Aravamudhan et al., 2015). Briefly, Mps1 was tagged with Frb, and the kinetochore subunit—either Ndc80 or Mtw1—was tagged with 2xFkbp12. When rapamycin is added to the growth medium (1 μg/ml), Mps1-Frb dimerizes with the Fkbp12-tagged protein. To quantify Bub1-GFP and Mad1-GFP recruited to the kinetochores, we treated log-phase cultures with rapamycin for 1 h before microscopy.
To quantify the Bub1 molecules in the cell in Supplemental Figure S3D, we expressed Bub1-Frb-GFP from the BUB1 locus using the endogenous Bub1 promoter. These cells also expressed a selected kinetochore subunit with the 2xFkbp12 fused at its C-terminus. These cells were treated with rapamycin for 45 min. This allows most of the Bub1-Frb-GFP in the nucleus to dimerize with the Fkbp12 domains and thus localize to the kinetochores. Cells were then prepared for microscopy.
Fitting equilibrium binding models
In fitting equilibrium binding models, we assume that the observed recruitment of SAC proteins by unattached kinetochores represents equilibrium binding between a soluble SAC protein and its receptor in the kinetochore. For the recruitment of Bub3-Bub1 complex, the receptor in the kinetochore is the phosphorylated MELT repeat in Spc105. For the recruitment of the Mad1-Mad2 heterotetramer, the receptor is the phosphorylated Bub3-Bub1 complex. We used two simple models to fit the experimental data. In these experiments, either the number of receptors (as in Figures 2C, 3F, and 4C) or the concentration of the soluble SAC protein was varied (as in Figure 3, C and E). The first model is based on saturation binding (Figures 2A and 3C):
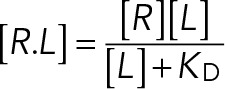 |
where [R] and [L] are the concentrations of the receptor and SAC protein, respectively, [R.L] is the concentration of the receptor–SAC protein complex, and KD is the dissociation constant for the receptor–SAC protein complex.
In cases in which we expected the limited abundance of SAC proteins to limit steady-state binding, we modeled the interaction as a bimolecular interaction governed by ligand depletion (Figures 1E, 2C, and 4C). In this case, the concentration of the ligand–receptor complex is given by
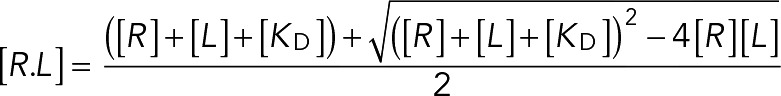 |
All data fits were performed using GraphPad Prism software.
Calyculin A treatment for inhibiting Glc7
G1-arrested cells were released into the cell cycle by washing out the α-factor. These cells were treated with nocodazole for ∼1 h to activate the SAC and additionally with 2.5 μM calyculin A (208851; EMD Millipore, Billerica, MA) for 1 h to inhibit Glc7. Then samples were drawn for immunoblotting to verify Glc7 inhibition and also for live-cell imaging to quantify Bub1 recruitment by the unattached kinetochores.
Immunoblotting and quantification
To detect Snf1 phosphorylation, samples were prepared as described previously (Orlova et al., 2008). Briefly, 2 OD (at λ600) of cells were lysed in a boiling water bath for 5 min before extraction of proteins with alkaline treatment. The sample was suspended in a final volume of 100 μl of SDS–PAGE sample buffer. The proteins in the extract were resolved on 8% SDS-polyacrylamide gels and transferred to nitrocellulose. The blot was then blocked with 5% bovine serum albumin and probed with the phosphospecific antibody rabbit α-AMPk(172T) (L4188; Cell Signaling, Danvers, MA) overnight at a 1:1000 dilution in Tris-buffered saline/Tween 20 (TBST) at 4°C. The blots were washed and treated with goat α-rabbit peroxidase-conjugated antibody at a 1:10,000 dilution in TBST for 1 h before treatment with enhanced chemiluminescence reagent for visualization of phosphorylated Snf1. To detect total protein, the samples were probed on another blot in parallel using α-histidine antibody (Sigma-Aldrich, St. Louis, MO) at a 1:2000 dilution for 1.5 h in 2% milk to detect the polyhistidine stretch in Snf1.
For the quantification of Bub1 abundance, whole lysates were prepared from cells expressing either Bub1-GFP or Ndc80-GFP from their respective endogenous promoters as described previously (Aravamudhan et al., 2014). Three OD (at λ600) of log-phase culture was lysed into sample buffer by repeated vortexing with glass beads and intermittent boiling. The proteins were separated on 8% SDS-polyacrylamide gels and transferred to polyvinylidene fluoride membrane. The blots were probed with a 1:1000 dilution of the mouse α-GFP antibody (GFP(B-2):sc-9996; Santa Cruz Biotechnology, Dallas, TX). Peroxidase-conjugated α-mouse immunoglobulin G (1:5000; A-4416; Sigma-Aldrich) treated with ECL was used to visualize the protein. The bands corresponding to GFP-tagged proteins (Bub1 or Ndc80) were quantified in ImageJ by manually integrating pixel intensities over equal areas covering each band of interest, and background was subtracted using intensities from an equal area immediately above or below the band of interest in each lane. The intensities were normalized relative to Ndc80-GFP, and protein abundance was estimated assuming 1160 molecules of Ndc80 per cell (Ghaemmaghami et al., 2003). Bub1-GFP in wild-type cells was not detectable (even when twofold more sample was loaded), even though fourfold less Ndc80-GFP could be detected under the same conditions (unpublished data). Therefore we estimate the abundance of bub1 to be <145 molecules/cell. Note that Bub1-GFP was expressed in these cells and could be readily detected by fluorescence microscopy at unattached kinetochores when the cells were treated with nocodazole (Figure 1 C).
Benomyl sensitivity testing
Tenfold serial dilutions of log-phase cultures were replica-spotted on YPD or plates containing 30 μg/ml benomyl. Plates were incubated at 32°C and photographed after 2 d.
Spindle length
Spindle length in metaphase with various degrees of Spc105 depletion was measured as described previously (Aravamudhan et al., 2014).
Supplementary Material
Acknowledgments
We thank Sue Biggins for sharing plasmids, Diane Fingar for sharing the pAMPK antibody, and Nate Krefman for helpful discussions. We also thank Mara Duncan and Edward Salmon for comments on the manuscript. This work was supported by National Institute of General Medical Sciences R01-GM-105948.
Abbreviations used:
- GFP
green fluorescent protein
- SAC
spindle assembly checkpoint.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0007) on May 11, 2016.
REFERENCES
- Aravamudhan P, Felzer-Kim I, Gurunathan K, Joglekar AP. Assembling the protein architecture of the budding yeast kinetochore-microtubule attachment using FRET. Curr Biol. 2014;24:1437–1446. doi: 10.1016/j.cub.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravamudhan P, Felzer-Kim I, Joglekar AP. The budding yeast point centromere associates with two Cse4 molecules during mitosis. Curr Biol. 2013;23:770–774. doi: 10.1016/j.cub.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravamudhan P, Goldfarb AA, Joglekar AP. The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat Cell Biol. 2015;17:868–879. doi: 10.1038/ncb3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Chen R-H, Brady DM, Smith D, Murray AW, Hardwick KG. The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol Biol Cell. 1999;10:2607–2618. doi: 10.1091/mbc.10.8.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin P, Nashchekina O, Walker R, Pines J. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol. 2013;15:1378–1385. doi: 10.1038/ncb2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke CA, Schaar B, Yen TJ, Earnshaw WC. Localization of CENP-E in the fibrous corona and outer plate of mammalian kinetochores from prometaphase through anaphase. Chromosoma. 1997;106:446–455. doi: 10.1007/s004120050266. [DOI] [PubMed] [Google Scholar]
- Dick AE, Gerlich DW. Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol. 2013;15:1370–1377. doi: 10.1038/ncb2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Ben-Jacob E, Barkai N. Evaluating putative mechanisms of the mitotic spindle checkpoint. Proc Natl Acad Sci USA. 2005;102:6332–6337. doi: 10.1073/pnas.0409142102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Gillett ES, Espelin CW, Sorger PK. Spindle checkpoint proteins and chromosome-microtubule attachment in budding yeast. J Cell Biol. 2004;164:535–546. doi: 10.1083/jcb.200308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Heinrich S, Geissen EM, Kamenz J, Trautmann S, Widmer C, Drewe P, Knop M, Radde N, Hasenauer J, Hauf S. Determinants of robustness in spindle assembly checkpoint signalling. Nat Cell Biol. 2013;15:1328–1339. doi: 10.1038/ncb2864. [DOI] [PubMed] [Google Scholar]
- Hill A, Bloom K. Genetic manipulation of centromere function. Mol Cell Biol. 1987;7:2397–2405. doi: 10.1128/mcb.7.7.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma Y, Sacristan C, Pachis ST, Adamopoulos A, Kuijt T, Ubbink M, von Castelmur E, Perrakis A, Kops GJ. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science. 2015;348:1264–1267. doi: 10.1126/science.aaa4055. [DOI] [PubMed] [Google Scholar]
- Hoffman DB, Pearson CG, Yen TJ, Howell BJ, Salmon ED. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol Biol Cell. 2001;12:1995–2009. doi: 10.1091/mbc.12.7.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, Fung E, Proctor M, Shokat KM, Zhang C, Davis RW, et al. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol. 2008;4:498–506. doi: 10.1038/nchembio.100. [DOI] [PubMed] [Google Scholar]
- Howell BJ, Moree B, Farrar EM, Stewart S, Fang G, Salmon ED. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr Biol. 2004;14:953–964. doi: 10.1016/j.cub.2004.05.053. [DOI] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Ji Z, Gao H, Yu H. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science. 2015;348:1260–1264. doi: 10.1126/science.aaa4029. [DOI] [PubMed] [Google Scholar]
- Joglekar AP, Bouck DC, Molk JN, Bloom KS, Salmon ED. Molecular architecture of a kinetochore-microtubule attachment site. Nat Cell Biol. 2006;8:581–585. doi: 10.1038/ncb1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar A, Chen R, Lawrimore J. A sensitized emission based calibration of FRET efficiency for probing the architecture of macromolecular machines. Cell Mol Bioeng. 2013;6:369–382. doi: 10.1007/s12195-013-0290-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmler S, Stach M, Knapp M, Ortiz J, Pfannstiel J, Ruppert T, Lechner J. Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. EMBO J. 2009;28:1099–1110. doi: 10.1038/emboj.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerscher O, Crotti LB, Basrai MA. Recognizing chromosomes in trouble: association of the spindle checkpoint protein Bub3p with altered kinetochores and a unique defective centromere. Mol Cell Biol. 2003;23:6406–6418. doi: 10.1128/MCB.23.18.6406-6418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Krefman NI, Drubin DG, Barnes G. Control of the spindle checkpoint by lateral kinetochore attachment and limited Mad1 recruitment. Mol Biol Cell. 2015;26:2620–2639. doi: 10.1091/mbc.E15-05-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V, Overlack K, Primorac I, van Gerwen S, Musacchio A. KI motifs of human Knl1 enhance assembly of comprehensive spindle checkpoint complexes around MELT repeats. Curr Biol. 2014;24:29–39. doi: 10.1016/j.cub.2013.11.046. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Larsen NA, Al-Bassam J, Wei RR, Harrison SC. Structural analysis of Bub3 interactions in the mitotic spindle checkpoint. Proc Natl Acad Sci USA. 2007;104:1201–1206. doi: 10.1073/pnas.0610358104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Biggins S. Mad1 kinetochore recruitment by Mps1-mediated phosphorylation of Bub1 signals the spindle checkpoint. Genes Dev. 2014;28:140–152. doi: 10.1101/gad.233700.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Ceto S, Ranish JA, Biggins S. Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr Biol. 2012;22:900–906. doi: 10.1016/j.cub.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle MW, Kim T, Hattersley N, Espeut J, Cheerambathur DK, Oegema K, Desai A. A Bub1-Mad1 interaction targets the Mad1-Mad2 complex to unattached kinetochores to initiate the spindle checkpoint. J Cell Biol. 2014;204:647–657. doi: 10.1083/jcb.201311015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25:R1002–R1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- Orlova M, Barrett L, Kuchin S. Detection of endogenous Snf1 and its activation state: application to Saccharomyces and Candida species. Yeast. 2008;25:745–754. doi: 10.1002/yea.1628. [DOI] [PubMed] [Google Scholar]
- Overlack K, Primorac I, Vleugel M, Krenn V, Maffini S, Hoffmann I, Kops GJ, Musacchio A. A molecular basis for the differential roles of Bub1 and BubR1 in the spindle assembly checkpoint. Elife. 2015;4:e05269. doi: 10.7554/eLife.05269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsky BA, Nelson CR, Biggins S. Protein phosphatase 1 regulates exit from the spindle checkpoint in budding yeast. Curr Biol. 2009;19:1182–1187. doi: 10.1016/j.cub.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primorac I, Weir JR, Chiroli E, Gross F, Hoffmann I, van Gerwen S, Ciliberto A, Musacchio A. Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. Elife. 2013;2:e01030. doi: 10.7554/eLife.01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RJ, Lusk CP, Dilworth DJ, Aitchison JD, Wozniak RW. Interactions between Mad1p and the nuclear transport machinery in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:4362–4374. doi: 10.1091/mbc.E05-01-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sear RP, Howard M. Modeling dual pathways for the metazoan spindle assembly checkpoint. Proc Natl Acad Sci USA. 2006;103:16758–16763. doi: 10.1073/pnas.0603174103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer F, Hieter P. Centromere DNA mutations induce a mitotic delay in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1992;89:8908–8912. doi: 10.1073/pnas.89.19.8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Kitamura E, Tanaka TU. Live-cell analysis of kinetochore-microtubule interaction in budding yeast. Methods. 2010;51:206–213. doi: 10.1016/j.ymeth.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Mukae N, Dewar H, van Breugel M, James EK, Prescott AR, Antony C, Tanaka TU. Molecular mechanisms of kinetochore capture by spindle microtubules. Nature. 2005;434:987–994. doi: 10.1038/nature03483. [DOI] [PubMed] [Google Scholar]
- Vleugel M, Omerzu M, Groenewold V, Hadders MA, Lens SM, Kops GJ. Sequential multisite phospho-regulation of KNL1-BUB3 interfaces at mitotic kinetochores. Mol Cell. 2015;57:824–835. doi: 10.1016/j.molcel.2014.12.036. [DOI] [PubMed] [Google Scholar]
- Vleugel M, Tromer E, Omerzu M, Groenewold V, Nijenhuis W, Snel B, Kops GJ. Arrayed BUB recruitment modules in the kinetochore scaffold KNL1 promote accurate chromosome segregation. J Cell Biol. 2013;203:943–955. doi: 10.1083/jcb.201307016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, von Mering C. Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics. 2015;15:3163–3168. doi: 10.1002/pmic.201400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne DJ, Funabiki H. Kinetochore function is controlled by a phospho-dependent coexpansion of inner and outer components. J Cell Biol. 2015;210:899–916. doi: 10.1083/jcb.201506020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen KW, Warren CD, Chen O, Kwok T, Hieter P, Spencer FA. Systematic genome instability screens in yeast and their potential relevance to cancer. Proc Natl Acad Sci USA. 2007;104:3925–3930. doi: 10.1073/pnas.0610642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- Zhang G, Lischetti T, Nilsson J. A minimal number of MELT repeats supports all the functions of KNL1 in chromosome segregation. J Cell Sci. 2014;127:871–884. doi: 10.1242/jcs.139725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.