

The anchor structure of CD16 affects its binding affinity in a ligand-specific manner. The ligand binding affinity decreases for human IgG1 but increases for murine IgG2a when the anchor is changed from full to partial to none. Removing N-glycosylation from CD16 also increases the ligand binding affinity.
Abstract
Isoforms of the Fcγ receptor III (FcγRIII or CD16) are cell surface receptors for the Fc portion of IgG and important regulators of humoral immune responses. Different ligand binding kinetics of FcγRIII isoforms are obtained in three dimensions by surface plasmon resonance and in two dimensions by a micropipette adhesion frequency assay. We show that the anchor structure of CD16 isoforms isolated from the cell membrane affects their binding affinities in a ligand-specific manner. Changing the receptor anchor structure from full to partial to none decreases the ligand binding affinity for human IgG1 (hIgG1) but increases it for murine IgG2a (mIgG2a). Removing N-glycosylation from the CD16 protein core by tunicamycin also increases the ligand binding affinity. Molecular dynamics simulations indicate that deglycosylation at Asn-163 of CD16 removes the steric hindrance for the CD16-hIgG1 Fc binding and thus increases the binding affinity. These results highlight an unexpected sensitivity of ligand binding to the receptor anchor structure and glycosylation and suggest their respective roles in controlling allosterically the conformation of the ligand binding pocket of CD16.
INTRODUCTION
In humans, type III cell surface receptors for the Fc portion of immunoglobulin (Ig) G (Fcγ receptor III or CD16) are encoded by two genes, A and B, which give two protein products, CD16a and CD16b, respectively (Kimberly et al., 2002; Nimmerjahn and Ravetch, 2008). CD16a is expressed on macrophages, mast cells, and natural killer cells as a transmembrane receptor (Ravetch and Perussia, 1989; Selvaraj et al., 1989). Expressed exclusively on neutrophils, CD16b is the only Fc receptor (FcR) anchored by a glycosyl-phosphatidylinositol (GPI) linker to the plasma membrane (Selvaraj et al., 1988). CD16b is polymorphic, with two alleles—neutrophil alloantigen 1 (NA1) and 2 (NA2). Despite the lack of a signaling component, CD16b plays an active role in triggering Ca2+ mobilization and neutrophil degranulation (Kimberly et al., 1990; Unkeless et al., 1995). Furthermore, in conjunction with FcγRIIa (CD32a), CD16b activates phagocytosis, degranulation, and oxidative burst, which lead to clearance of opsonized pathogens by neutrophils. Moreover, soluble forms of CD16 containing the Ig-like extracellular domains of the receptor circulate in plasma (Teillaud et al., 1994).
CD16 are highly glycosylated molecules, with five (CD16a), four (CD16bNA1), or six (CD16bNA2) N-glycosylation sites on their extracellular domains (Figure 1). CD16a on NK cells has a higher affinity for ligands than on monocytes due to differential cell type–specific glycosylation (high mannose– and complex-type oligosaccharides for NK cell CD16a but not monocyte CD16a), despite their identical protein cores (Edberg and Kimberly, 1997). Single-residue replacements of the four N-glycosylation sites (Asn-39, Asn-75, Asn-163, and Asn-170) of CD16bNA1 show that the N163Q mutant binds monomeric IgG with higher affinity than the other mutants (Drescher et al., 2003). In addition, preincubation of wild-type CD16bNA1-transfected cells with tunicamycin (an inhibitor of N-glycosylation) results in increased binding of monomeric IgG, whereas the same treatment of cells transfected with N163Q mutant of CD16bNA1 has no effect. These results suggest that glycosylation at Asn-163, which is shared by all three CD16 membrane isoforms and located in the ligand binding pocket (Sondermann et al., 2000; Radaev et al., 2001; Ferrara et al., 2011), regulates the affinity of CD16 for IgG (Drescher et al., 2003). However, the extent to which the glycosylation affects the ligand binding affinity and kinetics has not been quantified. The structural basis for the glycosylation effects also has not been elucidated.
FIGURE 1:
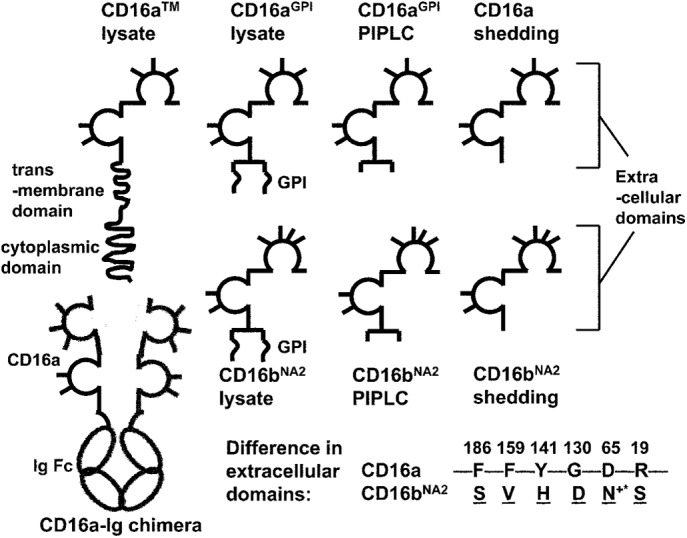
Schematics of solubilized CD16 isoforms with different anchor structures and soluble CD16a–Ig chimera. The extracellular domains of CD16 are depicted as two Ig-like globules, with the N-glycosylation sites shown as sticks. An additional glycan is part of the GPI anchor, which acts as a linker between the C-terminus of the peptide and the phosphatidylinositol group. The amino acids in extracellular domains that differ between CD16a and CD16bNA2 are listed. +*The gained glycosylation site in CD16bNA2 due to the change D65 → N65.
The cocrystal structures of CD16bNA2 and CD16a in complex with an Fc fragment of human IgG1 (hFc1) have been independently solved by several groups (Sondermann et al., 2000; Radaev et al., 2001; Ferrara et al., 2011). Although the crystals obtained have different space group symmetries, the structures are almost identical, showing a 1:1 stoichiometry for binding. CD16 adopts the characteristic heart-shaped domain arrangement as described for soluble FcγRIIb and other soluble FcRs. Compared with the standalone structure, CD16 further opens its interdomain angle by 10° when it is liganded with hFc1. Concurrently, hFc1 opens asymmetrically upon complex formation, upon which both CH2 domains bend away from the C2 axis of the homodimeric hFc1, but one CH2 domain is dislocated more than the other.
Ligand binding kinetics of CD16 has been studied using surface plasma resonance (SPR; Galon et al., 1997; Maenaka et al., 2001; Li et al., 2007). In SPR, one binding partner is immobilized onto a sensor surface over which the other binding partner flows in a fluidic phase to interact in three dimensions. Soluble aglycosylated CD16NA2 produced by Escherichia coli used in two studies yielded drastically different kinetic rates (Galon et al., 1997; Maenaka et al., 2001), despite the fact that the respective materials from the two groups produced the same crystal structure (Sondermann et al., 2000; Radaev et al., 2001). One group also compared glycosylated with aglycosylated CD16bNA2 and showed that they have similar binding affinity for human IgG1 (hIgG1; Galon et al., 1997). The similarity in three-dimensional (3D) affinity is at odds with the observation that the glycosylation of CD16 greatly affects its ligand binding (Edberg and Kimberly, 1997; Drescher et al., 2003).
Ligand binding of CD16 triggers signaling and effector functions such as phagocytosis and antibody-dependent cellular cytotoxicity (Nagarajan et al., 1995b). Cell surface CD16 binds small immune complex in three dimensions and IgG-opsonized surfaces in two dimensions (Nagarajan et al., 1995b). Ligand binding kinetics of CD16 has also been measured in two dimensions by micropipette adhesion assay, which analyzes receptor–ligand interactions across the junctional gap between two apposing surfaces (Chesla et al., 1998, 2000; Williams et al., 2000a, b, 2001; Shashidharamurthy et al., 2009). This assay can examine effects of membrane organization and cellular environment of the interacting molecules that cannot be addressed by SPR. The differences between two-dimensional (2D) and 3D binding have been highlighted by studies of binding of T-cell receptors (TCRs) and coreceptors with peptide-major histocompatibility complex (pMHC) molecules (Huang et al., 2010; Huppa et al., 2010; Adams et al., 2011; Jiang et al., 2011; Sabatino et al., 2011). Indeed, the micropipette assay shows that, compared with transmembrane (TM) CD16a (CD16aTM), CD16aGPI binds faster and with higher affinity to human and rabbit IgGs but slower and with lower affinity to murine IgG2a (mIgG2a; Chesla et al., 2000). Previous study suggests that the membrane anchor influences ligand binding by conformational change of CD16 (Chesla et al., 2000).
In the study, we further explored the effect of anchor structure of CD16 on its ligand binding using three different methods to generate soluble CD16s with different anchor structures, allowing us to isolate the effect of linker segment from the membrane anchor itself on the intrinsic binding parameters. We also tested how glycosylation of the receptor affected ligand binding, by using tunicamycin treatment to block the addition of sugar moieties to the protein core. Our results show that affinity of CD16 binding to hIgG1 correlates with receptor anchor structures, and this correlation is inverted upon switching ligand from hIgG1 to mIgG2a, suggesting that long-range conformational change on the receptor anchor could propagate to the ligand binding epitope. Aglycosylated CD16 has higher affinity for hIgG1 than the glycosylated forms, suggesting an important role of posttranslational modification in regulating receptor binding affinity. Molecular dynamics (MD) simulations show that deglycosylation of CD16 removes the steric hindrance for CD16–hIgG1 binding, resulting in increased binding affinity.
RESULTS
Solubilized CD16 captured on microspheres specifically binds IgG coated on red blood cells
To test for binding specificity, adhesion frequencies of IgG coated on red blood cells (RBCs) to solubilized CD16 of different forms and anchor structures (Figure 1) captured by 214.1 precoated on microspheres were compared with several controls. Adhesion frequencies were measured with a 2-s contact duration. As exemplified using microspheres incubated with various CD16 lysates (Figure 2A), the ∼50% adhesion frequencies to hIgG1-coated RBCs were reduced to ∼10% by the anti-CD16 antibody, 3G8. Furthermore, use of microspheres incubated with bovine serum albumin (BSA) instead of CD16aTM lysate or RBCs not coated with anything (but undergoing the same CrCl3 procedure except adding IgG) reduced the adhesion frequencies to the background level (∼2%).
FIGURE 2:
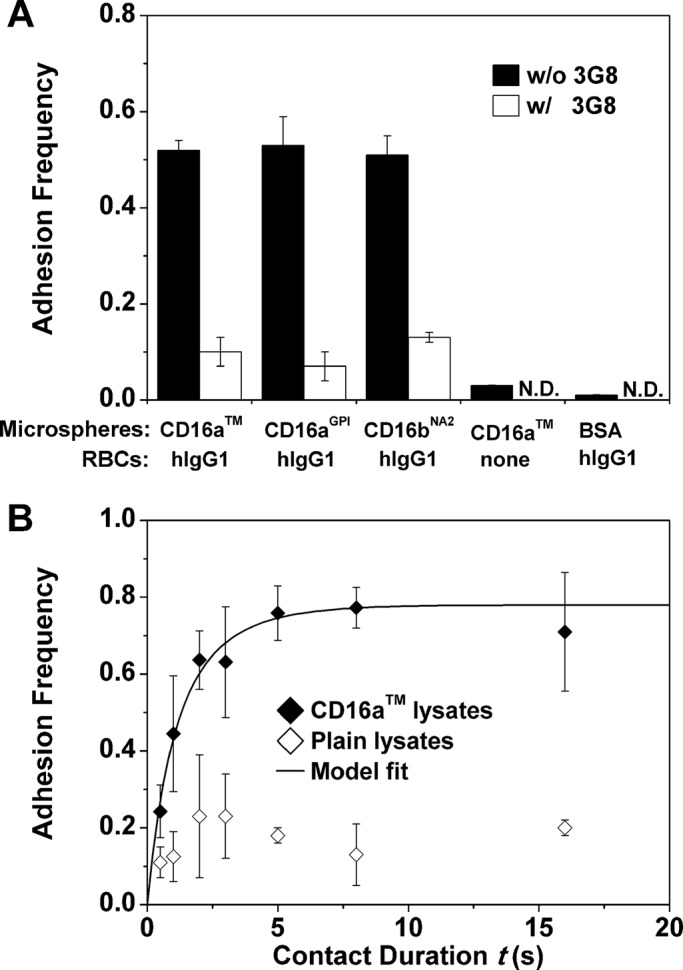
Microspheres bearing CD16 bound RBCs coated with hIgG1 specifically. (A) Adhesion frequency of CD16 lysate–coated microspheres to hIgG1-coated RBCs was substantially higher than the nonspecific adhesions, controlled using a blocking anti-CD16 (3G8) or microspheres incubated with BSA (instead of lysates of CD16 expressing CHO cells) or RBCs not coated with anything. N.D., not done. Adhesion frequencies were measured with a 2-s contact duration. (B) Adhesion frequency vs. contact duration (t) binding curves. The two sets of data (points) were obtained using RBCs coated with hIgG1 to contact 214.1-precoated microspheres incubated with lysates from CHO cells expressing CD16aTM (filled diamonds) or plain CHO cells (open diamonds). For the former, the specific adhesion frequency Pa after removing the nonspecific adhesion frequency (Eq. 1) is shown and fitted by Eq. 2 (curve). Data are mean ± SEM of five RBC-microsphere pairs with 100 contacts each per bar or point.
The kinetic information is embedded in the curves of adhesion frequency versus contact duration, as exemplified by the data in Figure 2B, measured with CD16aTM lysate–coated microspheres and hIgG1-coated RBCs using seven contact durations, t, from 0.5 to 16 s. Five microsphere–RBC pairs were tested at each contact duration, and 100 contacts were repeated for each pair to calculate a total adhesion frequency Pt (number of adhesions observed divided by the 100 contacts). Nonspecific adhesion frequency, Pn, was controlled using the 214.1-coated microspheres incubated with lysates of plain CHO cells (Figure 2B). The specific adhesion frequency Pa was then calculated using the following equation (Williams et al., 2001):
|
|
The Pa versus t binding curve was fitted by Eq. 2 together with the separately measured receptor and ligand densities (mr and ml, respectively; Chesla et al., 1998):
|
|
to estimate Ka and koff, the 2D affinity (in μm2) and off-rate (in s−1), respectively. The 2D on-rate can be calculated from kon = Kakoff. The Ka value is lumped with the contact area Ac in the curve fit, which is called collectively the effective 2D affinity. We kept Ac constant in all experiments.
CD16aGPI lysate binds hIgG1 with a higher affinity than CD16aTM lysate
Previously we showed that CD16aGPI expressed on CHO cells had a higher 2D affinity for total human IgG than CHO cell CD16aTM (Chesla et al., 2000). This result was confirmed by a new set of micropipette experiments (Figure 3A). Here we measured binding frequencies at multiple contact durations and then fitted the data to Eq. 2 to obtain affinities and off-rates as shown in Figure 2B. We showed that the differential 2D affinities observed previously using CHO cell CD16 and total human IgG were preserved despite the fact that in the new experiments, the CD16a membrane isoforms were isolated from the cell surface and captured by 214.1 on the microspheres to interact with hIgG1. The higher AcKa values of solubilized than cell surface receptors may partly be due to a higher Ac value for the former than the latter because microspheres have a smoother surface than CHO cells (Wu et al., 2007), resulting in greater effective contact area for the former than the latter. On the other hand, CD16aGPI lysate and CD16aTM lysate had comparable 2D off-rates for dissociation from hIgG1 (Figure 3B), consistent with previous findings for cell-surface CD16a membrane isoforms (Chesla et al., 2000).
FIGURE 3:
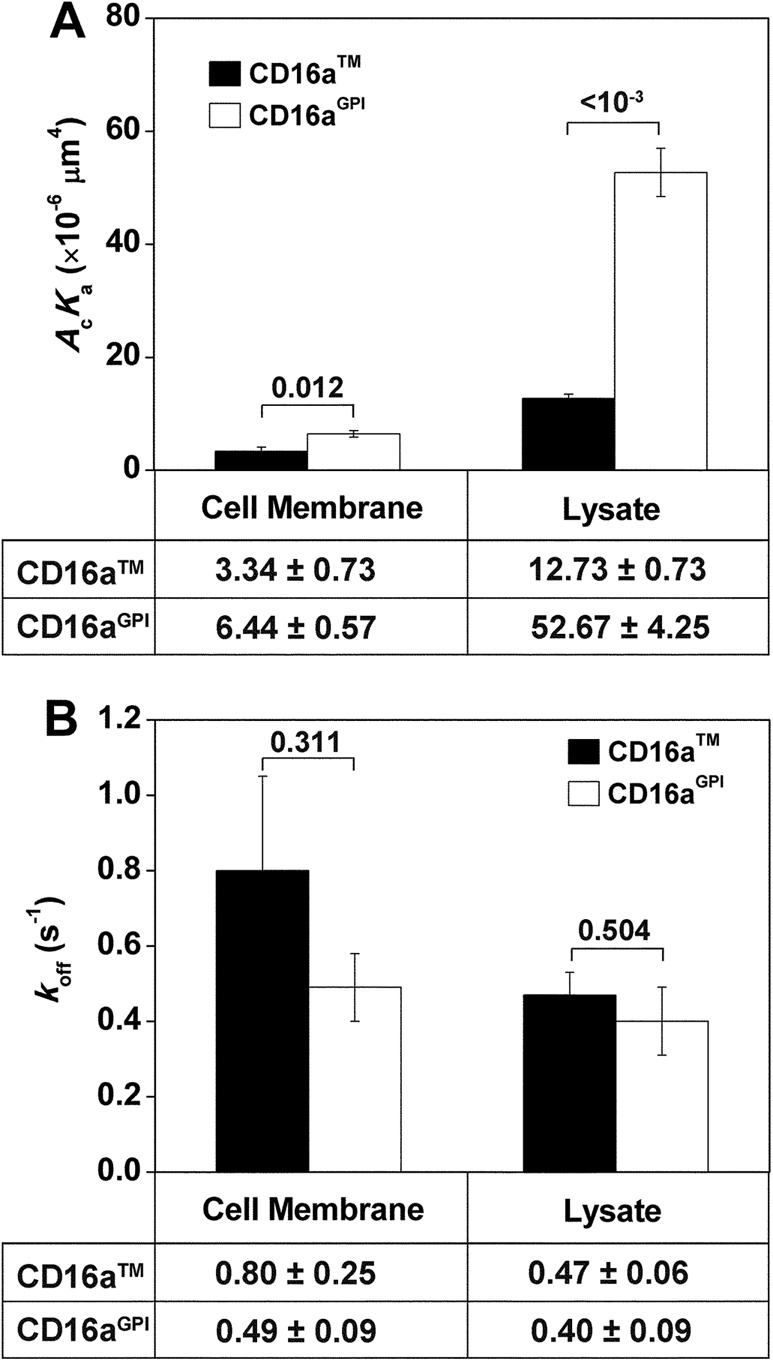
Comparison of 2D affinities (A) and off-rates (B) of CD16aTM and CD16aGPI for hIgG1. CD16 molecules were expressed on CHO cell surface or captured on 214.1-precoated microspheres from CHO cell lysates. Data are presented as mean ± SEM from fitting Eq. 2 to specific adhesion frequencies measured at multiple contact durations (for CD16 on CHO cell surface, six contact durations ranging from 0.25 to 8 s, three RBC–CHO cell pairs with 100 contacts each per contact duration; for CD16 lysates, seven contact durations ranging from 0.5 to 16 s, five RBC–microsphere pairs with 100 contacts each per contact duration) as in Figure 2B. Data values are below the plots; p values from Student’s t test are above the data bars.
CD16 anchor structure affects 2D binding affinity for hIgG1
The findings that hIgG1 bound with a higher 2D affinity to CD16aGPI than CD16aTM even after the receptors were captured from the cell lysates on the microspheres (Figure 3A) exclude differential diffusivities, flexibilities, heights, orientations, and organizations on the membrane, as well as any other cell-associated factors between the two isoforms, as possible causes for the affinity difference. We therefore focused on the structural differences in the membrane anchor per se. We hypothesized that the membrane anchor affects ligand binding by inducing conformational changes in the ligand binding site (Chesla et al., 2000). Because the membrane anchor is one Ig domain away from the ligand binding site, the proposed anchor effect on binding affinity must be exerted allosterically. Perturbing the structure along this allosteric pathway of regulation is therefore predicted to alter the binding affinity.
To test this hypothesis, we used lysis, phosphatidylinositol-specific phospholipase C (PIPLC) treatment, and spontaneous shedding to obtain different anchor structures of solubilized receptors from CD16aGPI or CD16bNA2-expressing CHO cells. Lysing the cells keeps the molecule’s original GPI anchor. PIPLC treatment enzymatically cleaves the diacylglycerol moiety of the GPI anchor. Note that PIPLC does not affect N-glycans on CD16. Spontaneous shedding results from the action of metalloproteinases bound to the cell membrane, which produces molecules with only the extracellular domain (Lanier et al., 1989). The micropipette adhesion assay was applied to measure 2D affinities between hIgG1 and these molecules at an 8-s contact duration, long enough for the interaction to reach equilibrium (Figure 2B). Specific adhesion frequencies, Pa, were obtained by removing background adhesion frequencies from total adhesion frequencies using Eq. 1 and then substituting into the following equation to calculate the effective 2D affinities:
|
|
This equation is a transformation of the equilibrium (i.e., t → ∞) version of Eq. 2.
Changing the anchor structure of CD16aGPI from full to partial to none resulted in a progressive decrease in its affinity for hIgG1 (Figure 4A, middle). The same result was seen with CD16bNA2, also a GPI-anchored molecule (Figure 4A, right). Further confirmation was obtained using CD16aTM, as the molecule with the full anchor structure plus cytoplasmic tail (lysate) showed a much higher affinity for hIgG1 than the molecule with no anchor (shedding; Figure 4A left). The PIPLC treatment was not used because PIPLC cannot cleave the polypeptide transmembrane anchor of CD16aTM. Student’s t test confirmed that the affinity differences within each group of CD16 were significant (p values in Figure 4A). These results support our hypothesis regarding allosteric regulation of ligand binding affinity by receptor anchor.
FIGURE 4:
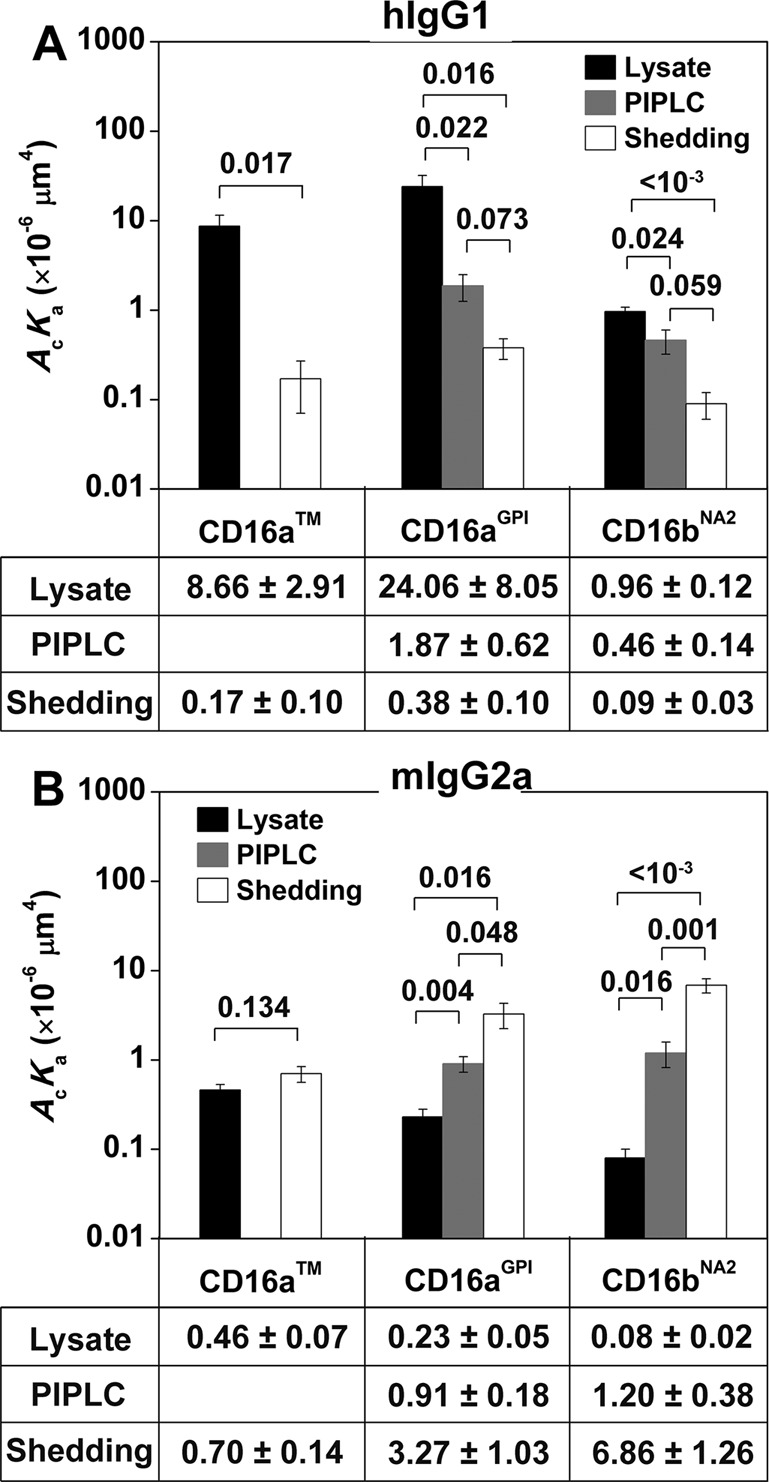
Comparisons of 2D affinities of hIgG1 (A) and mIgG2a (B) for CD16. CD16 molecules are captured on 214.1-precoated microspheres from CHO cell lysates, supernatants of CHO cells treated with PIPLC, or supernatants of CHO cells subjected to shedding treatment. Adhesion frequencies were measured with an 8-s contact duration and converted to 2D effective affinities using Eq. 3. Data are mean ± SEM of 5–15 RBC–microsphere pairs with 100 contacts each per bar. Data values are below the plots; p values from Student’s t test are above data bars.
The anchor effect on ligand binding is inverted when hIgG1 is replaced by mIgG2a
The correlation between CD16a anchor structure and its affinity was inverted when hIgG1 was changed to mIgG2a for all membrane isoforms of CD16 (CD16aTM, CD16aGPI, and CD16bNA2), such that for each CD16 membrane isoform, the solubilized receptor from lysates with the full anchor structure had the lowest affinity, that from shedding with no anchor had the highest affinity, and that cleaved from PIPLC treatment (for CD16aGPI and CD16bNA2 only) with partial GPI anchor had intermediate affinity (Figure 4B). Again, Student’s t test confirmed the significance of the differences in 2D affinities (p values in Figure 4B).
Glycosylation of CD16 molecules affects their ligand binding affinities
The effect of the receptor anchor structure on the ligand binding affinity may be changed by perturbations in the structure of the ligand binding site, as it resides on the other end of the proposed allosteric pathway of binding affinity regulation. One possible structure is the glycan attached to Asn-163 of CD16, which is inside the binding pocket of the CD16–hFc1 complex (Sondermann et al., 2000; Radaev et al., 2001; Ferrara et al., 2011). We thus prepared deglycosylated CHO cell CD16 molecules by adding tunicamycin, a widely used N-glycosylation inhibitor, to culture medium of CD16-expressing CHO cells for 40 h. This treatment did not alter CD16 expression significantly, as revealed by fluorescence-activated cell sorting (FACS; unpublished data). The micropipette adhesion frequency assay was used to estimate 2D affinities and off-rates of CD16aTM, CD16aGPI, and CD16bNA2 for hIgG1. As expected, for all three CD16 isoforms, deglycosylation increased their effective 2D affinities for hIgG1 (Figure 5A). By comparison, the 2D off-rates were comparable or slightly different between aglycosylated and glycosylated CD16 molecules (Figure 5B).
FIGURE 5:

Effects of tunicamycin treatment on CD16–hIgG1 binding. (A) The 2D affinities and (B) off-rates of CD16 isoforms expressed on CHO cell-surface and soluble CD16–Ig chimera for hIgG1 obtained from fitting Eq. 3 to specific adhesion frequencies measured at six contact durations ranging from 0.25 to 8 s (three RBC–CHO cell or RBC–RBC pairs with 50 or 100 contacts each per contact duration) as in Figure 2B. (C) Representative Scatchard plot analysis for saturation binding of CLBFcgran-1 to CD16aTM. (D) The 3D affinities of CD16 isoforms expressed on CHO cell surface for CLBFcgran-1 obtained by Scatchard plot analysis as illustrated in C. In A, B, and D, data both without and with tunicamycin treatment are presented as mean ± SEM. Data values are below the plots; p-values from Student’s t test are above data bars.
To exclude the possibility that the increase of effective 2D affinities of CD16 for hIgG1 with tunicamycin treatment was due to a cellular effect, we measured 2D binding of a soluble CD16a–Ig chimera for hIgG1. In agreement with the results of CHO cell CD16, the effective 2D binding affinity of hIgG1 was fourfold higher for CD16a–Ig secreted from CHO cells with tunicamycin treatment than without (Figure 5A, far right), confirming that the affinity increases in the cell-surface CD16 for hIgG1 were due to the change in the receptor molecules. The two-orders-of-magnitude higher effective 2D affinities of hIgG1 for soluble CD16a–Ig than for CHO cell-surface CD16a, regardless of whether they were for glycosylated or deglycosylated molecules, are consistent with the increase by one to two orders of magnitude in effective 2D affinities of hIgG1 for CD16a lysates on microspheres (Figure 3A); both can be explained by the increases in effective contact area, Ac. This is consistent with our previous report that CD16bNA2 reconstituted on RBCs has a ∼50-fold-higher effective 2D affinity for hIgG than the same CD16bNA2 reconstituted or transfected on CHO cells (Williams et al., 2001).
Finally, we measured 3D affinities of an anti-CD16 monoclonal antibody (mAb; CLBFcgran-1) for different CD16 isoforms using saturation binding experiments and Scatchard plot analysis (Figure 5C). Consistently, deglycosylation also increased the 3D affinity of CLBFcgran-1 for CD16 (Figure 5D). These results support our hypothesis that glycosylation perturbs the structure of CD16.
Deglycosylation of CD16 enhances CD16–hFc1 binding
To understand the structural basis for the effects of CD16 glycosylation on its ligand binding, we performed all-atom, explicit-solvent MD simulations for both glycosylated and aglycosylated CD16a liganded with hFc1, which contains two glycans on Asn-297. The starting structure of the simulation of the glycosylated CD16a-hFc1 complex was a crystal structure (Protein Data Bank 3SGJ; Ferrara et al., 2011) in which two N-glycans are attached to Asn-46 and Asn-163. The starting structure of the aglycosylated CD16a-hFc1 complex was obtained by removing in silico the glycans of CD16a in the crystal structure. After 10 ns in the simulations, the root mean square deviations of the Cα atoms of the complexes reached a plateau, suggesting that the systems were equilibrated (Supplemental Figure S1). The 40-ns period after equilibration was used for further analysis.
To measure how glycosylation of CD16 affects the binding, we calculated the contact times of interaction for residue pairs between hFc1 and CD16. Two residues are defined as in contact if any pair of their heavy atoms of side chains (Cα atom for glycine) are within 4.5 Å. Then the contact time is calculated as the fraction of the total time that the two residues are in contact. The difference in contact time between the aglycosylated and glycosylated complexes is shown in Figure 6. Positive or negative value indicates that the interaction is strengthened or weakened upon deglycosylation, respectively. Some interactions between the N-terminal loops of hFc1 and CD16 are weakened (blue spheres in Figure 6C). However, more interactions between the CH2 domain of hFc1 and CD16 are strengthened (red spheres in Figure 6C). Of interest, the strengthened interactions are mostly located near the glycan at Asn-163 of CD16 (green sticks in Figure 6C). This result suggests that deglycosylation of CD16 clears the steric hindrance for the CD16–hFc1 binding.
FIGURE 6:

Deglycosylation of CD16 enhances the CD16-hFc1 binding. (A) Difference in contact time between aglycosylated and glycosylated CD16-hFc1 complexes for residue pairs that interact between chain A of hFc1 and CD16. Two residues are defined as in contact if any pair of their heavy atoms of side chains (Cα atom for glycine) are within 4.5 Å distance. Then the contact time is calculated as the fraction of the total time that the two residues are in contact. (B) Difference in contact time between aglycosylated and glycosylated CD16-hFc1 complexes for residue pairs that interact between chain B of hFc1 and CD16. In A and B, only pairs with >0.1 absolute difference are shown. The calculations were done on the last 40 ns of the simulations, with one snapshot per 10 ps. (C) Structure of the glycosylated CD16–hFc1 complex (PDB 3SGJ). Chains A and B of hFc1 are shown in pink and gray, respectively. CD16 is in green. Sticks represent glycans attached to proteins. Red spheres show residues whose interactions strengthened upon deglycosylation; blue spheres show residues whose interactions weakened.
DISCUSSION
The Fc portions of antibodies can modulate immune responses through their interactions with Fc receptors. Different Fc receptors have different cell-specific expression patterns, signaling mechanisms, and ligand binding affinities, which enable specific cellular functions (Nimmerjahn and Ravetch, 2010). Previously we showed that different membrane anchor forms (TM vs. GPI) of the same FcγRIII molecule could modulate their ligand binding affinities (Chesla et al., 2000). Here we extended this work by showing that the anchor structure of CD16 affects the ligand binding affinity even in an acellular system. We also demonstrated that glycosylation provides another important means for Fc receptors to modulate their ligand binding affinities. In addition, results from our MD simulations suggest a structural basis for deglycosylation to increase the ligand binding affinity.
A significant new finding of the present work is the preservation of the previously observed anchor effect (Chesla et al., 2000) after replacement of the CD16-expressing CHO cells by solubilized CD16-bearing microspheres (Figure 3). A concern for using solubilized CD16 is that the exposed membrane anchor could induce nonspecific interactions. To test this possibility, we used an anti-CD16 antibody, 3G8, to block the CD16–IgG binding. The binding was reduced by fourfold to fivefold (Figure 2A), indicating that the nonspecific binding due to exposed anchor is negligible. Also note that the same mAb (214.1) was used to capture different solubilized CD16 molecules, therefore presenting the same molecular segment to the IgG ligand for binding. In spite of this, changing the anchor structure, which represents a molecular segment extending away from the 214.1 capture point on the other side of the ligand binding site, still affects ligand binding affinity (Figure 4). This allows us to rule out the differential diffusivities, flexibilities, heights, orientations, and organizations on the membrane, as well as any other cell-associated factors of these molecules, as possible causes for the observed anchor effects.
Another important new finding is the preservation of the ligand specificity of the anchor effect, such that the changes of affinity of CD16 for hIgG1 with changes of anchor structure are opposite to that for mIgG2a: CD16 with full membrane anchor has highest affinity for hIgG1 but lowest affinity for mIgG2a, whereas CD16 without anchor has the lowest affinity for hIgG1 but the highest affinity for mIgG2a (Figure 4). The observation further supports that the anchor effect is caused by CD16 itself rather than its cellular environment. To dissect the ligand specificity of the anchor effect, we compared the amino acid sequences of hIgG1 and mIgG2a (Supplemental Figure S2). The two sequences are highly conserved, with >60% identity. However, in the regions directly interacting with CD16, three residues are different. As in the CD16–hIgG1 complex structure, His-268 and Glu-269 of the hFc1 B chain interact with Lys-132 of CD16 (Supplemental Figure S3). His-268 and Glu-269 are changed to Glu and Asp, respectively, in mIgG2a. This change increases the negative charge of IgG and thus may strengthen the interaction with Lys-132 of CD16. Moreover, Ala-327 of the hFc1 B chain is in contact with His-135/136. When Ala-327 is replaced by Asp in mIgG2a, additional attractive electrostatic interactions are expected between Asp-327 and His-135/136. Therefore both changes in the binding interface increase interactions between IgG and CD16, which may contribute to the higher affinity of mIgG2a for CD16 than hIgG1. Note that the CD16 in the complex crystal structure is a soluble form, that is, it does not have an anchor and corresponds to the shedding form used in our micropipette experiments. We speculate that the binding interface is changed through an allosteric mechanism when CD16 has an anchor, and the change depends on the anchor form. For the PIPLC-cleaved form of CD16, the interacting residues between CD16 and IgG may all be conserved between hIgG1 and mIgG2a, so that their affinities for CD16 are similar. However, for CD16 with a full anchor, other, nonconserved residues may interact with CD16, and these interactions are favored for hIgG1, so that hIgG1 has higher affinity for CD16 than mIgG2a.
Although our findings may seem surprising, many examples of allosteric regulation of ligand binding have been reported. In interactions between selectins and ligands (Lou et al., 2006; Lou and Zhu, 2007), between platelet glycoprotein Ibα and von Willebrand factor (Yago et al., 2008), and between bacterial FimH receptor and mannose ligand (Le Trong et al., 2010), force-induced conformational changes may give rise to catch bonds (Dembo et al., 1988; Marshall et al., 2003). In integrins, allosteric conformational changes are related to inside-out and outside-in signaling (Luo et al., 2007) and may cause catch bonds (Kong et al., 2009; Chen et al., 2010, 2012; Xiang et al., 2011). Closer to our findings, the GPI-anchored protein Thy-1 has been shown to lose reactivity with several mAbs and a polyclonal antibody after anchor cleavage by phospholipase C, indicating that the GPI anchor affects the protein conformation (Barboni et al., 1995; Kukulansky et al., 1999).
Notwithstanding the foregoing examples, that truncating the membrane anchor of a receptor may affect its ligand binding has been underappreciated. It is a common practice to make recombinant soluble receptors with membrane anchors truncated and replaced by different tags for purification by affinity chromatography. Almost all structural determinations of extracellular domains of cell surface receptors by crystallography and nuclear magnetic resonance use molecules made in this way. Molecules so made are also widely used for kinetics measurements by SPR (Galon et al., 1997; Maenaka et al., 2001; Li et al., 2007). It is an implicit but usually untested assumption that the soluble receptors have identical structures and ligand binding properties as the native molecules on the cell membrane. This assumption has been challenged by work revealing drastic differences between 3D kinetics of TCR-pMHC binding measured by SPR using soluble molecules and 2D kinetics of the same interactions measured in situ with cell surface molecules (Huang et al., 2010; Huppa et al., 2010; Adams et al., 2011; Jiang et al., 2011; Sabatino et al., 2011). The findings of the present work also serve as reminder for caution to investigators who use soluble molecules for structural and functional studies.
To obtain direct evidence for propagation of conformational changes from the membrane anchor to the ligand binding site may require structural comparison among CD16 molecules with different anchor structures, which are currently not available. We reasoned that perturbing the structures that display the conformational changes at the ligand binding site might alter the ligand-specific binding affinity. We chose the N-glycan attached to Asn-163 of CD16, which resides in the binding pocket of the CD16–hFc1 complex (Sondermann et al., 2000; Radaev et al., 2001; Ferrara et al., 2011). Targeting glycosylation also has interest in its own right because a substantial number of the recombinant soluble receptors used in structure determination and SPR measurements are produced in aglycosylated forms by E. coli (Sondermann et al., 2000; Maenaka et al., 2001; Radaev et al., 2001). It is also an implicit but usually untested assumption that the aglycosylated receptors have identical structures and ligand binding properties as the native (and often glycosylated) molecules on the cell membrane. Using tunicamycin to deglycosylate CHO cell CD16 isoforms, we found about threefold increase in the effective 2D binding affinity. This is consistent with the observation that wild-type CD16bNA1-transfected cells treated with tunicamycin exhibit increased binding of monomeric IgG (Drescher et al., 2003). Of interest, tunicamycin treatment also increases the binding of another Fc receptor, FcαR (CD89), to IgA (Xue et al., 2010). It may be a common property that deglycosylation of FcR causes increased binding of Ig. Of greater interest, deglycosylation of one N-glycosylation site among others, Asn-163 of CD16 or Asn-58 of CD89, contributes most to the increase of the respective binding to IgG or IgA (Drescher et al., 2003; Xue et al., 2010). In fact, Asn-163 of CD16 and Asn-58 of CD89 are located in the binding interfaces of the CD16–IgG (Sondermann et al., 2000; Radaev et al., 2001; Ferrara et al., 2011) and CD89–IgA complexes (Herr et al., 2003), respectively.
Two previous SPR studies reported similar 3D affinities but drastically different kinetics for hIgG1 of recombinant aglycosylated soluble CD16bNA2 containing the extracellular region of the receptor (Galon et al., 1997; Maenaka et al., 2001). The off-rate measured in this work is similar to that found by Maenaka et al. (2001). On the other hand, Galon et al. (1997) reported similar 3D affinities for both glycosylated and aglycosylated soluble CD16bNA2, contradicting our finding that glycosylated and aglycosylated CD16 have different 2D affinities for hIgG1. This contradiction highlights the intrinsic difference between 2D and 3D binding, which is also observed in the binding between TCR and pMHC ligands (Huang et al., 2010). The difference in 2D affinity of CD16 for IgG is only severalfold among natural forms of CD16 (TM vs. GPI and glycosylated vs. deglycosylated), in contrast to several orders of difference in 2D affinity of TCR for different pMHC ligands. However, in the physiological situation in which CD16 on cell surface binds a large array of IgG on an immune complex, the difference in 2D affinity can be substantially amplified to a large difference in 2D avidity. Therefore we believe that the difference in 2D affinity of the CD16-IgG binding has a large effect on the binding of CD16 to immune complexes. In vivo, the change of the CD16–IgG binding affinity by posttranslational modification of CD16 such as glycosylation may be a way to regulate immune responses. In fact, CD16 encoded by the same gene could undergo differential cell type–specific glycosylation (Edberg and Kimberly, 1997; Drescher et al., 2003), and so the CD16-expressing cells may be selectively activated in an immune response. The importance of posttranslational processes is also observed in other molecular systems, such as CD8, for which nonsialylated glycoforms are present in immature thymocytes but virtually absent in mature thymocytes. This glycosylation difference is linked to the difference in ligand binding affinity between CD8 on mature and immature thymocytes (Moody et al., 2003).
Our MD simulations suggest that deglycosylation at Asn-163 of CD16 removes the steric hindrance for the CD16–hFc1 binding and therefore enhances the binding affinity (Figure 6). This provides a structural explanation for the glycosylation effects on the CD16–IgG binding shown by us (Figure 5) and others (Drescher et al., 2003).
In summary, we explored the effect of anchor structure and glycosylation of CD16 on ligand binding using three different methods to solubilize CD16 molecules with distinct anchor forms. The results extended our previous study (Chesla et al., 2000) and showed that the membrane anchor structure itself and glycosylation can regulate allosterically these intrinsic binding parameters.
MATERIALS AND METHODS
Cells and antibodies
Transfected CHO cells expressing human CD16aTM, CD16aGPI, and CD16bNA2, as well as untransfected control CHO cells, were cultured as described previously (Chesla et al., 1998, 2000). The expressions of various forms of CD16s were periodically checked with flow cytometry. The anti-CD16 nonblocking mAb 214.1 (murine IgG1; Fleit et al., 1992) was a generous gift from Howard Fleit (State University of New York, Stony Brook, NY). The anti-CD16 adhesion blockade mAb CLBFcgran-1 and 3G8 were purified from hybridomas (Selvaraj et al., 1988). HIgG1 was kindly provided by Adrian Whitty (Biogen, Boston, MA) and was purchased from Sigma-Aldrich (St. Louis, MO). Fluorescein isothiocyanate–labeled goat anti-human Fc-specific and goat anti-mouse Fc-specific antibodies (Sigma-Aldrich) and phycoerythrin (PE)-labeled goat anti-human Fc-specific and goat anti-mouse Fc-specific antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) were used to estimate the site density of hIgG1, CLBFcgran-1, and mIgG2a. The mouse anti-human IgG Fc mAb 7QD was purchased from General Bioscience (Brisbane, CA).
Preparation of solubilized CD16 and soluble CD16a–Ig chimera
One gram of each CD16-expressing CHO cell pellet was resuspended in lysis buffer containing 1% Triton X-100 and protease inhibitors and incubated for 30 min at 4°C (Nagarajan et al., 1995a). Protease inhibitors included the serine protease inhibitors phenylmethylsulfonyl fluoride (1 mM) and aprotinin (5 mg/ml) and the cysteine protease inhibitor iodoacetamide (1 mM). The lysate was clarified by centrifugation at 100,000 × g for 1 h at 4°C. Supernatant was collected and stored at –80°C for further use. For CD16b and CD16aGPI, 1% octyl glucoside was added to lyse the cells and preserve the GPI anchor. To prepare PIPLC cleavage of GPI-anchored CD16, 107 cells were incubated in 1 ml phosphate-buffered saline (PBS)/5 mM EDTA, pH 7.4, with 0.2 unit of PIPLC for 1 h at 37°C. To prepare shed CD16, 107 CD16-expressing CHO cells were incubated in 1 ml of Hanks balanced salt solution at 37°C for 3 h with gentle mixing. The CD16 molecules so generated are schematically shown in Figure 1 in different forms as labeled by lysate, PIPLC, and shedding. CD16a–Ig chimera was generated as described (Li et al., 2002) and is also depicted in Figure 1. Briefly, the chimeric CD16a–Ig cDNA was transfected into CHO cells, which were then cultured using CHO serum-free medium. The culture supernatant was collected, and CD16a-Ig was purified using a Sepharose protein G column by affinity chromatography.
Generation of CHO cells expressing aglycosylated CD16
Tunicamycin blocks N-glycosylation synthesis by inhibiting the transfer of N-acetylgucosamine-1-phosphate to dolicholmonophosphate. At high concentrations, tunicamycin treatment drastically reduces the expression level of CD16. Twofold serial dilution of tunicamycin was performed to determine the optimal concentration (78 ng/ml) that blocks the glycosylation of CD16 and yet does not significantly alter its expression level. CHO cells transfected to express cell-surface CD16 or CD16a-Ig were rinsed three times using culture medium containing 78 ng/ml tunicamycin (Sigma-Aldrich) and cultured in the same medium for 40 h at 37°C. Cells expressing CD16 membrane isoforms were then rinsed three times with clean culture medium, detached, and analyzed by FACS for CD16 expression or by micropipette for IgG binding. Supernatant from the CD16a-Ig–expressing cell culture was collected, and CD16a–Ig was purified using Sepharose protein G column by affinity chromatography.
Coupling of capturing antibody to microspheres
After being washed twice in carbonate buffer, 10 million carboxylated microspheres were washed three times in phosphate buffer (pH 4.5) and resuspended in 120 μl of phosphate buffer. The same volume of 2% carbodiimide solution was added dropwise to the microsphere suspension, and the mixture was incubated for 3–4 h at room temperature with agitation. Unreacted carbodiimide was removed by three washes with phosphate buffer and resuspended in 200 μl of borate buffer. Next 20–40 μg of 214.1 or 7QD was added to the microsphere mixture and incubated overnight at room temperature with mixing. Microspheres were collected, and the antibody concentration in the supernatant was tested by the Micro BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL) to estimate the efficiency of coupling. The pellet was resuspended in 0.2 M borate buffer, and 50 μl of 0.1 M methanolamine was added and mixed gently for 30 min at room temperature to block unreacted sites on the microspheres. Microspheres were then incubated in 10 mg/ml BSA solution for 30 min at room temperature. Finally, microspheres were collected and stored in PBS with 1% BSA at 4°C for future use.
Chromium chloride coupling of antibody to RBCs
HIgG1, mIgG2a, or F(ab′)2 of mAb 7QD was covalently coupled to the membranes of RBCs using a previously described CrCl3 method (Chesla et al., 1998). A 1% CrCl3 solution was prepared, properly aged at pH 5, and diluted in 20 mM acetate-buffered saline, pH 5.5, at ratios ranging from 1:6700 to 1:17,000. Fresh RBCs were washed five times in saline and resuspended to 2% hematocrit. IgG was added to each 250-μl sample and mixed. An equal volume of diluted CrCl3 solution was added dropwise with constant agitation. After 5 min, the reaction was stopped by addition of 0.5 ml PBS/5 mM EDTA/1% BSA. Cells were subsequently washed and stored in EAS45 as described previously (Dumaswala et al., 1996). An aliquot from each sample was examined under light microscopy for aggregation. Samples were assayed for coating density and uniformity by flow cytometry.
Site-density determination
Site densities of proteins on RBCs (CrCl3-coupled IgG or soluble CD16–Ig captured by precoated 7QD) and microspheres (solubilized CD16 captured by precoated 214.1) were determined by quantitative fluorescence immunoassay (Chesla et al., 1998, 2000; Williams et al., 2000a, b, 2001; Shashidharamurthy et al., 2009). Samples were prepared for flow cytometry as described (Chesla et al., 2000). The mean fluorescence intensities of the RBCs or microspheres were compared with standard calibration beads (Bangs Laboratories, Fishers, IN, and Becton Dickinson, San Jose, CA) to determine the mean number of fluorophores per cell or microsphere, which was then converted into labeled protein per cell.
Micropipette adhesion frequency assay
Two-dimensional affinities and kinetic rates of CD16 for IgG were measured by the micropipette adhesion frequency assay as previously described (Chesla et al., 1998). Briefly, 103 CHO cells expressing membrane CD16, microspheres captured with solubilized CD16, or RBCs captured with CD16–Ig and 104 ligand-coated RBCs were respectively added to separate corners of a cell chamber sufficiently far apart to avoid mixing. The cell chamber was filled with RPMI (Mediatech, Manassas, VA) plus 1% BSA, 5 mM EDTA, and 0.04% sodium azide and mounted on the stage of an inverted microscope (Axiovert 100; Zeiss, Oberkochen, Germany). A single CD16-bearing CHO cell, microsphere, or RBC and a single IgG-coated RBC were respectively aspirated by two apposing micropipettes and aligned via micromanipulation. One pipette was mounted to a computer-driven piezoelectric translator to move the two cells into contact for a predetermined area and duration. On pipette retraction, the two cells were either immediately separated (i.e., no adhesion, scored 0) or remained bound, which stretched the RBC for a short time before it was detached by force (i.e., adhesion, scored 1). This adhesion test cycle was repeated 100 times to estimate adhesion frequency, and three or five pairs of cells were used to obtain a mean and SEM of adhesion frequency for each contact duration, which was varied from 0.5 to 16 s.
Antibody saturation binding experiment
The affinities of the anti-CD16 mAb CLBFcgran-1 for CD16 isoforms expressed on CHO cells were measured by saturation binding with twofold serial dilution. CLBFcgran-1 at varying concentrations was incubated with CD16-expressing cells for 30 min in triplicate wells containing FACS buffer. Cells were then washed and stained with PE-conjugated goat anti-mouse Fc specific antibody for an additional 30 min. Cells were washed, and the expression of CD16 was checked via flow cytometry. Controls were performed both with no staining and with secondary antibody staining alone without CLBFcgran-1.
Molecular dynamics simulation
The starting structure for the glycosylated CD16-hFc1 complex was the published cocrystal structure downloaded from the Protein Data Bank (PDB code 3SGJ; Ferrara et al., 2011). The starting structure for the aglycosylated CD16-hFc1 complex was obtained by removing in silico the glycans on CD16 in the crystal structure. The simulation systems were prepared using LEaP in AmberTools14 (Case et al., 2014). The Amber ff12SB (Maier et al., 2015) and GLYCAM06 (Kirschner et al., 2008) force fields were used for proteins and glycans, respectively. Each starting structure was placed into a truncated octahedral periodic box of TIP3P water molecules with a 15-Å minimal distance between the edges of the water box and the solute. Sodium and chloride ions were added to neutralize the systems and produce a 150 mM ionic strength. MD simulations were performed using NAMD (Phillips et al., 2005). The particle mesh Ewald method (Darden et al., 1993) was used to treat long-range electrostatic interactions. The cutoff for nonbonded interactions was set to 12 Å, with a switching function starting from 10 Å. SHAKE (Ryckaert et al., 1977) was used to constrain lengths of bonds involving hydrogen. Time-step was 2 fs. The systems were energy minimized using the conjugate gradient method in two steps: first with heavy atoms of the proteins fixed, and then with heavy atoms of the proteins harmonically restrained (force constants of 5 kcal/mol Å2). After minimization, the systems were gradually heated from 0 to 310 K during 120-ps canonical ensemble (NVT)-MD simulations with harmonic restraints (force constants of 5 kcal/mol Å2) on heavy atoms of the proteins. Subsequent isothermal isobaric ensemble (NPT)-MD simulations were performed for 5 ns with the same harmonic restraints. Then the harmonic restraints were reduced to zero in four steps with respective force constants of 1, 0.1, 0.01, and 0 kcal/mol Å2. Each step contains a 1-ns NPT-MD simulation. Finally, NPT-MD production simulations were performed for 50 ns each. The simulations were run on XSEDE (Towns et al., 2014).
VMD (Humphrey et al., 1996) and PyMOL (version 1.7) were used for visualization and analysis. Two residues were defined as in contact if any pair of their heavy atoms of side chains (Cα atom for glycine) were within 4.5 Å. Then the contact time was calculated as fraction of total time the two residues were in contact. Only the last 40 ns was used to calculate contact time.
Supplementary Material
Acknowledgments
We thank Jizhong Lou and Fabien Cailliez for helping with MD simulations, Fang Kong for helping with data analysis, and Stephen C. Harvey for providing computational resources. This work was supported by National Institutes of Health Grant HL132019 and used the Extreme Science and Engineering Discovery Environment, which is supported by National Science Foundation Grant ACI-1053575.
Abbreviations used:
- BSA
bovine serum albumin
- 2D
two dimensional
- 3D
three dimensional
- FcγR
Fcγ receptor
- GPI
glycosyl-phosphatidylinositol
- hFc1
Fc fragment of hIgG1
- hIgG1
human IgG1
- Ig
immunoglobulin
- mIgG2a
mouse IgG2a
- NA1
neutrophil alloantigen 1
- NA2
neutrophil alloantigen 2
- PIPLC
phosphatidylinositol-specific phospholipase C
- pMHC
peptide-major histocompatibility complex
- RBC
red blood cell
- SPR
surface plasma resonance
- TCR
T-cell receptor
- TM
transmembrane.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0470) on August 31, 2016.
REFERENCES
- Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, Chen W, Levin AM, Connolly JM, Zhu C, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity. 2011;35:681–693. doi: 10.1016/j.immuni.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboni E, Rivero BP, George AJ, Martin SR, Renoup DV, Hounsell EF, Barber PC, Morris RJ. The glycophosphatidylinositol anchor affects the conformation of Thy-1 protein. J Cell Sci. 1995;108:487–497. doi: 10.1242/jcs.108.2.487. [DOI] [PubMed] [Google Scholar]
- Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Gohlke H, et al. AMBER 14. San Francisco: University of California; 2014. [Google Scholar]
- Chen W, Lou J, Evans EA, Zhu C. Observing force-regulated conformational changes and ligand dissociation from a single integrin on cells. J Cell Biol. 2012;199:497–512. doi: 10.1083/jcb.201201091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Lou J, Zhu C. Forcing switch from short- to intermediate- and long-lived states of the alphaA domain generates LFA-1/ICAM-1 catch bonds. J Biol Chem. 2010;285:35967–35978. doi: 10.1074/jbc.M110.155770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesla SE, Li P, Nagarajan S, Selvaraj P, Zhu C. The membrane anchor influences ligand binding two-dimensional kinetic rates and three-dimensional affinity of FcgammaRIII (CD16) J Biol Chem. 2000;275:10235–10246. doi: 10.1074/jbc.275.14.10235. [DOI] [PubMed] [Google Scholar]
- Chesla SE, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle Mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- Dembo M, Torney DC, Saxman K, Hammer D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc R Soc Lond B Biol Sci. 1988;234:55–83. doi: 10.1098/rspb.1988.0038. [DOI] [PubMed] [Google Scholar]
- Drescher B, Witte T, Schmidt RE. Glycosylation of FcgammaRIII in N163 as mechanism of regulating receptor affinity. Immunology. 2003;110:335–340. doi: 10.1046/j.1365-2567.2003.01743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaswala UJ, Wilson MJ, Jose T, Daleke DL. Glutamine- and phosphate-containing hypotonic storage media better maintain erythrocyte membrane physical properties. Blood. 1996;88:697–704. [PubMed] [Google Scholar]
- Edberg JC, Kimberly RP. Cell type-specific glycoforms of Fc gamma RIIIa (CD16): differential ligand binding. J Immunol. 1997;159:3849–3857. [PubMed] [Google Scholar]
- Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleit HB, Kobasiuk CD, Peress NS, Fleit SA. A common epitope is recognized by monoclonal antibodies prepared against purified human neutrophil FcgRIII (CD16) Clin Immunol Immunopathol. 1992;62:16–24. doi: 10.1016/0090-1229(92)90018-j. [DOI] [PubMed] [Google Scholar]
- Galon J, Robertson MW, Galinha A, Mazieres N, Spagnoli R, Fridman WH, Sautes C. Affinity of the interaction between Fc gamma receptor type III (Fc gammaRIII) and monomeric human IgG subclasses. Role of Fc gammaRIII glycosylation. Eur J Immunol. 1997;27:1928–1932. doi: 10.1002/eji.1830270816. [DOI] [PubMed] [Google Scholar]
- Herr AB, Ballister ER, Bjorkman PJ. Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. Nature. 2003;423:614–620. doi: 10.1038/nature01685. [DOI] [PubMed] [Google Scholar]
- Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Huang J, Edwards LJ, Liu B, Zhang Y, Beal CD, Evavold BD, Zhu C. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity. 2011;34:13–23. doi: 10.1016/j.immuni.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberly RP, Ahlstrom JW, Click ME, Edberg JC. The glycosyl phosphatidylinositol-linked Fc gamma RIIIPMN mediates transmembrane signaling events distinct from Fc gamma RII. J Exp Med. 1990;171:1239–1255. doi: 10.1084/jem.171.4.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberly RP, Wu J, Gibson AW, Su K, Qin H, Li X, Edberg JC. Diversity and duplicity: human FCgamma receptors in host defense and autoimmunity. Immunol Res. 2002;26:177–189. doi: 10.1385/IR:26:1-3:177. [DOI] [PubMed] [Google Scholar]
- Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. 2009;185:1275–1284. doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukulansky T, Abramovitch S, Hollander N. Cleavage of the glycosylphosphatidylinositol anchor affects the reactivity of thy-1 with antibodies. J Immunol. 1999;162:5993–5997. [PubMed] [Google Scholar]
- Lanier LL, Phillips JH, Testi R. Membrane anchoring and spontaneous release of CD16 (FcR III) by natural killer cells and granulocytes. Eur J Immunol. 1989;19:775–778. doi: 10.1002/eji.1830190431. [DOI] [PubMed] [Google Scholar]
- Le Trong I, Aprikian P, Kidd BA, Forero-Shelton M, Tchesnokova V, Rajagopal P, Rodriguez V, Interlandi G, Klevit R, Vogel V, et al. Structural basis for mechanical force regulation of the adhesin FimH via finger trap-like beta sheet twisting. Cell. 2010;141:645–655. doi: 10.1016/j.cell.2010.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Jiang N, Nagarajan S, Wohlhueter R, Selvaraj P, Zhu C. Affinity and kinetic analysis of Fcg receptor IIIa (CD16a) binding to IgG ligands. J Biol Chem. 2007;282:6210–6221. doi: 10.1074/jbc.M609064200. [DOI] [PubMed] [Google Scholar]
- Li P, Nagarajan S, Zhu C, Selvaraj P. Recombinant CD16A-Ig forms a homodimer and cross-blocks the ligand binding functions of neutrophil and monocyte Fcgamma receptors. Mol Immunol. 2002;38:527–538. doi: 10.1016/s0161-5890(01)00088-8. [DOI] [PubMed] [Google Scholar]
- Lou J, Yago T, Klopocki AG, Mehta P, Chen W, Zarnitsyna VI, Bovin NV, Zhu C, McEver RP. Flow-enhanced adhesion regulated by a selectin interdomain hinge. J Cell Biol. 2006;174:1107–1117. doi: 10.1083/jcb.200606056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J, Zhu C. A structure-based sliding-rebinding mechanism for catch bonds. Biophys J. 2007;92:1471–1485. doi: 10.1529/biophysj.106.097048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenaka K, van der Merwe PA, Stuart DI, Jones EY, Sondermann P. The human low affinity Fc gamma receptors IIa, IIb, and III bind IgG with fast kinetics and distinct thermodynamic properties. J Biol Chem. 2001;276:44898–44904. doi: 10.1074/jbc.M106819200. [DOI] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- Moody AM, North SJ, Reinhold B, Van Dyken SJ, Rogers ME, Panico M, Dell A, Morris HR, Marth JD, Reinherz EL. Sialic acid capping of CD8beta core 1-O-glycans controls thymocyte-major histocompatibility complex class I interaction. J Biol Chem. 2003;278:7240–7246. doi: 10.1074/jbc.M210468200. [DOI] [PubMed] [Google Scholar]
- Nagarajan S, Anderson M, Ahmed SN, Sell KW, Selvaraj P. Purification and optimization of functional reconstitution on the surface of leukemic cell lines of GPI-anchored Fc gamma receptor III. J Immunol Methods. 1995a;184:241–251. doi: 10.1016/0022-1759(95)00095-r. [DOI] [PubMed] [Google Scholar]
- Nagarajan S, Chesla S, Cobern L, Anderson P, Zhu C, Selvaraj P. Ligand binding and phagocytosis by CD16 (Fc gamma receptor III) isoforms. Phagocytic signaling by associated zeta and gamma subunits in Chinese hamster ovary cells. J Biol Chem. 1995b;270:25762–25770. doi: 10.1074/jbc.270.43.25762. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Antibody-mediated modulation of immune responses. Immunol Rev. 2010;236:265–275. doi: 10.1111/j.1600-065X.2010.00910.x. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radaev S, Motyka S, Fridman WH, Sautes-Fridman C, Sun PD. The structure of a human type III Fcgamma receptor in complex with Fc. J Biol Chem. 2001;276:16469–16477. doi: 10.1074/jbc.M100350200. [DOI] [PubMed] [Google Scholar]
- Ravetch JV, Perussia B. Alternative membrane forms of Fc gamma RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J Exp Med. 1989;170:481–497. doi: 10.1084/jem.170.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints—molecular-dynamics of N-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- Sabatino JJ, Jr., Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med. 2011;208:81–90. doi: 10.1084/jem.20101574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj P, Carpen O, Hibbs ML, Springer TA. Natural killer cell and granulocyte Fc gamma receptor III (CD16) differ in membrane anchor and signal transduction. J Immunol. 1989;143:3283–3288. [PubMed] [Google Scholar]
- Selvaraj P, Rosse WF, Silber R, Springer TA. The major Fc receptor in blood has a phosphatidylinositol anchor and is deficient in paroxysmal nocturnal haemoglobinuria. Nature. 1988;333:565–567. doi: 10.1038/333565a0. [DOI] [PubMed] [Google Scholar]
- Shashidharamurthy R, Zhang F, Amano A, Kamat A, Panchanathan R, Ezekwudo D, Zhu C, Selvaraj P. Dynamics of the interaction of human IgG subtype immune complexes with cells expressing R and H allelic forms of a low-affinity Fc gamma receptor CD32A. J Immunol. 2009;183:8216–8224. doi: 10.4049/jimmunol.0902550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- Teillaud JL, Bouchard C, Astier A, Teillaud C, Tartour E, Michon J, Galinha A, Moncuit J, Mazieres N, Spagnoli R, et al. Natural and recombinant soluble low-affinity Fc gamma R: detection, purification, and functional activities. Immunomethods. 1994;4:48–64. doi: 10.1006/immu.1994.1007. [DOI] [PubMed] [Google Scholar]
- Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D, Peterson GD, et al. XSEDE: accelerating scientific discovery. Comput Sci Eng. 2014;16:62–74. [Google Scholar]
- Unkeless JC, Shen Z, Lin CW, DeBeus E. Function of human Fc gamma RIIA and Fc gamma RIIIB. Semin Immunol. 1995;7:37–44. doi: 10.1016/1044-5323(95)90006-3. [DOI] [PubMed] [Google Scholar]
- Williams TE, Nagarajan S, Selvaraj P, Zhu C. Concurrent and independent binding of Fcgamma receptors IIa and IIIb to surface-bound IgG. Biophys J. 2000a;79:1867–1875. doi: 10.1016/S0006-3495(00)76436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TE, Nagarajan S, Selvaraj P, Zhu C. Quantifying the impact of membrane microtopology on effective two-dimensional affinity. J Biol Chem. 2001;276:13283–13288. doi: 10.1074/jbc.M010427200. [DOI] [PubMed] [Google Scholar]
- Williams TE, Selvaraj P, Zhu C. Concurrent binding to multiple ligands: kinetic rates of CD16b for membrane-bound IgG1 and IgG2. Biophys J. 2000b;79:1858–1866. doi: 10.1016/S0006-3495(00)76435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Xiao B, Jia X, Zhang Y, Lu S, Chen J, Long M. Impact of carrier stiffness and microtopology on two-dimensional kinetics of P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) interactions. J Biol Chem. 2007;282:9846–9854. doi: 10.1074/jbc.M609219200. [DOI] [PubMed] [Google Scholar]
- Xiang X, Lee CY, Li T, Chen W, Lou J, Zhu C. Structural basis and kinetics of force-induced conformational changes of an aA domain-containing integrin. PLoS One. 2011;6:e27946. doi: 10.1371/journal.pone.0027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Zhao Q, Zhu L, Zhang W. Deglycosylation of FcalphaR at N58 increases its binding to IgA. Glycobiology. 2010;20:905–915. doi: 10.1093/glycob/cwq048. [DOI] [PubMed] [Google Scholar]
- Yago T, Lou J, Wu T, Yang J, Miner JJ, Coburn L, Lopez JA, Cruz MA, Dong JF, McIntire LV, et al. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest. 2008;118:3195–3207. doi: 10.1172/JCI35754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.