

Despite the increasingly widespread use of single-molecule localization microscopy (SMLM) in biology, the extent to which the spatial organization of proteins influences signaling is not easy to quantify. Clus-DoC is a novel analysis method that combines cluster detection and colocalization analysis for SMLM data.
Abstract
Advances in fluorescence microscopy are providing increasing evidence that the spatial organization of proteins in cell membranes may facilitate signal initiation and integration for appropriate cellular responses. Our understanding of how changes in spatial organization are linked to function has been hampered by the inability to directly measure signaling activity or protein association at the level of individual proteins in intact cells. Here we solve this measurement challenge by developing Clus-DoC, an analysis strategy that quantifies both the spatial distribution of a protein and its colocalization status. We apply this approach to the triggering of the T-cell receptor during T-cell activation, as well as to the functionality of focal adhesions in fibroblasts, thereby demonstrating an experimental and analytical workflow that can be used to quantify signaling activity and protein colocalization at the level of individual proteins.
INTRODUCTION
Most biological processes are regulated by complex networks of protein–protein interactions that ultimately lead to specific cellular behaviors. The process of signal transduction is often mediated by heterogeneous and dynamic protein complexes rather than through linear cascades of biochemical interactions as frequently depicted in textbooks. An in-depth understanding of the molecular mechanisms underlying the regulation of cell behavior requires better knowledge of the molecular composition and molecular interactions of multiscale molecular structures and signaling complexes. Conventional biochemical, genetic, structural, and imaging approaches have provided important insights into biological processes but do not provide a molecular picture of the key nanometer-scale organization. During the past 30 yr, fluorescence microscopy has emerged as a valuable tool to unravel biomolecular interactions in cellular processes by facilitating the systematic study of protein colocalization in cells. However, the spatial resolution of conventional fluorescence microscopy is limited by diffraction to ∼250 nm, whereas molecular interactions occur at a scale of ∼10 nm. Superresolution and single-molecule imaging techniques have revolutionized the way in which we can address these complex biological questions. Constant improvements in methodology are increasingly pushing the limits of resolution, turning the focus back on how biologists can use these tools.
Several techniques can locate single fluorescent molecules in intact cells down to a few nanometers (Moerner, 2012). Collectively known as single-molecule localization microscopy (SMLM), these techniques include photoactivated localization microscopy (PALM; Betzig et al., 2006) and direct stochastic optical reconstruction microscopy (dSTORM; Heilemann et al., 2008; van de Linde et al., 2011). SMLM techniques reconstruct a molecular image of the sample by exploiting fluorophores that exist in both a dark and a bright state, such as photoactivatable or photoswitchable fluorescent proteins or dyes. A sparse subset of fluorophores can be photoconverted to the bright state, imaged, and then bleached, and the location of these molecules can be determined with high precision using center-of-mass algorithms (Henriques et al., 2010). Repeating this cycle over several thousand frames yields the localizations of all of the imaged molecules in the sample, making SMLM an effective approach for quantifying the distribution of proteins in cells (Owen et al., 2010).
Unlike conventional diffraction-limited microscopy or ensemble superresolution microscopy, SMLM techniques produce not just a “pretty image” but also a map of the coordinates of all detected individual points (Endesfelder and Heilemann, 2014). This allows a level of quantification that goes beyond the conventional way of analyzing images by measuring averages, such as intensity correlation–based colocalization of two proteins in an image using Pearson’s r (Pearson, 1896; Manders et al., 1992) or Manders’ overlap coefficient (Manders et al., 1993). By retaining the spatial information of each molecule, local parameters can be extracted to establish hierarchical information—at the level of the whole cell, at the level of protein clusters or complexes, and at the level of molecules.
Point pattern analysis methods evaluate the spatial distribution of points in an image through different approaches. A global overview of the distribution of points can be obtained using Ripley’s K (Ripley, 1979; Perry, 2004; Owen et al., 2010) or pair correlation analysis (Sengupta et al., 2011; Veatch et al., 2012), allowing researchers to determine whether the proteins are dispersed, clustered, or randomly distributed within the region of interest. Clusters are defined as regions of high density separated by regions of lower density. For the identification of clusters, hierarchical clustering methods such as density-based spatial clustering of applications with noise (DBSCAN; Ester et al., 1996) or OPTICS (Ankerst et al., 1999) provide additional segmentation of clusters, providing quantitative information at the level of individual clusters. Ripley’s K can also be extended to allow cluster detection (Owen et al., 2010). Similarly, several methods have been developed to investigate the colocalization of two proteins imaged by SMLM, including coordinate-based colocalization (CBC) analysis (Malkusch et al., 2012) and coclustering methods (Rossy et al., 2014). CBC analysis has also been applied to single-color data to correlate localization distributions from the same protein with each other and determine the degree of clustering for each individual point (Tarancón Díez et al., 2014). Voronoi tessellation is another strategy that has recently been implemented for cluster detection and colocalization analysis in SMLM data (Levet et al., 2015; Andronov et al., 2016).
Whereas cell biologists are exploiting SMLM techniques and producing large amounts of finely detailed images, the development of analysis tools that are able to extract relevant quantitative information is lagging behind. Although many algorithms underlying point pattern analysis methods have been published, they have not often been implemented in biological applications. There is thus an urgent need to make these analytical methods more accessible in order to address specific biological questions. Malkusch and Heilemann (2016) released an analysis toolbox called Lama: The LocAlization Microscopy Analyzer, which comprises a collection of algorithms for postprocessing and data analysis for SMLM data sets. This is a useful tool because it provides a variety of previously published routines for extracting quantitative information on localization precision, spatial distributions, cluster size, cluster composition, and colocalization within the same package.
Here we propose a new approach that combines two analysis techniques of cluster detection (Ester et al., 1996) and colocalization (Malkusch et al., 2012) to develop a multilevel analysis that goes beyond what these methods can achieve separately. We call this combined analysis strategy cluster detection with degree of colocalization (Clus-DoC). Clus-DoC can subclassify the data for a deeper analysis at the level of different cluster populations. By unifying different tools into one analysis, we can achieve a more in-depth analysis of colocalization in the context of protein clustering. This type of approach is applicable to various biological systems; here we demonstrate its capabilities in relation to T-cell signaling and focal adhesions.
RESULTS AND DISCUSSION
Acquisition of SMLM data
To achieve nanometer-scale resolution, SMLM exploits the properties of photoactivatable or photoswitchable fluorophores to reconstruct a single-molecule image (Betzig et al., 2006; Rust et al., 2006). The basic principle behind SMLM is the stochastic photoactivation of small numbers of fluorescent molecules and the localization of each activated molecule using center-of-mass algorithms. Thus, in a typical SMLM acquisition, several thousand frames need to be captured in order to image sufficient molecules to produce a high-resolution image. We typically acquire 20,000 frames (Figure 1A) and achieve a localization precision of 20-30 nm. Reconstructed SMLM images contain single-molecule localizations, with each point representing one detected molecule (Figure 1, B and C). SMLM methods generate data sets of molecular coordinates at nanometer resolution rather than traditional fluorescence intensity images. One of the advantages of conventional fluorescence microscopy that is retained in SMLM is the capability for multicolor imaging. Because most cellular functions involve multiple components, multicolor SMLM is an essential strategy to address how interactions between different proteins determine biological function. Although several methods have been proposed to quantify dual-color SMLM data, none of them satisfyingly incorporates an evaluation of the spatial distribution with the quantification of colocalization between the two colors, making it difficult to evaluate the link between protein clustering and function.
FIGURE 1:
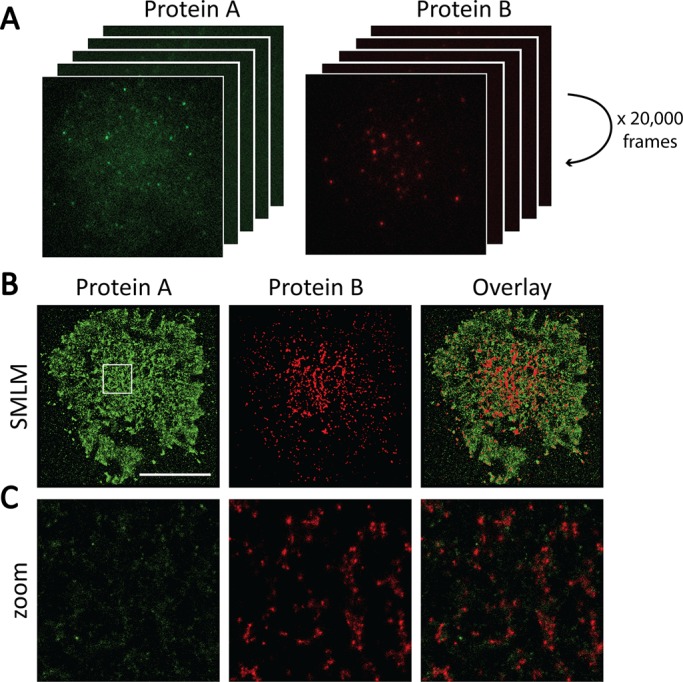
SMLM acquisition and image reconstruction. (A) For SMLM, a large number of frames are acquired for each cell. In each frame, only sparse subsets of molecules are fluorescent and can be accurately localized. If sufficient frames are collected (e.g., 20,000 frames in our example), a superresolved image of the sample can be reconstructed from the localizations of all molecules detected during the acquisition. (B) Reconstructed images of a two-color SMLM acquisition. Scale bar, 10 μm. (C) Enlargement of the 4 μm × 4 μm region highlighted in white in B. Each individual dot represents one detected molecule.
It is important to note several challenges when using SMLM for quantitative measurements. For example, sources of error include overcounting of fluorophores due to photoblinking (Annibale et al., 2010; Lee et al., 2012; Veatch et al., 2012) or limited detection efficiency related to incomplete photoconversion of fluorochromes (Annibale et al., 2011; Durisic et al., 2012). Therefore individual detected localizations do not always represent biological molecules at a 1:1 ratio. Photoblinking occurs when a fluorophore goes into a long-lived dark state instead of getting photobleached and then returns to the bright state in a later frame. To account for the photoblinking phenomenon, several “grouping” methods have been proposed that use a threshold in time and space to assign several localizations to one molecule (Annibale et al., 2011). However, the optimal grouping parameters to correct for photoblinking vary between fluorophores and experimental settings, and quantification remains difficult. Here we used ungrouped data to avoid grouping artifacts and to retain high numbers of data points. For obtaining absolute numbers of molecules, grouped data should be used.
Single-molecule cluster detection and colocalization analysis
For cluster detection, we implemented DBSCAN (Ester et al., 1996) and applied it to the table of coordinates generated from SMLM. DBSCAN has been used in biology to investigate the role of RAF multimers in cell growth (Nan et al., 2013) and the spatial organization of RNA polymerase during bacterial transcription (Endesfelder et al., 2013). DBSCAN is a propagative cluster detection method that links points that are closely packed together and detects outliers. Points are considered part of a cluster if they fulfill two criteria of connectivity: they need to have a number of neighbors equal to or greater than MinPts (minimum number of neighbors) within a radius r (Figure 2A). The analysis starts with an arbitrarily selected starting point in the image. If this point has MinPts neighbors or more within r, a cluster is started, and this point and its neighbors are part of the cluster. If the neighbors themselves have MinPts neighbors or more, the cluster is propagated; if not, these points are cluster boundary points. A new starting point that has not been visited is then arbitrarily selected to continue the analysis. If a point does not have more than MinPts neighbors and is not a neighbor for another point, it is classified as an outlier, that is, a nonclustered point, corresponding to isolated molecules or noise. Some of the advantages of using DBSCAN over other point pattern analysis methods are that DBSCAN can detect arbitrarily shaped clusters (rather than being biased toward circular clusters, as in Ripley’s K cluster analysis), it is rapid, and it is robust to outliers. In our implementation of the DBSCAN analysis, the outputs include a cluster map in which clustered molecules are in green, nonclustered molecules are in gray, and cluster contours are in black, as well as quantification of certain cluster properties, such as cluster size (Table 1). The user can select the values for the parameters MinPts and search radius, r. A good starting point for choosing r can be the median localization precision (σ, in nanometers). In our experimental conditions, σ = 21.3 nm for PSCFP2 and 20.6 nm for Alexa Fluor 647. We therefore typically use MinPts = 3 and r = 20 nm and define a cluster as having 10 localizations or more.
FIGURE 2:

Principles underlying DBSCAN and DoC analysis methods. (A) DBSCAN is a propagative cluster detection method in which connectivity between molecules is established if the number of neighbors is above a certain threshold (e.g., 3 in the diagram) within a radius r (e.g., 20 nm). The connection is propagated if the parameters are fulfilled (green dots) and stops when the parameters are no longer fulfilled (yellow dots). This method can also identify isolated points and noise (blue dots). This analysis yields cluster maps (4 μm × 4 μm) in which molecules in clusters are colored green or red and molecules outside clusters are in gray. Cluster contours are highlighted with black lines. (B) The DoC analysis is a coordinate-based colocalization analysis. From the molecular coordinates of both channels, the local density of each channel is calculated at increasing radius sizes around each molecule, providing density gradients for both channels. The two gradients of density are tested for correlation, resulting in a DoC score for each molecule. DoC scores range from −1 to +1, with −1 indicating segregation, 0 indicating random distribution, and +1 indicating colocalization. This analysis yields colocalization maps (4 μm × 4 μm) for both proteins in which each molecule is color coded according to its DoC score.
TABLE 1:
Output parameters from DoC, DBSCAN, and Clus-DoC.
| Algorithm | Output parameter |
|---|---|
| DoC | DoC score for each localization for protein A and protein B |
| Percentage of colocalized points for protein A and protein B (threshold based) | |
| DBSCAN plus contour mapping | Percentage of localizations in clusters |
| Number of clusters in ROI | |
| Number of localizations per cluster | |
| Circularity of clusters | |
| Size of clusters | |
| Absolute molecular density within clusters | |
| Relative molecular density within clusters | |
| Clus-DoC | Distinguishes colocalized versus noncolocalized clusters for protein A or protein B: |
| - Number of molecules per cluster | |
| - Circularity of clusters | |
| - Size of clusters | |
| - Absolute or relative density within clusters | |
| - Distribution of total or colocalized molecules between colocalized and noncolocalized clusters | |
| Distinguishes cluster populations based on density/size/number of localizations per cluster: | |
| - Average DoC score | |
| - Distribution of DoC scores | |
| - Percentage of colocalized molecules within each cluster population | |
| - Distribution of colocalized molecules between the subpopulations | |
| - Cluster morphology parameters | |
| Isolates nonclustered molecules: | |
| - Average DoC score | |
| - Proportion of total molecules and colocalized molecules that are not in clusters | |
| - Percentage of nonclustered molecules that are colocalized |
To quantify the extent of colocalization between two proteins at the single-molecule level, we implemented a coordinate-based colocalization method (Malkusch et al., 2012). Intensity-based approaches can be used on SMLM data but require histogramming or blurring of localizations, thereby losing the resolution of single-molecule localizations. By applying colocalization algorithms directly to the coordinates of single molecules, information is retained at the single-molecule level. This approach relies on the comparison of the spatial distribution of surrounding localizations from both species around each individual point (Figure 2B). For each molecule of protein A, the number of molecules of protein A and protein B within circles of increasing radius is calculated, respectively, giving the density gradients of protein A and protein B around this molecule. These density gradients are then corrected by the density at the maximum radius respectively for A and B. The two distributions are compared by calculating a rank correlation coefficient (using Spearman correlation), in which the colocalization coefficient is weighted by a value proportional to the distance to the nearest neighbor to avoid long-distance effects (Malkusch et al., 2012). Each molecule is thus assigned a DoC score ranging from −1 (anticolocalized or segregated), through 0 for noncorrelated distributions (no colocalization), to +1 (perfectly colocalized). Our implementation of this analysis yields colocalization maps for protein A or protein B that retain the spatial location of molecules and color codes them according to their DoC score, that is, their level of colocalization with the other species. Thus this method enables the visualization of the distribution of colocalization within an image and quantifies the DoC score on a per-molecule basis within the image. We take this analysis one step further by using a DoC threshold for colocalization, allowing us to calculate the percentage of colocalized molecules for protein A and for protein B (Table 1). We chose the threshold based on the distribution of DoC scores when a single-molecule data set was doubled and shifted in one direction by 10 nm (Malkusch et al., 2012). This resulted in the majority of the localizations (>90%) having a DoC score >0.4, with a peak of the DoC distribution at 1, indicating high colocalization, despite the 10-nm shift. Because in practice SMLM at best achieves 10-nm precision, it is therefore reasonable to assume that a 10-nm shift recapitulates experimental conditions. Therefore a threshold of 0.4 was chosen to distinguish colocalized from noncolocalized molecules. The DoC threshold is a user-specified variable that can be changed to suit the experimental conditions. The choice of the DoC threshold value can be confirmed through simulations that match the experimental conditions and specific biological scenario. We can therefore characterize the extent of protein A colocalization with protein B and the extent of protein B colocalization with protein A separately.
Clus-DoC: a combined cluster detection and colocalization analysis
Protein clustering is known to be a common strategy for cells to spatially concentrate signaling proteins for efficient signal transduction. To assess the influence of cluster properties (such as cluster geometry or cluster density) on signaling, we combined the DoC analysis with the DBSCAN cluster detection analysis in a method we call Clus-DoC (Figure 3). Here the first step in the analysis is to calculate the DoC score of each molecule for both channels using the coordinate-based colocalization method described earlier (green in Figure 3). In the next step, individual clusters are detected using DBSCAN for all localizations—this determines, for each given point, whether it is part of a cluster or not—and the cluster contours are defined using a two-dimensional (2D) histogram smoothing method (blue in Figure 3). From these two steps, localizations are simultaneously classified into clusters and assigned a DoC score. This allows us to perform a second level of classification (red in Figure 3) in which clusters are separated into colocalized clusters (containing a user-defined number and DoC threshold, i.e., >10 localizations with a DoC score >0.4) and noncolocalized clusters. These two populations of clusters are then further analyzed to extract cluster properties. Alternatively, clusters can be subclassified according to cluster characteristics, such as size, density, or number of molecules per cluster, and these cluster populations can be interrogated regarding average DoC scores or distribution of DoC scores. Thus this combined approach not only provides cluster parameters for individual clusters, but it also facilitates a cluster reclassification according to a specific parameter (such as the degree of colocalization). In all cases, it is also possible to compare the DoC distributions for clustered and nonclustered molecules.
FIGURE 3:

Analysis workflow for Clus-DoC. The input for the analysis consists in table(s) of x, y-coordinates of all molecules. The DoC module (green) assigns DoC scores to each molecule. The DBSCAN module (blue) detects clusters and defines cluster contours. Finally, the Clus-DoC module (red) links the two previous modules by merging the information on cluster detection and DoC to subsequently distinguish subpopulations of clusters or nonclustered molecules. The outputs include several maps (colocalization, cluster, and density maps). Clustering and colocalization parameters are also extracted, providing parameterized information on the organization of proteins in the sample.
In addition to the outputs of DBSCAN or DoC analysis performed separately (such as the distribution of DoC scores per molecule; Figure 4A), the outputs from Clus-DoC can be represented as maps (visualization of the data, Figure 4, B–D) and colocalization parameters (Table 1). For the example shown in Figure 4A, we can see that only a certain fraction of protein A molecules are colocalized with protein B, whereas the majority of protein B molecules are colocalized with protein A (visualized in the colocalization maps in Figure 4B). This type of data warrants a more in-depth analysis, since perhaps some specific spatial distribution of protein A is preferred for association of protein B. The analysis also yields cluster maps for both channels (Figure 4C), whereas the density maps in Figure 4D plot the relative density distributions of the proteins.
FIGURE 4:
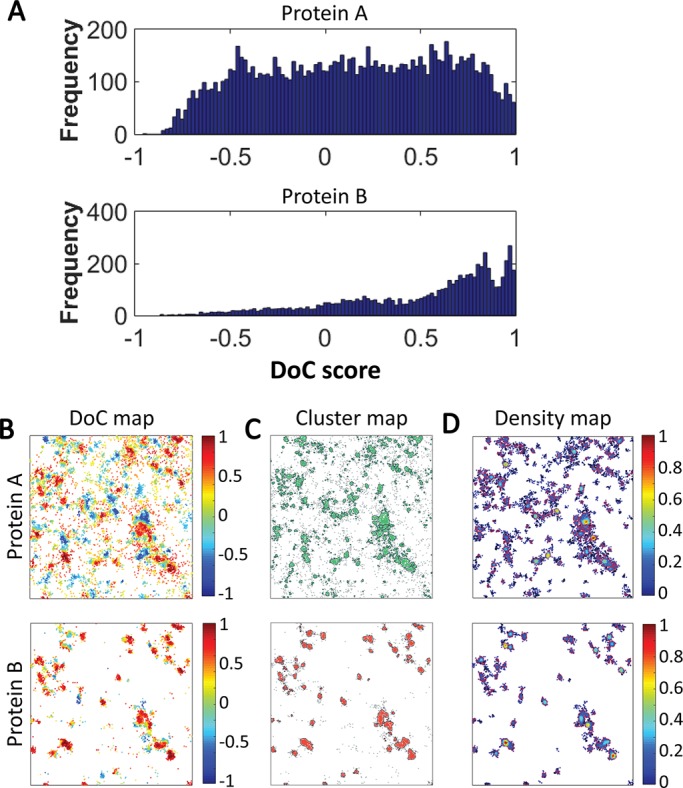
Output data from Clus-DoC analysis. (A) Frequency distributions of DoC scores of all molecules for protein A (top) and protein B (bottom). (B) Colocalization maps for both channels in which molecules are color coded according to their DoC scores. (C) Cluster maps for both channels in which clustered molecules are green or red and nonclustered molecules are gray. Cluster contours are in black. (D) Density maps for both channels in which the color scale represents normalized relative density. All maps are 4 μm × 4 μm.
To make the Clus-DoC analysis more accessible, we generated a graphical user interface (GUI) with which the user can load the data, select regions of interest (ROIs), and run the Clus-DoC analysis (Figure 5). The dimensions of the ROI can be defined by the user. The GUI runs in MATLAB and can load several cells to allow batch processing. Input data consist of an ASCII text file containing x, y-coordinates and channel information for each localization event.
FIGURE 5:

GUI for Clus-DoC analysis. Image of the GUI for the Clus-DoC analysis, which can be used to load data sets and then run a global clustering analysis (Ripley’s K), cluster detection (DBSCAN), or Clus-DoC analysis. Here the two-color data of one cell are displayed (in green and red), and one ROI is selected (blue square). The number of points from each channel within that ROI and the dimensions of the ROI (in nanometers) are displayed at the top.
Traditionally, biological approaches have viewed on/off processes as occurring at the level of a whole cell. However, it is becoming increasingly obvious that cellular processes are localized to specific sites within a cell and that signaling can be turned on or off differentially at different locations. The potential of Clus-DoC lies in its ability to address the functional state of individual structures (e.g., clusters, fibers) to determine which spatial parameters drive a particular structure to be active.
Application to T-cell signaling
The complexity of signal transduction is well illustrated by the example of T-cell signaling, in which even the first step in signal initiation remains controversial. Phosphorylation of the T-cell receptor (TCR) complex by the kinase Lck is one of the earliest detectable biochemical events in T-cell signaling, but the precise mechanism by which extracellular ligation of the TCR results in intracellular phosphorylation remains contested. Several SMLM studies reported that at least a proportion of TCRs were organized into small clusters that were 30–300 nm in diameter, termed “nanoclusters,” that were remodeled upon T-cell activation (Lillemeier et al., 2010; Sherman et al., 2011). However, little is known about the signaling activity of these clusters and whether they are equivalent in terms of function.
Using two-color SMLM, we simultaneously imaged the distribution of total TCR and phosphorylated TCR (pTCR) in activated T-cells. We imaged CD3ζ (a marker of the TCR complex) fused to the photoswitchable fluorescent protein PSCFP2 and pTCR using an antibody conjugated to Alexa Fluor 647 (Figure 6A). We also imaged TCR and the phosphatase CD45 (Figure 6, B and C). Using DBSCAN, we detected TCR clusters and, as expected, found that the TCR showed similar spatial patterns in both experiments, as quantified in Supplemental Figure S1. However, visual inspection of the maps suggested that TCR colocalized much more with pTCR than with CD45 (Figure 6, B and C). Using the DoC analysis, we next quantified the degree of colocalization of pTCR and CD45 with TCR and found that CD45 molecules had a much lower average DoC score than with pTCR (Figure 6D). In fact, pTCR molecules colocalized quite highly with TCR (as expected), whereas the average DoC score for CD45 molecules was negative, indicating no colocalization and perhaps a tendency toward segregation. Similarly, by using a threshold of 0.4 to distinguish colocalized from noncolocalized molecules, we found that the percentage of CD45 molecules that colocalized with TCR was significantly lower than the percentage of pTCR molecules that colocalized with TCR (Figure 6E). Using these two colocalization parameters, we are able to distinguish between proteins that do and do not colocalize with a given protein cluster or structure.
FIGURE 6:

Clus-DoC analysis applied to T-cell receptor triggering. (A) Reconstructed SMLM images of TCR (green) and phosphorylated TCR (pTCR; red) in activated Jurkat cells. Scale bar, 10 μm. (B) Left, TCR (green) and pTCR (red) localizations from 4 μm × 4 μm region highlighted in white in A. Right, TCR (green) and CD45 (red) localizations in a representative 4 μm × 4 μm region. (C) Colocalization maps for TCR relative to pTCR (left) and CD45 (right). TCR molecules are color coded according to their DoC scores. (D) Average DoC scores for pTCR and CD45 relative to TCR. (E) Percentage of colocalized pTCR and CD45 molecules relative to TCR. (F) Average cluster diameter for noncolocalized and colocalized TCR clusters relative to pTCR. (G) Average cluster circularity for noncolocalized and colocalized TCR clusters relative to pTCR. Circularity is measured as the ratio of perimeter to area, such that a perfect circle has a circularity score of 1. ****p < 0.0001 (unpaired t test for D and E and paired t test for F and G).
We then proceeded to analyze the TCR-pTCR data with Clus-DoC to separate TCR clusters into two populations: clusters that were colocalized with pTCR (i.e., phosphorylated clusters) and clusters that were not colocalized with pTCR. We then compared the morphology of these two subpopulations of TCR clusters and found that phosphorylated TCR clusters were larger and less circular than nonphosphorylated clusters (Figure 6, F and G), suggesting that phosphorylation and thus signal initiation may be dependent on the geometry of the clusters.
With Clus-DoC, we can also isolate nonclustered molecules and assess their DoC scores (Supplemental Figure S2 for TCR and CD45). We found that for TCR and CD45, nonclustered molecules were randomly distributed relative to each other (DoC scores peaking around 0; Supplemental Figure S2A). We also assessed the distribution of total or colocalized molecules between colocalized clusters, noncolocalized clusters, and outside clusters and found that the majority of TCR molecules were present in noncolocalized clusters (Supplemental Figure S2B), whereas colocalized molecules were as abundant in colocalized clusters as outside clusters (Supplemental Figure S2C). Clus-DoC also quantifies the average DoC score between these different populations (Supplemental Figure S2D).
Application to focal adhesion complexes
Focal adhesions are integrin-based structures that form at the cell–substrate interface, where they serve to connect the cell membrane to the extracellular matrix (ECM). Focal adhesions function as signaling hubs and traction points to help cells to sense or anchor to the ECM. Tyrosine phosphorylation of the proteins that make up these adhesions, such as paxillin, regulates adhesion dynamics from formation to maturation and turnover (Nobes and Hall, 1995; Wozniak et al., 2004). The phosphorylation of paxillin appears as a major switch in the assembly and turnover of focal adhesions. SMLM studies revealed that phosphorylated paxillin (p-paxillin) is abundant in the adhesion structures located at the cell periphery but is mainly absent from adhesions in the cell center (Zaidel-Bar et al., 2007). However, the distribution of p-paxillin nanoclusters within the adhesion structures remains unknown.
To examine the distribution of paxillin and p-paxillin within focal adhesion complexes, we imaged paxillin fused to the photoswitchable fluorescent protein mEos2 and p-paxillin using an antibody conjugated to Alexa Fluor 647 in fibroblasts on fibronectin (Figure 7, A–C). As expected, paxillin and p-paxillin were highly colocalized within focal adhesions, with 53 and 62% of paxillin and p-paxillin being colocalized with the other, respectively (Figure 7, D and E). We also quantified the distribution of DoC scores for nonclustered molecules (Supplemental Figure S3). The average DoC scores and the percentage of colocalized molecules were lower for nonclustered molecules than for total molecules (Supplemental Figure S3, A and B, and Figure 7, D and E), suggesting a preference for paxillin to be phosphorylated when in clusters. We then divided paxillin clusters into those that were or were not colocalized with p-paxillin (Figure 7F) and found that colocalized clusters were denser.
FIGURE 7:
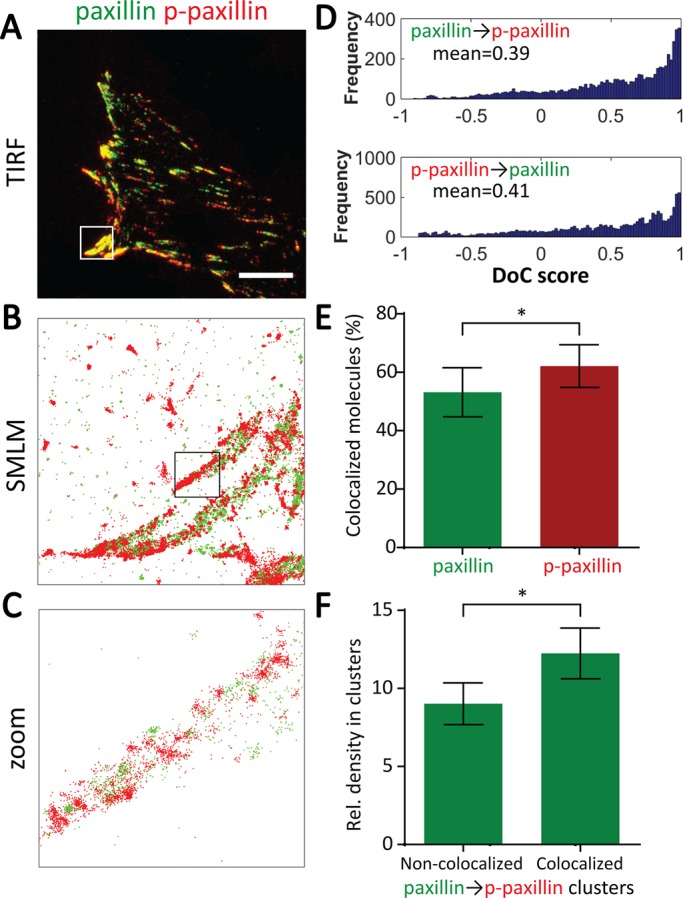
Clus-DoC analysis applied to focal adhesions. (A) Two-color TIRF image of paxillin (green) and phosphorylated paxillin (p-paxillin; red) in MEF cells on fibronectin. Scale bar, 10 μm. (B) SMLM data of paxillin (green) and p-paxillin (red) from 6 μm × 6 μm region highlighted in white in A. (C) Zoom of 1 μm × 1 μm region highlighted in black in B. (D) Frequency distributions of DoC scores of all molecules for paxillin (top) and p-paxillin (bottom), with mean DoC scores indicated. (E) Percentage of paxillin molecules colocalized with p-paxillin and percentage of p-paxillin molecules colocalized with paxillin. (F) Relative density of paxillin molecules in paxillin nanoclusters that are or are not colocalized with p-paxillin. *p ≤ 0.05 (paired t test).
With the application of Clus-DoC to two biological systems, we showed that Clus-DoC provides a more in-depth characterization of signaling complexes and protein colocalization than DBSCAN or DoC alone would allow.
Conclusion
We demonstrated that a combined cluster detection and colocalization analysis method (Clus-DoC) is a powerful tool to extract quantitative and biologically relevant information from SMLM data sets. Clus-DoC provides several advantages over existing analysis methods. It is fast (6-min processing time on a standard desktop PC for a data set of 10 regions corresponding to ∼500,000 localizations) and retains single-molecule information rather than providing ensemble measurements. Clus-DoC provides outputs at the level of molecules (e.g., distribution of DoC scores), at the level of clusters (e.g., size, density, morphology), and at the level of cells (e.g., comparisons across different conditions). In addition to being quantitative, Clus-DoC also generates maps for visualization of the data. Of importance, it has the potential to subclassify clusters according to different parameters (colocalization status, size, density, number of molecules, etc.), which provides deeper insight into the links between protein organization and function. By providing a user-friendly GUI to run Clus-DoC, we make this method easily accessible to any biologist with access to an SMLM-capable instrument. In addition, analysis parameters such as the region selection, region size, and cluster detection variables are controllable by the user. In the future, Clus-DoC will be extended to the analysis of multicolor SMLM data with three or more channels or with three-dimensional information. The software project, with new features and issue fixes as added, is open-source and continually maintained in the authors’ Git repository.
We believe that Clus-DoC can be applied to address a variety of biological questions in different fields. Because it quantifies the molecular structure and the degree of colocalization at the same time, this approach is well suited to characterizing the molecular composition and organization of higher-order molecular complexes or comparing the affinity of different proteins for a common structure (such as actin-binding proteins; Malkusch et al., 2012). Furthermore, Clus-DoC facilitates the identification of active signaling complexes (as shown here for TCR clusters) to probe the functional aspects of a molecular system. Similar mechanisms might be at play across different types of immune cells, such as natural killer cells, for which nanoclusters of inhibitory receptors were found to become smaller and denser upon cell activation (Pageon et al., 2013). This type of approach would also be beneficial for comparing the association of specific proteins with vesicles of different sizes (Caetano et al., 2015) or studying the mechanism of viral budding from infected cells (Malkusch et al., 2013).
In conclusion, we have developed a combined cluster detection and colocalization analysis (Clus-DoC) to probe the links between protein clustering and colocalization. We provide this analysis as a user-friendly GUI, making it accessible to biologists. With the increasing availability of commercial SMLM microscopes for imaging single molecules in intact cells, our analysis strategy may facilitate new insights into different cellular processes.
MATERIALS AND METHODS
Sample preparation for activated T-cells
Jurkat E6.1 T-cells (American Type Culture Collection, Manassas, VA) were maintained in RPMI 1640 (Life Technologies, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, and 1 mM penicillin and streptomycin (all from Invitrogen, Carlsbad, CA) and transfected by electroporation (NEON; Invitrogen) to express CD3ζ fused to PSCFP2. Clean glass coverslips were coated with anti-CD3ε (16-0037; eBioscience, San Diego, CA) and anti-CD28 (16-0289; eBioscience) antibodies for at least 1 h at 37°C. Cells were activated on antibody-coated coverslips for 10 min at 37°C and then fixed with 4% paraformaldehyde (vol/vol) in phosphate-buffered saline (PBS) for 20 min at room temperature. Cells were permeabilized with 0.1% (vol/vol) Triton X-100 for 4 min at room temperature, blocked in 5% bovine serum albumin (wt/vol) in PBS, and then labeled with primary antibody against human CD3ζ phosphorylated at Tyr-142 directly conjugated to Alexa Fluor 647 (558489; BD Biosciences, San Jose, CA) overnight at 4°C or against CD45 (ab10559; Abcam, Cambridge, UK) for 1 h at room temperature, followed by staining with Alexa Fluor 647–conjugated goat antibody specific to the rabbit F(ab′)2 fragment (111-606-047; Jackson ImmunoResearch).
Sample preparation for imaging focal adhesions
Mouse embryonic fibroblasts (MEFs) were cultured in high-glucose DMEM (Life Technologies) supplemented with 10% FCS at 37°C and 5% CO2. MEF cells were transfected by Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions to express paxillin-mEos2. Transfected cells were plated onto 25 μg/ml fibronectin–coated clean cover glasses and incubated for 3 h at 37°C. The cells were then fixed for 15 min in freshly made warm 4% paraformaldehyde. For immunostaining against phosphorylated paxillin, cells were permeabilized for 10 min in 0.5% Triton X-100, followed by blocking with 5% FCS for an additional 40 min. Cells were then labeled with primary antibody from rabbit against mouse paxillin phosphorylated at Tyr-118 (Life Technologies), followed by staining with Alexa Fluor 647–coupled secondary antibody specific to the rabbit F(ab′)2 fragment (111-606-047; Jackson ImmunoResearch). Cells were washed in 10 mM cysteamine in PBS three times, for 5 min each time, to decrease the floating background for dSTORM imaging.
SMLM imaging and dual-color image acquisition
SMLM image sequences were acquired on a total internal reflection fluorescence (TIRF) microscope (ELYRA; Zeiss, Jena, Germany) with a 100× oil-immersion objective (numerical aperture 1.46) and 1.6× Optivar. Photoconversion of PSCFP2 or mEos2 was achieved with 8 μW of 405-nm laser radiation, and the green-converted PSCFP2/red-converted mEos2 was imaged with 18 mW of 488/561-nm light, respectively. For Alexa Fluor 647, 15 mW of 633-nm laser illumination was used for imaging. An oxygen-scavenging PBS-based buffer (containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 25 mM glucose, 5% glycerol, 0.05 mg/ml glucose oxidase, and 0.025 mg/ml horseradish peroxidase, supplemented with 50 mM cysteamine; all from Sigma-Aldrich, St. Louis, MO) was used for dSTORM imaging. For each cell, 20,000 images were acquired with a cooled, electron-multiplying charge-coupled device camera (iXon DU-897; Andor, Belfast, UK) using an exposure time of 30 ms and a pixel size of 100 nm in the image space. Raw fluorescence intensity images were analyzed with the software Zen 2011 SP3 (Zeiss MicroImaging), generating reconstructed SMLM images, as well as tables containing the x, y-coordinates of each molecule detected during the acquisition.
Data processing
Data in the form of an ASCII text file containing spatial coordinate and channel identification information for each detected localization are loaded into the GUI application. An additional file containing user-defined ROIs is also loaded. These ROIs define subregions within the cell area within which each analysis will be applied. ROI area was typically 4000 nm × 4000 nm, and care was taken that the ROI border fell entirely within the cell boundary.
An average clustering value across an entire ROI is measured using the linearized form of Ripley’s K function, L(r) −r, which relates to Ripley’s K function as 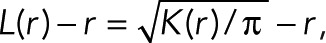 where K(r) is Ripley’s K function at radius r. This is performed on a single user-given square ROI, with the start, end, and step size of r also user defined. An additional parameter is supported to randomly subsample the points within a ROI to a user-defined maximum value. This improves processing speed for dense point fields and allows clustering behaviors between ROIs to be compared at identical point densities.
where K(r) is Ripley’s K function at radius r. This is performed on a single user-given square ROI, with the start, end, and step size of r also user defined. An additional parameter is supported to randomly subsample the points within a ROI to a user-defined maximum value. This improves processing speed for dense point fields and allows clustering behaviors between ROIs to be compared at identical point densities.
Points are segmented into clusters using the DBSCAN algorithm. Before this clustering, the user has the option of segmenting noise localizations from the ROI. Here a “noise” point is defined as a point whose L(r) − r value at a user-given radius (typically 50 nm) is below that of the value expected for a spatially random distribution of that density of points. Given user-specified values for the search radius and minimum number of points per cluster, a custom-written DBSCAN implementation (written in C++ and compiled using MEX) segments the points within each ROI into clusters or as the outliers or noise component. Data are extracted on a per-cluster basis, including number of points per cluster, cluster border, area, and circularity based on the cluster contour, cluster density, and, where included with DoC scores, the number of points within the cluster above and below the DoC threshold. Those clusters containing at least one point with a DoC score above the threshold are further subsegmented into points above and below the threshold, with the number, area, and density measures for these subpopulations extracted.
The cluster contour is defined by generating a 2D image of the histogram of localization points within the cluster. This 2D histogram has bin widths of 1 nm in each dimension. This histogram rendering is smoothed by a user-defined value (typically 15 nm). The value of this smoothed rendering at each localization point is evaluated and a threshold taken at the lowest value associated with a data point. The outline of this thresholded region is taken as the contour of the cluster for measuring cluster circularity, diameter, and area and for counting the number of molecules per cluster.
A DoC score is calculated for each point within a ROI for a range of discrete search radii from 0 to a maximum value. Both the step size and maximum radius (RMax) are user specified, typically 10 and 500 nm, respectively. The search for points within this radius is accelerated by a MEX function performing a k-dimensional tree search algorithm. The density of points at each search radius for channel i is correlated against the values from channel j using a pairwise linear correlation of Spearman’s ρ coefficient. This correlation, Sij, is converted into a DoC score by first calculating the cross-channel nearest-neighbor distance at each point in i to j, Nij, by the expression
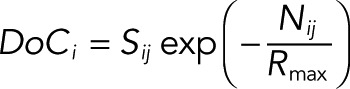 |
This is repeated for both channels, performing the correlation and nearest-neighbor searches in the opposite direction.
Once calculated, this DoC score can be compared with a user-defined threshold (typically 0.4) to segment points that are colocalized, clusters that are or are not colocalized (given a minimum number of points per cluster with a DoC score above the threshold), or to segment points within a cluster based on DoC score.
Generation of GUI
A MATLAB GUI was written to provide a graphical front end to the underlying clustering and DoC functions. This interface allows users to load and visualize SMLM dual-color data sets, executes the described functions, and exports plots and tabulated results. Additional functionalities, such as further data analysis, the ability to specify ROIs and imported binary masks and to do batch processing, are supported.
Codes are written for 64-bit MATLAB R2014b or above under a Windows operating system. All processing completes within minutes on a standard desktop PC for a single SMLM data set of ∼106 points.
The latest version of the source codes for the underlying functions and GUI application, complete with new functionalities and bug fixes, are available at the authors’ Git repository (https://github.com/PRNicovich/ClusDoC).
Statistical analysis
All statistical analysis was performed using GraphPad software (Prism, San Diego, CA). Statistical significance between data sets was determined by performing two-tailed Student’s t tests. Graphs show mean values, and error bars represent the SEM. In statistical analysis, p > 0.05 is indicated as not significant (n.s.), whereas statistically significant values are indicated as follows: *p ≤ 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
Abbreviations used:
- Clus-DoC
cluster detection with degree of colocalization
- DBSCAN
density-based spatial clustering of applications with noise
- GUI
graphical user interface
- SMLM
single-molecule localization microscopy
- TCR
T-cell receptor.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0478) on August 31, 2016.
REFERENCES
- Andronov L, Orlov I, Lutz Y, Vonesch J-L, Klaholz BP. ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Sci Rep. 2016;6:24084. doi: 10.1038/srep24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankerst M, Breunig MM, Kriegel H-P, Sander J. OPTICS: ordering points to identify the clustering structure. ACM SIGMOD Record. 1999;28:49–60. [Google Scholar]
- Annibale P, Scarselli M, Kodiyan A, Radenovic A. Photoactivatable fluorescent protein mEos2 displays repeated photoactivation after a long-lived dark state in the red photoconverted form. J Phys Chem Lett. 2010;1:1506–1510. [Google Scholar]
- Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Identification of clustering artifacts in photoactivated localization microscopy. Nat Methods. 2011;8:527–528. doi: 10.1038/nmeth.1627. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Caetano FA, Dirk BS, Tam JHK, Cavanagh PC, Goiko M, Ferguson SSG, Pasternak SH, Dikeakos JD, de Bruyn JR, Heit B. MIiSR: molecular interactions in super-resolution imaging enables the analysis of protein interactions, dynamics and formation of multi-protein structures. PLoS Comput Biol. 2015;11:e1004634. doi: 10.1371/journal.pcbi.1004634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durisic N, Godin AG, Wever CM, Heyes CD, Lakadamyali M, Dent JA. Stoichiometry of the human glycine receptor revealed by direct subunit counting. J Neurosci. 2012;32:12915–12920. doi: 10.1523/JNEUROSCI.2050-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endesfelder U, Finan K, Holden SJ, Cook PR, Kapanidis AN, Heilemann M. Multiscale spatial organization of RNA polymerase in Escherichia coli. Biophys J. 2013;105:172–181. doi: 10.1016/j.bpj.2013.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endesfelder U, Heilemann M. Art and artifacts in single-molecule localization microscopy: beyond attractive images. Nat Methods. 2014;11:235–238. doi: 10.1038/nmeth.2852. [DOI] [PubMed] [Google Scholar]
- Ester M, Kriegel H-P, Sander J, Xu X. 1996. A density-based algorithm for discovering clusters in large spatial databases with noise. In: Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining, Menlo Park, CA: AAAI, 226–231.
- Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chemie (Int Ed Engl) 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- Henriques R, Lelek M, Fornasiero EF, Valtorta F, Zimmer C, Mhlanga MM. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods. 2010;7:339–340. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- Lee S-H, Shin JY, Lee A, Bustamante C. Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM) Proc Natl Acad Sci USA. 2012;109:17436–17441. doi: 10.1073/pnas.1215175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levet F, Hosy E, Kechkar A, Butler C, Beghin A, Choquet D, Sibarita J-B. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat Methods. 2015;12:1065–1071. doi: 10.1038/nmeth.3579. [DOI] [PubMed] [Google Scholar]
- Lillemeier BF, Mörtelmaier MA, Forstner MB, Huppa JB, Groves JT, Davis MM. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkusch S, Endesfelder U, Mondry J, Gelléri M, Verveer PJ, Heilemann M. Coordinate-based colocalization analysis of single-molecule localization microscopy data. Histochem Cell Biol. 2012;137:1–10. doi: 10.1007/s00418-011-0880-5. [DOI] [PubMed] [Google Scholar]
- Malkusch S, Heilemann M. Lama: The LocAlization Microscopy Analyzer. 2016. Available at http://user.uni-frankfurt.de/~malkusch/lama.html (accessed 1 July 2016) [DOI] [PMC free article] [PubMed]
- Malkusch S, Muranyi W, Müller B, Kräusslich H-G, Heilemann M. Single-molecule coordinate-based analysis of the morphology of HIV-1 assembly sites with near-molecular spatial resolution. Histochem Cell Biol. 2013;139:173–179. doi: 10.1007/s00418-012-1014-4. [DOI] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Verbeek F, Aten J. Measurement of colocalization of objects in dual-color confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Moerner WE. Microscopy beyond the diffraction limit using actively controlled single molecules. J Microsc. 2012;246:213–220. doi: 10.1111/j.1365-2818.2012.03600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Collisson EA, Lewis S, Huang J, Tamgüney TM, Liphardt JT, McCormick F, Gray JW, Chu S. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci USA. 2013;110:18519–18524. doi: 10.1073/pnas.1318188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Owen DM, Rentero C, Rossy J, Magenau A, Williamson D, Rodriguez M, Gaus K. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. J Biophotonics. 2010;3:446–454. doi: 10.1002/jbio.200900089. [DOI] [PubMed] [Google Scholar]
- Pageon SV, Cordoba S-P, Owen DM, Rothery SM, Oszmiana A, Davis DM. Superresolution microscopy reveals nanometer-scale reorganization of inhibitory natural killer cell receptors upon activation of NKG2D. Sci Signal. 2013;6:ra62. doi: 10.1126/scisignal.2003947. [DOI] [PubMed] [Google Scholar]
- Pearson K. Mathematical contributions to the theory of evolution III. Regression, heredity and panmixia. Philos Trans R Soc Lond B Biol Sci. 1896;187:253–318. [Google Scholar]
- Perry GLW. SpPack: spatial point pattern analysis in Excel using Visual Basic for Applications (VBA) Environ Model Softw. 2004;19:559–569. [Google Scholar]
- Ripley BD. Tests of randomness for spatial point patterns. J R Stat Soc. 1979;B 41:368–374. [Google Scholar]
- Rossy J, Cohen E, Gaus K, Owen DM. Method for co-cluster analysis in multichannel single-molecule localisation data. Histochem Cell Biol. 2014;141:605–612. doi: 10.1007/s00418-014-1208-z. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman E, Barr V, Manley S, Patterson G, Balagopalan L, Akpan I, Regan CK, Merrill RK, Sommers CL, Lippincott-Schwartz J, Samelson LE. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor. Immunity. 2011;35:705–720. doi: 10.1016/j.immuni.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarancón Díez L, Bönsch C, Malkusch S, Truan Z, Munteanu M, Heilemann M, Hartley O, Endesfelder U, Fürstenberg A. Coordinate-based co-localization-mediated analysis of arrestin clustering upon stimulation of the C-C chemokine receptor 5 with RANTES/CCL5 analogues. Histochem Cell Biol. 2014;142:69–77. doi: 10.1007/s00418-014-1206-1. [DOI] [PubMed] [Google Scholar]
- van de Linde S, Löschberger A, Klein T, Heidbreder M, Wolter S, Heilemann M, Sauer M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc. 2011;6:991–1009. doi: 10.1038/nprot.2011.336. [DOI] [PubMed] [Google Scholar]
- Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, Baird BA. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One. 2012;7:e31457. doi: 10.1371/journal.pone.0031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–119. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci. 2007;120:137–148. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.