

Spatial targeting of signaling components to activated receptors on the plasma membrane is key for initiating signal transduction. The actin cytoskeleton restricts antigen-stimulated colocalization of IgE-FcεRI with membrane-anchored signaling partner Lyn kinase, and this regulation is mediated by organization of plasma membrane lipids.
Abstract
The allergic response is initiated on the plasma membrane of mast cells by phosphorylation of the receptor for immunoglobulin E (IgE), FcεRI, by Lyn kinase after IgE-FcεRI complexes are cross-linked by multivalent antigen. Signal transduction requires reorganization of receptors and membrane signaling proteins, but this spatial regulation is not well defined. We used fluorescence localization microscopy (FLM) and pair-correlation analysis to measure the codistribution of IgE-FcεRI and Lyn on the plasma membrane of fixed cells with 20- to 25-nm resolution. We directly visualized Lyn recruitment to IgE-FcεRI within 1 min of antigen stimulation. Parallel FLM experiments captured stimulation-induced FcεRI phosphorylation and colocalization of a saturated lipid-anchor probe derived from Lyn’s membrane anchorage. We used cytochalasin and latrunculin to investigate participation of the actin cytoskeleton in regulating functional interactions of FcεRI. Inhibition of actin polymerization by these agents enhanced colocalization of IgE-FcεRI with Lyn and its saturated lipid anchor at early stimulation times, accompanied by augmented phosphorylation within FcεRI clusters. Ising model simulations provide a simplified model consistent with our results. These findings extend previous evidence that IgE-FcεRI signaling is initiated by colocalization with Lyn in ordered lipid regions and that the actin cytoskeleton regulates this functional interaction by influencing the organization of membrane lipids.
INTRODUCTION
Stimulated signaling in mast cells that results in the allergic immune response is initiated by spatial colocalization of signaling components in the plasma membrane. Cross-linking of immunoglobulin E (IgE) bound to its high-affinity receptor, FcεRI, by multivalent antigen induces formation of IgE-FcεRI clusters and consequent association with the Src-family tyrosine kinase Lyn, which is anchored to the inner leaflet of the plasma membrane by saturated acyl chains. Lyn phosphorylates immunoreceptor tyrosine–based activation motifs (ITAMs) in cytoplasmic segments of FcεRI subunits as the first transmembrane signaling step, and this serves to recruit and activate Syk tyrosine kinase of the Syk/Zap70 family (Paolini et al., 1991; Jouvin et al., 1994; Gilfillan and Rivera, 2009). Phosphorylation of multiple Syk substrates, including the scaffold protein LAT and phospholipase Cγ, leads to downstream Ca2+ mobilization and secretion of proinflammatory molecules, among other cellular responses (Siraganian, 2003; Kraft and Kinet, 2007; Holowka et al., 2012).
Details of the spatial rearrangement of signaling proteins that mediates cell signaling triggered by FcεRI clustering and the dynamic regulation of signaling during the early phases of the stimulated response are open questions. Cumulative evidence supports the view that functional coupling of Lyn with IgE-FcεRI occurs within relatively ordered lipid regions of the plasma membrane (Holowka et al., 2005), whose properties and composition resemble the liquid-ordered (Lo) phase of isolated plasma membrane vesicles (Baumgart et al., 2007; Levental and Levental, 2015) but are small, dynamic structures in intact cells. These ordered regions would include Lo-preferring lipids (saturated and monounsaturated lipids, sphingomyelin, cholesterol), as well as proteins that preferentially associate with the Lo phase, including those anchored to the membranes by saturated lipids, such as Lyn. A wide range of experiments provides the following simplified picture (Holowka and Baird, 2015): Lyn kinases prefer an ordered lipid environment, and they transactivate within those regions but also diffuse in and out. Transmembrane tyrosine phosphatases partition more readily into disordered lipid (Ld-like) environments and deactivate Lyn encountered within those environments. Monomeric FcεRI has some preference for an ordered lipid environment that is substantially enhanced by antigen cross-linking of multiple IgE-FcεRI. In the resting state, the plasma membrane is dynamic, but the counterbalance of Lyn tyrosine kinase and transmembrane tyrosine phosphatase yields subthreshold phosphorylation of FcεRI. Stimulation by antigen-mediated clustering of IgE-FcεRI stabilizes ordered lipid regions, preferentially enriching Lyn and excluding phosphatases (Young et al., 2003, 2005; Heneberg et al., 2010). In this manner, Lyn-mediated phosphorylation of FcεRI surpasses the threshold sufficient for Syk recruitment and consequent activation of signaling pathways leading to cellular responses.
Lipid compositional heterogeneity, which plays a key role in this picture of stimulated IgE-FcεRI signaling, can also dynamically couple with other membrane and membrane-associated structures. In particular, the actin cytoskeleton demonstrably plays a role in actively organizing the distribution and mobility of plasma membrane lipids and order-preferring proteins in multiple cell types (Murase et al., 2004; Chichili and Rodgers, 2007; Goswami et al., 2008), including FcεRI in mast cells (Andrews et al., 2008) and related B-cell receptors (Treanor et al., 2010; Mattila et al., 2013). The actin cytoskeleton is also known to influence the strength of FcεRI signaling, and de novo actin polymerization that follows receptor cross-linking likely participates in feedback inhibition after initiation of the cellular response (Pfeiffer et al., 1985; Urata and Siraganian, 1985; Narasimhan et al., 1990; Pierini et al., 1997; Frigeri and Apgar, 1999; Holowka et al., 2000; Tolarová et al., 2004; Torigoe et al., 2004). Many studies found that treatment with drugs that inhibit actin polymerization—for example, cytochalasins and latrunculins—enhance FcεRI signaling responses at the earliest stages of the cascade (Pierini et al., 1997; Frigeri and Apgar, 1999; Tolarová et al., 2004). Thus an intact actin cytoskeleton appears to be involved in setting a threshold for cell activation by controlling the sensitivity of transmembrane signaling initiated by clustering of IgE-FcεRI.
A possible mechanism for this control is that the actin cytoskeleton modulates the organization of ordered lipid regions in a way that regulates the activity of order-preferring signaling proteins such as Lyn and/or their functional colocalization with FcεRI. Latrunculin treatment modulates the activation state of the Src-family tyrosine kinase Lck (Chichili et al., 2012), but regulation of the specific activity of Lyn by actin in mast cells has not been established. Nonetheless, the cytoskeleton may regulate the localization of Lyn to FcεRI clusters, as previously suggested (Frigeri and Apgar, 1999; Holowka et al., 2000; Tolarová et al., 2004).
Direct measurement of Lyn/FcεRI colocalization in mast cells at the earliest stages of signaling has posed a major challenge to testing this hypothetical mechanism due to the small dimensions of signaling-competent IgE-FcεRI clusters, which can be as small as dimers or trimers (Holowka et al., 2007), well below the diffraction limit of light (Wilson et al., 2000; Veatch et al., 2012b; Shelby et al., 2013). Detection of Lyn colocalized with FcεRI by conventional fluorescence microscopy has been limited to large, stably cross-linked FcεRI structures consisting of thousands of receptors and/or relatively long times after antigen addition (Sheets et al., 1999; Holowka et al., 2000; Pyenta et al., 2001, 2003; Wu et al., 2004; Larson et al., 2005; Torres et al., 2008). Lyn association with IgE receptors has been measured by a number of biochemical techniques, such as Western blotting and ultracentrifugation of detergent-solubilized cells (Pribluda et al., 1994; Yamashita et al., 1994; Field et al., 1997), but these approaches cannot capture the overall spatial organization of Lyn on the plasma membrane. Studies using electron microscopy from our lab and others have measured Lyn/FcεRI colocalization within minutes of receptor clustering (Wilson et al., 2000; Veatch et al., 2012a), but these methods involve sample preparations that limit specific labeling of proteins of interest.
Here we describe our use of superresolution fluorescence localization microscopy (FLM; Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006) to image the colocalization of Lyn and FcεRI with nanoscale resolution. The improved spatial resolution afforded by FLM in conjunction with our quantitative analysis using pair-correlation functions (Sengupta et al., 2011; Veatch et al., 2012b) enables us to examine Lyn association with FcεRI in resting cells and within the first few minutes after receptor cross-linking. In addition, we test for effects of actin cytoskeleton perturbation using the actin polymerization inhibitors latrunculin A and cytochalasin D and demonstrate a regulatory role for F-actin in the functional coupling of Lyn to FcεRI. Our results bring us closer to understanding the regulation of molecular interactions in the earliest stages of antigen-mediated FcεRI signaling and the mechanisms that set the threshold for cell triggering.
RESULTS
Two-color FLM reveals nanoscale spatial colocalization of IgE-FcεRI and Lyn kinase
We used two-color FLM to directly observe nanoscale coupling of Lyn kinase with IgE-FcεRI complexes before and during the first few minutes after antigen is added to cross-link IgE-FcεRI and activate rat basophilic leukemia (RBL-2H3) mast cells. For this purpose we labeled FcεRI with IgE that is specific for 2,4-dinitrophenyl (DNP) and is conjugated with the far-red organic dye Dyomics 654 (Dy654). In addition to endogenous Lyn, RBL-2H3 cells were transiently transfected to also express fluorescent Lyn with the photoconvertible protein mEos3.2 fused to its C-terminus. Cells were stimulated with the multivalent antigen DNP–bovine serum albumin (BSA) for defined time intervals before chemical fixation and FLM imaging as described in Materials and Methods. For these imaging experiments in fixed cells, we achieve localization precisions of ∼20 nm for Dy654 and 25 nm for mEos3.2.
Figure 1A shows two-color FLM images of IgE-FcεRI (red) and Lyn (green) in representative cells stimulated for 0, 1, 3, 6, and 12 min at 37°C. Single-color images of these representative cells are shown separately in Supplemental Figure S1. The distribution of IgE-FcεRI clearly changes with stimulation time: clusters are evident at the earliest stimulation time point and become more pronounced with continued exposure to antigen. However, antigen-dependent rearrangement of Lyn is less striking. Although we can find fields of view in two-color images of stimulated cells where IgE-FcεRI clusters appear to colocalize with concentrated areas of Lyn, the effect of antigen stimulation on Lyn/IgE-FcεRI colocalization is not readily assessed visually.
FIGURE 1:

Pair-correlation analysis of two-color FLM images measures stimulation time-dependent nanoscale colocalization of IgE-FcεRI and Lyn. Cells expressing Lyn-mEos3.2 and sensitized with Dy654 IgE are stimulated with 500 ng/ml DNP-BSA for 0, 1, 3, 6, or 12 min, fixed, and imaged as described in Materials and Methods. (A) Representative two-color FLM images of cells for each stimulation time point. Regions indicated by white boxes are magnified in the insets. The map of single-probe localizations obtained through FLM imaging is convolved with 2D Gaussian functions with radii of 50 nm in whole images and 20 nm in insets for display. (B) Individual and average pair cross-correlation functions for cells fixed at each stimulation time point. Individual Lyn/IgE-FcεRI cross-correlation functions are fitted to Eq. 1 as described in Materials and Methods. Fits of these individual cross-correlation functions to Eq. 1 are used to extract the fit parameters A (amplitude) and ξ (correlation length, in nanometers) for each correlation function. Measured individual cross-correlations with fits are shown in light colors; averages of the individual cross-correlation functions measured at a given time point are shown in black (eight or nine cells per time point). (C) Fit parameters A and ξ generated by the fit of individual Lyn/IgE-FcεRI cross-correlation functions averaged and plotted as a function of stimulation time. Error bars represent SEM.
We quantified colocalization of IgE-FcεRI and Lyn using pair-correlation analysis of the spatial map of single-molecule localizations collected in each channel (Sengupta et al., 2011; Veatch et al., 2012a, b; Shelby et al., 2013). In general, pair-correlation functions measure the average probability of finding a second probe within radius r away from a given probe, normalized by this probability for a random distribution of probes at the same average density. Pair cross-correlation functions measure spatial correlation between probes of two different colors in a two-color image and are used to quantify colocalization between the two species. Cross-correlation functions calculated from multiple two-color images of IgE-FcεRI and Lyn for each of the stimulation time points are shown in Figure 1B. The values of cross-correlation functions at small radii increase with stimulation time, indicating that Lyn and IgE-FcεRI become increasingly coenriched in structures with these dimensions. We fitted cross-correlation functions to a single-exponential function (see Eq. 1 in Materials and Methods; Veatch et al., 2012a, b; Shelby et al., 2013) and extract fit parameters that describe physical properties of coclusters. The amplitude of the exponential fit approximates the value of the correlation function c(r) at r = 0 and quantifies the coenrichment of the two species in correlated structures relative to their average density on the membrane. For example, an amplitude value of 2 indicates that the density of Lyn very close to the average labeled receptor is, on average, twofold higher than the average density of Lyn across the entire membrane. In other words, the probability of finding labeled Lyn closely associated with a labeled receptor is twofold higher than one would expect from a random distribution of Lyn. The correlation length of the exponential fit, ξ, is a measure of the average radius of correlated structures. Fits are shown with measured cross-correlation functions plotted in Figure 1B. Auto-correlations of labels in individual color channels are also tabulated to evaluate antigen-dependent changes in the distributions of IgE-FcεRI and Lyn independently and are shown in Supplemental Figure S1.
Averaged fit parameters (amplitude and ξ) were determined from cross-correlation functions for IgE-FcεRI and Lyn in multiple cells for each stimulation time point (Figure 1C). In unstimulated cells, IgE-FcεRI and Lyn appear to colocalize weakly over relatively long distances, as indicated by small amplitudes (close to 1) and large values of ξ (close to 150 nm). After stimulation, the amplitude of cross-correlations increases monotonically with time to values >3 in 12 min. The value of ξ falls rapidly to <100 nm within the first 5 min of stimulation, indicating that IgE-FcεRI and Lyn become colocalized in smaller, denser structures. Concurrently, over the 12-min-stimulation time course, IgE-FcεRI clusters increase in density, as quantified by IgE-FcεRI auto-correlations (Supplemental Figure S1).
Antigen-induced spatial colocalization of Lyn and IgE-FcεRI coincides with initiation of transmembrane signaling
To relate FLM measurements of Lyn colocalization with IgE-FcεRI to a functional readout of the first stages of transmembrane signaling, we measured tyrosine phosphorylation correlated with IgE-FcεRI. RBL-2H3 cells were sensitized with Dy654 IgE, stimulated, and fixed as for two-color experiments in Figure 1. Here tyrosine-phosphorylated proteins at the plasma membrane were fluorescently labeled in the fixed cells using anti-phosphotyrosine (4G10) primary and Alexa Fluor 488 (A488)–labeled secondary antibodies. A488 labels were imaged in FLM experiments with a typical localization precision of 25 nm. Figure 2A shows representative FLM images of Dy654 IgE and A488 anti-phosphotyrosine in an unstimulated cell and a cell stimulated for 6 min. In unstimulated cells, the phosphotyrosine signal corresponds to low-level tyrosine phosphorylation due to basal tyrosine kinase activity countered by phosphatases, and it does not visually colocalize with IgE-FcεRI. After antigen stimulation, which activates FcεRI coupling to Lyn and other signaling components, there is a clear concentration of labeled phosphotyrosine colocalized with IgE-FcεRI clusters, likely due at least in part to phosphorylation of ITAMs on FcεRI. Accumulation of newly phosphorylated signaling molecules that coredistribute with antigen–cross-linked IgE-FcεRI, such as Lyn, Syk, or LAT (Veatch et al., 2012a), likely also contribute to the signal.
FIGURE 2:
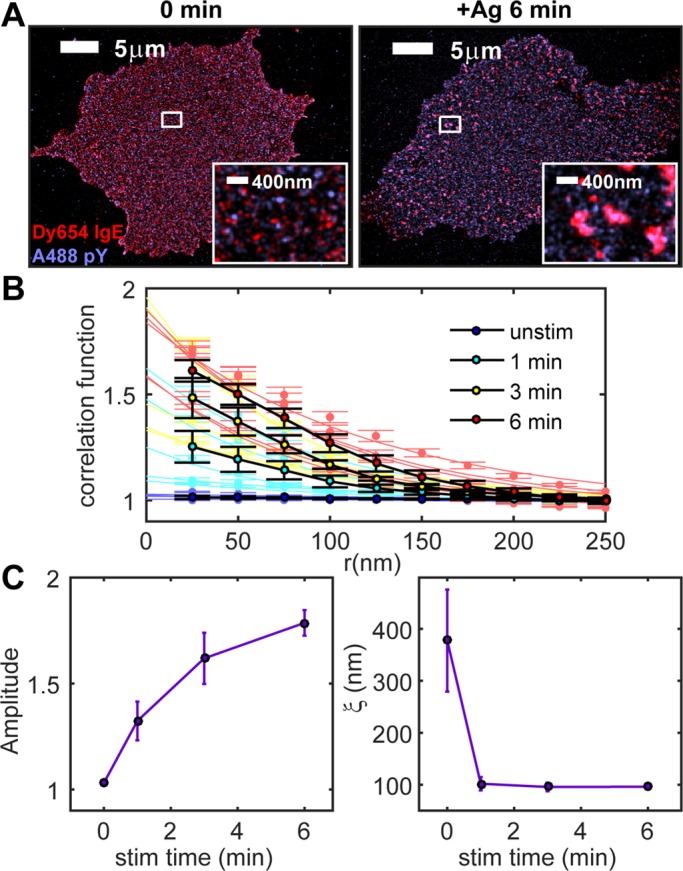
Phosphotyrosine/IgE-FcεRI cross-correlation amplitude increases with stimulation time. (A) Two-color FLM images of A488 anti-phosphotyrosine and Dy654 IgE in cells fixed after stimulation for 0 (left) or 6 min (right). Regions indicated by white boxes are magnified in the insets. (B) Individual and average pair cross-correlation functions for cells imaged for each stimulation time point. Individual cross-correlation functions are fitted to Eq. 1 as in Figure 1. Measured individual cross-correlations with fits are shown in light colors; average cross-correlation functions are shown in black (six cells per time point). (C) Cross-correlation fit parameters A and ξ as a function of stimulation time for A488 phosphotyrosine/Dy654 IgE-labeled cells stimulated for 0, 1, 3, or 6 min and averaged over multiple cells for each time point.
Cross-correlation functions that quantify the relative accumulation of phosphotyrosine in the vicinity of IgE-FcεRI were fitted to Eq. 1 to extract the physical properties of coclusters. Phosphotyrosine/IgE-FcεRI cross-correlations (Figure 2, B and C) follow stimulation-dependent trends similar to those of Lyn/IgE-FcεRI (Figure 1, B and C). Before antigen stimulation, cross-correlation functions have very small amplitudes (close to 1) and correlation lengths >300 nm, suggesting a nearly random distribution of basal phosphorylated tyrosine and IgE-FcεRI. In stimulated cells, the amplitude increases steadily, approaching 2, and values for ξ after stimulation are ∼100 nm, similar to those for Lyn/IgE-FcεRI (Figure 1C). Thus both phosphotyrosine and Lyn appear to be organized in structures that spatially correlate with IgE-FcεRI clusters, have similar dimensions, and form with similar time dependence after addition of antigen. Differences in the shapes of these curves—amplitude increases more quickly and ξ falls more quickly for phosphotyrosine/IgE-FcεRI than for Lyn/IgE-FcεRI—may reflect the fact that the latter measures the accumulation of labeled protein, whereas the former measures the consequence of catalytic activity.
Inhibition of F-actin polymerization enhances stimulated coupling of Lyn with IgE-FcεRI
We investigated how the actin cytoskeleton might influence IgE-FcεRI coupling with Lyn and consequent phosphorylation that initiates signaling. We observed with live-cell total internal reflection fluorescence (TIRF) microscopy that the membrane-proximal cytoskeleton undergoes dynamic remodeling within seconds after antigen stimulation (Supplemental Figure S3). Substantial portions of cortical F-actin first rapidly disassociate from the plasma membrane in response to antigen and then apparently undergo repolymerization to reappear at the plasma membrane (Wilson et al., 2016). In spite of this dramatic behavior, no changes were observed in the cross-correlation of IgE-FcεRI with the bulk F-actin labeled with the fluorescent actin-binding peptide LifeAct-mEos3.2 (Riedl et al., 2008; Supplemental Figure S4). We measured colocalization of Lyn with IgE-FcεRI in experiments in which actin polymerization is inhibited. Samples were prepared for Lyn-mEos3.2/Dy654 IgE two-color imaging as in Figure 1, except that these experiments also included 5-min incubation with 1 μM cytochalasin D or 1 μM latrunculin A preceding antigen stimulation in the continued presence of the actin-disrupting drug. These treatment conditions were shown previously to provide maximal enhancement effects on stimulated degranulation and enhance early events in FcεRI signaling (Frigeri and Apgar, 1999). Two-color FLM images of cells stimulated for 6 min in the presence and absence of latrunculin or cytochalasin (Figure 3A) show no clear differences in Lyn/IgE-FcεRI colocalization. However, comparison of Lyn/IgE-FcεRI cross-correlations for the various treatment conditions as a function of stimulation time reveal that Lyn/IgE-FcεRI interactions are enhanced when actin polymerization is inhibited (Figure 3B). Average cross-correlation functions for unstimulated cells and cells stimulated for 1 min, with and without drug treatment, highlight some of these differences (Figure 3C). Cytochalasin treatment causes sustained enhancement of cross-correlation amplitudes as early as 1 min (red; Figure 3C, middle) and for the first 6 min after stimulation (red; Figure 3B, top). Enhancement due to latrunculin is shorter lived, extending through 3 min, but is also larger in magnitude at these earlier time points (green; Figure 3, B, top, and C, middle). Of interest, latrunculin also causes a small but significant increase in the cross-correlation amplitude for Lyn/IgE-FcεRI in unstimulated cells (green; Figure 3, B and C, top).
FIGURE 3:

Inhibition of actin polymerization enhances Lyn/IgE-FcεRI cross-correlation amplitude. (A) Representative Lyn-mEos3.2/Dy654 IgE FLM images of cells stimulated for 6 min without (top) or with treatment with cytochalasin (middle) or latrunculin (bottom). (B) Lyn/IgE-FcεRI cross-correlation fit parameters A and ξ as a function of stimulation time for RBL-2H3 cells expressing Lyn-mEos3.2 and sensitized with Dy654 IgE that were stimulated for 0, 1, 3, 6, or 12 min with and without treatment with actin-disrupting drugs. Fit parameters are averaged over multiple cells for each time point (eight or nine cells per time point for untreated cells, nine cells per time point for cytochalasin-treated cells, and seven cells per time point for latrunculin-treated cells). Lyn/IgE-FcεRI cross-correlation function fit parameters for untreated cells are reproduced from Figure 1. Error bars denote SEM. (C) Average cross-correlation functions for the three treatment conditions for unstimulated cells (top), cells stimulated for 1 min (middle), and the average integral of the cross-correlation function minus 1 from 0 to 300 nm for 0- and 1-min stimulated time points (bottom).
Lyn and IgE-FcεRI are weakly correlated over longer distances for both cytochalasin and latrunculin treatments in unstimulated cells (Figure 3C, top) as quantified by ξ (Figure 3B, bottom). This could reflect some opening of the cortical actin meshwork that influences the organization of both Lyn and IgE-FcεRI within the membrane. For cells stimulated 1–12 min, Lyn/IgE-FcεRI correlated structures have similar values for ξ (∼100 nm), regardless of cytoskeletal perturbation (Figure 3B, bottom).
The integral of the correlation function contains contributions from both the amplitude and length scale of cross-correlations and is proportional to the total number of cross-correlated molecules per cluster, on average. The integral of the cross-correlation function over a radius of 250 nm is significantly larger for unstimulated and 1-min-stimulated cells when treated with either cytochalasin and latrunculin (Figure 3C, bottom), suggesting that a given IgE-FcεRI molecule is spatially correlated with significantly more Lyn molecules over this length scale under these conditions. In contrast, inhibition of actin polymerization has no apparent significant effect on IgE-FcεRI or Lyn auto-correlation (Supplemental Figure S5), suggesting that changes in Lyn/IgE-FcεRI cross-correlations are caused by changes in the capacity of Lyn and IgE-FcεRI to interact and not by differences in receptor cluster formation. Thus, for unstimulated cells and within 1 min of stimulation, IgE-FcεRI can interact with a larger pool of Lyn in the membrane of cells when actin polymerization has been inhibited.
Inhibition of actin polymerization enhances antigen-dependent tyrosine phosphorylation correlated with IgE-FcεRI
We carried out two-color phosphotyrosine/IgE-FcεRI FLM experiments to evaluate the functional consequence of cytoskeletal perturbation and resulting enhancement of Lyn association with IgE-FcεRI. We stimulated Dy654 IgE-sensitized RBL-2H3 cells fixed and labeled for phosphotyrosine as in Figure 2, except that cells were first treated with cytochalasin or latrunculin as in Figure 3. We found that perturbation of actin polymerization leads to relative increases in phosphotyrosine/IgE-FcεRI cross-correlation amplitudes (Figure 4A, top), consistent with enhancement of Lyn/IgE-FcεRI colocalization under the same conditions (Figure 3, B and C). Latrunculin enhances colocalization more prominently in unstimulated cells and at earlier times after stimulation (1- and 3-min time points; Figure 4A, top and inset), whereas the difference between cytochalasin-treated and untreated cells is greater after 6 min of stimulation. Enhancement of the cross-correlation amplitude in unstimulated cells by latrunculin (Figure 4A, top, inset) is consistent with results in Figure 3C (top) and with the possibility that non–cross-linked IgE-FcεRI has a somewhat increased capacity to interact with and be phosphorylated by Lyn when the actin cytoskeleton is disrupted.
FIGURE 4:
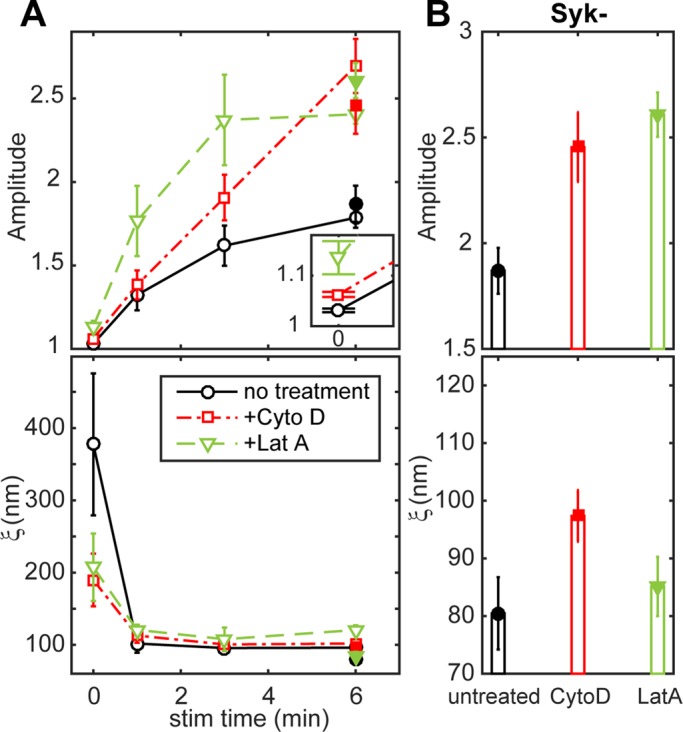
IgE-FcεRI-correlated phosphotyrosine is enhanced due to inhibition of actin polymerization. (A) Phosphotyrosine/IgE-FcεRI cross-correlation fit parameters A and ξ as a function of stimulation time for RBL-2H3 cells sensitized with Dy654 IgE and stimulated for 0, 1, 3, or 6 min with and without treatment with actin-disrupting drugs, followed by immunolabeling with A488 4G10 anti-phosphotyrosine (A488 pY). Fit parameters are averaged over multiple cells for each time point (six cells per time point for untreated cells, seven cells per time point for cytochalasin and latrunculin-treated cells). pY/IgE-FcεRI cross-correlation function fit parameters are also plotted and are reproduced from Figure 2. Inset, values for A at the unstimulated time point on an expanded scale. Equivalent data for Syk-negative cells shown in B are plotted in A (filled symbols) for direct comparison to wild-type RBL-2H3 cells. (B) Average pY/IgE-FcεRI cross-correlation fit parameters for A488 pY/Dy654 IgE two-color images of Syk-negative cells that were stimulated for 6 min with and without treatment with actin-disrupting drugs. Fit parameters are averaged over multiple cells for each treatment condition (four cells for all treatment conditions). All error bars denote SEM.
Values of the cross-correlation length, ξ, for phosphotyrosine/IgE-FcεRI in stimulated cells are very similar regardless of treatment with the actin-disrupting drugs (Figure 4A, bottom), consistent with the results for Lyn/IgE-FcεRI (Figure 3A, bottom). The value of ξ for phosphotyrosine/IgE-FcεRI in unstimulated, untreated cells has a high degree of uncertainty because the phosphotyrosine label is both less dense than in stimulated cells and the distribution of this basal phosphorylation appears nearly random (amplitude close to 1). The values for the unstimulated latrunculin- and cytochalasin-treated cells suggest a weak correlation between IgE-FcεRI and phosphotyrosine over a range of ∼200 nm (Figure 4A, bottom), similar to the corresponding Lyn/IgE-FcεRI cross-correlations (Figure 3B, bottom).
To distinguish contributions of stimulated Lyn kinase activity from downstream tyrosine phosphorylation activities, we evaluated responses in a Syk-negative variant of the RBL-2H3 cell line. These cells do not express Syk kinase at a detectable level and do not exhibit antigen-dependent Ca2+ or degranulation responses. However, antigen does stimulate FcεRI phosphorylation in Syk-negative cells, albeit at lower levels, consistent with severance of signaling downstream of Syk (Zhang et al., 1996). We found that Syk-negative cells pretreated with latrunculin or cytochalasin and stimulated with antigen for 6 min have larger phosphotyrosine/IgE-FcεRI cross-correlation amplitudes than untreated cells (Figure 4B, top) and are the same, within error, to values from wild-type RBL-2H3 cells (Figure 4A, top, 6 min, solid symbols). Values of ξ for treated and untreated Syk-negative cells after 6 min of stimulation are also similar to those in RBL-2H3 cells (Figure 4, B, bottom, and A, bottom, 6 min, solid symbols). These comparative results support the view that actin dependence of the tyrosine phosphorylation colocalized with IgE-FcεRI is present at the level of Lyn kinase phosphorylation, upstream of Syk-dependent signaling or feedback inhibition. They provide further evidence that inhibition of actin polymerization enhances phosphorylation by facilitating Lyn recruitment to clustered FcεRI at the earliest stages of stimulated signaling.
Colocalization of clustered IgE-FcεRI with lipid-anchored mEos3.2 is actin-dependent for saturated but not for unsaturated fatty acyl lipid anchors
We tested the current model that colocalization of Lyn with antigen-clustered IgE-FcεRI depends on its anchorage to the inner leaflet of the plasma membrane by saturated fatty acids, which prefer an ordered lipid environment. We compared minimal lipid-anchored fluorescent constructs consisting of mEos3.2 fused with a small segment of protein with selected acylation sites. The palmitoylated, myristoylated lipid anchor of Lyn (PM) incorporates the N-terminal tail of Lyn, including its palmitoylation and myristoylation sites (Pyenta et al., 2001). A second lipid anchor, which is similar to the membrane anchorage motif of K-Ras, consists of a geranylgeranyl modification (instead of a farnesyl modification) and polybasic sequence (GG), and has a greater preference for a disordered lipid environment (Pyenta et al., 2001). We imaged stimulated cells transfected with the PM-mEos3.2 or the mEos3.2-GG fusion construct and sensitized with Dy654 IgE (Figure 5A). FLM images were quantified using cross-correlation analysis. The amplitude parameter shows that, like Lyn-mEos3.2 (Figure 1C), PM-mEos3.2 becomes increasingly colocalized with IgE-FcεRI with increasing stimulation time, albeit at a somewhat slower rate (Figure 5B, top; solid lines, circles). PM-mEos3.2 association with IgE-FcεRI is also enhanced by treatment with latrunculin (green), especially at early stimulation time points, (Figure 5B, top; solid lines, triangles). In contrast to PM-mEos3.2, mEos3.2-GG is substantially less enriched in stimulated IgE-FcεRI clusters (Figure 5B, top; dashed lines, circles). The relatively low level of enrichment that we observe for mEos3.2-GG is on the order of the expected contribution from bleedthrough of Dy654 into the red emission channel (Supplemental Figure S2). The distribution of mEos3.2-GG relative to IgE-FcεRI is also not significantly affected by latrunculin treatment (Figure 5B, top; dashed lines, triangles). The cross-correlation length, ξ, for GG/IgE-FcεRI (Figure 5B, bottom; dashed lines) does not appear to change much with stimulation, whereas ξ decreases upon stimulation for PM/IgE-FcεRI (Figure 5B, bottom; solid lines), similar to that for Lyn/IgE-FcεRI (Figure 1C, right). Together these data indicate that the order-preferring PM lipid anchorage of Lyn is sufficient for nanoscale colocalization with clustered IgE-FcεRI that is attenuated by the actin cytoskeleton, whereas the disorder-preferring GG lipid anchorage does not confer these properties. The lack of an effect of latrunculin on GG/IgE-FcεRI cross-correlations also suggests that the effects of latrunculin on PM/IgE-FcεRI codistributions are not attributable to latrunculin-induced changes in membrane topology, which would also affect GG.
FIGURE 5:
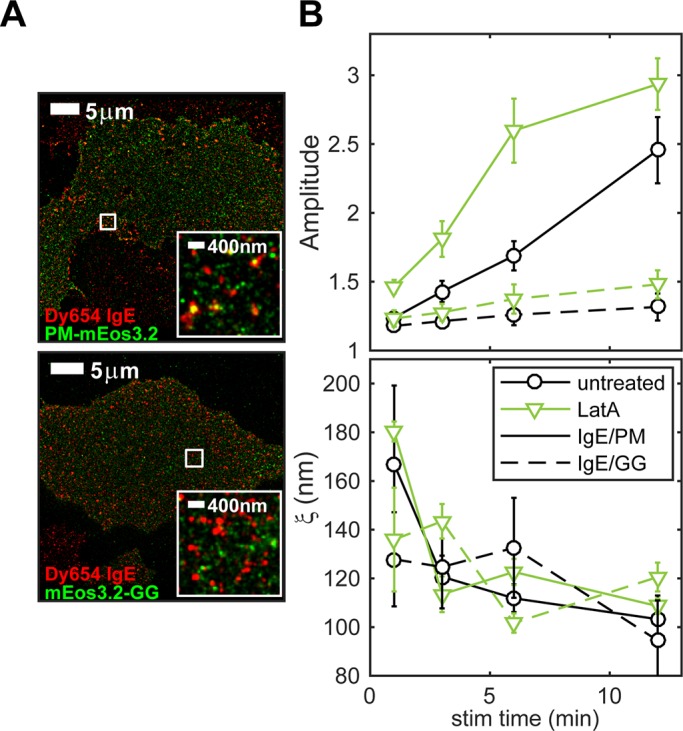
Lipid anchorage of mEos3.2 constructs determines antigen-induced recruitment to IgE/FcεRI and the effects of cytoskeletal perturbation. (A) Representative two-color images of cells expressing PM-mEos3.2 (top) or mEos3.2-GG (bottom) sensitized with Dy654 IgE and stimulated for 6 min. (B) Average PM/IgE-FcεRI and GG/IgE-FcεRI cross-correlation fit parameters A and ξ as a function of stimulation time in RBL-2H3 cells. Cells were stimulated for 1, 3, 6, or 12 min with and without treatment with 1 μM latrunculin. Fit parameters are averaged over multiple cells for each time point (seven cells per time point for cells expressing both constructs and for both treatment conditions). Error bars denote SEM.
Two-dimensional Ising model simulations recapitulate the effects of actin disruption on FcεRI interactions with order-preferring components
To place our results in a theoretical framework, we conducted simulations of a modified two-dimensional (2D) Ising model adapted from Machta et al. (2011), which combines critical fluctuations of ordered (Lo) and disordered (Ld) lipid compositions together with connectivity of one or the other phase to the cortical actin cytoskeleton. The previous simulations approximating physiological temperatures (1.05Tc) showed that when one phase is pinned to the actin meshwork, it forms rough “channels” along the actin filaments, surrounding the other phase within the resulting corral. These features in the relative distributions of the two phases represent regions of high probability for partitioning of components preferring one or the other phase (Machta et al., 2011). Subsequent experiments with F-actin and supported lipid bilayers of defined composition and pinning sites yielded results that support this model (Honigmann et al., 2014; Andrade et al., 2015).
To evaluate our FLM results in terms of this simple model, we first assumed that the ordered components in the plasma membrane preferentially connect with the cortical actin meshwork, as suggested previously (Viola and Gupta, 2007; Head et al., 2014). As depicted in Figure 6A, our simulation considers two types of dynamic membrane components: order (Lo) preferring (dark gray) and disorder (Ld) preferring (light gray). The ordered components are assigned a larger tendency to colocalize with an underlying actin meshwork, which remains static. FcεRI (red) and PM (green) are each represented as a small subset of the order-preferring population. We simulated a range of sizes for the static corrals from smaller (Figure 6A, top) to larger (Figure 6A, bottom) to incorporate potential effects of latrunculin- or cytochalasin-like perturbation of the cortical actin meshwork, which tends to increase the dimensions of actin-based corrals that confine the diffusion of membrane proteins and lipids (Kusumi et al., 2012; Fujiwara et al., 2016).
FIGURE 6:

Actin disruption enhances FcεRI receptor-PM interactions in simulations of the 2D Ising model. (A) Simulation snapshots (left) and time-averaged positions (right) of receptors (FcεRI) and PM (lipid anchor of Lyn) in simulations of the 2D Ising model coupled to a static actin network and performed as described in Materials and Methods. Receptors (red), PM (green), and actin (not shown) all prefer to be in contact with the same type of components, referred to as ordered lipids (Lo, dark gray pixels in simulation snapshots). Receptors are clustered within actin corrals by a Gaussian-shaped potential that does not directly affect the localization of other components in clustered simulations. Simulations were conducted over a range of corral sizes. Representative simulations with small (top) and large (bottom) average corral sizes are shown. The sizes indicated represent the square root of the median corral area. (B) Pair cross-correlation function, c(r), tabulated between receptors and PM in the absence (top) and presence (bottom) of receptor clustering. The curves on the right (blurred c(r)) are convolved with a Gaussian-shaped filter with σ = 24 nm in each channel to facilitate comparison with experimental results (Supplemental Figure S7). (C) Simulation snapshots (left) and time-averaged positions (right) of receptors and PM, equivalent to A for the case in which the disordered (Ld) components (light gray pixels) preferentially associate with the actin meshwork, whereas receptors and PM still associate with Lo components (dark gray pixels). (D) Summary of amplitude values, c(r = 0), from blurred receptor-PM cross-correlation functions over the range of median corral sizes and for the three types of membrane-actin interactions: order preferring (blue), disorder preferring (red), and locally randomized preference (green). Example simulation images, snapshots, and correlation functions from simulations of the latter two cases are shown in Supplemental Figure S9. Data from simulations run in the absence of actin are plotted as a single point (black diamond). The horizontal dashed line is set by the y-axis value of this point. The vertical dashed line indicates the diameter of receptor clusters (100 nm).
Using a large number of simulation snapshots (Figure 6A, left), we consider cross-correlations of FcεRI and PM in the unstimulated, unclustered state (Figure 6B, top) and after antigen binds to form FcεRI clusters of 100 nm diameter in the center of a corral (Figure 6B, bottom), as described in Materials and Methods. Raw cross-correlation curves are tabulated from the locations of discrete pixels of the simulation snapshots (Figure 6B, left), and cross-correlations are blurred by a Gaussian filter with σ = 24 nm to approximate the effect of the point spread function of our FLM measurements (Figure 6B, right). In raw cross-correlations, cross-correlation lengths increase for both the unclustered and clustered receptors as corral dimensions increase. Differences in correlation length are converted into differences in cross-correlation amplitude by Gaussian blurring, and the resulting cross-correlations show increasing amplitudes as the corral dimensions increase. Plotted as a function of the corral dimension (Figure 6D), these amplitudes increase sharply in the regime of 50–100 nm and continue to increase more gradually as the corrals essentially disappear in the regime of no actin meshwork.
We also considered the reverse case in which disordered membrane components preferentially associate with the actin meshwork, with all other simulation parameters remaining the same (Figure 6C and Supplemental Figure S9, A and B). In this reversed situation, the amplitudes of Gaussian-blurred FcεRI/PM cross-correlation functions also increase as corral dimensions increase in the regime of relatively small corral sizes of 50–200 nm, with a shallower slope and a shift up in amplitude compared with the first case (Figure 6D, red [Ld] compared with blue [Lo]). Also in contrast to the Lo case, the Ld curves display a local maximum at a corral size around 300 nm, and they begin to decrease at larger corral sizes, converging to the same value as for the Lo case as the corrals disappear (Figure 6D). Cross-correlation amplitudes from simulations in which the phase preference of actin is randomized along the length of the filament (Figure 6D, green, and Supplemental Figures S8 and S9, C and D) depend only weakly on corral size. These comparative simulations show that collective coupling to the actin meshwork by one type of membrane component preferentially over the other significantly affects the capacity of clustered Lo-preferring receptors to accumulate other ordered components when the actin meshwork is modulated. We consider similarities and differences between these two simulation cases together with experimental results in the next section.
DISCUSSION
FLM provides a quantitative nanoscale view of the key initiation event of the FcεRI signaling cascade: receptor association with and phosphorylation by Lyn tyrosine kinase. A sufficiently stable, functional interaction between IgE-FcεRI and Lyn requires antigen engagement, and previous evidence suggests that a threshold is set, in part, by constraints in the plasma membrane, including those caused by attachment to the actin cytoskeleton. Single-particle tracking studies demonstrate that the diffusion of membrane proteins and lipids is largely confined within corrals imposed by interaction with the underlying actin meshwork (Kusumi et al., 2005). Simulations (Ehrig et al., 2011; Machta et al., 2011) and recent experiments in model systems (Honigmann et al., 2014; Andrade et al., 2015) indicate that similar interactions could modulate the organization of plasma membrane lipids. Attachment to the actin meshwork by either order-preferring or disorder-preferring components may consequently affect the distribution of the other components that are not directly attached. We imaged cells before and during the first few minutes of antigen stimulation to focus on the earliest events of signaling, when the original cytoskeletal constraints are altered in response to signaling pathways initiated as Syk is recruited to Lyn-phosphorylated FcεRI. Application of FLM techniques enabled direct imaging of Lyn/IgE-FcεRI coupling before and at these earliest stimulation times, which were not previously accessible using conventional optical microscopy.
Lyn, stimulated tyrosine phosphorylation, and an order-preferring lipid anchor colocalize with antigen-clustered IgE-FcεRI
Pair cross-correlation analysis of FLM images shows increased Lyn/IgE-FcεRI colocalization by 1 min after antigen addition, and the amplitude of the cross-correlation continues to increase over the next 10 min (Figure 1). Previous studies using immunoprecipitation to measure the time course of antigen-stimulated interactions showed a rapid but transient increase in Lyn bound to FcεRI that peaks within 1–2 min after antigen stimulation (Yamashita et al., 1994; Honda et al., 1997). A possible explanation for this difference from our imaging results is that immunoprecipitation methods are less likely to capture Lyn that is not stably or directly bound to FcεRI. The cross-correlations we observe as long as 12 min after antigen addition suggest that this association may be mediated through other structures in the plasma membrane—for example, local enrichment of more ordered lipids—or through assembled complexes of signaling proteins, rather than through direct binding. Previous transmission electron microscopy (EM) evaluation of membrane sheets prepared from RBL cells showed coclusters of Lyn and IgE-FcεRI that rearrange within 2 min of activation with antigen (Wilson et al., 2000). However, participation of the actin cytoskeleton may be obscured by the rip-off procedure used in preparation of membrane sheets in this study. We previously used scanning EM of intact cells and quantified nanoscale clustering of Lyn and coclustering with IgE-FcεRI using pair-correlation functions (Veatch et al., 2012a). We observed stimulated coclustering within the first few minutes after addition of DNP-BSA, consistent with our FLM results reported here.
With FLM, we observe colocalization of IgE-FcεRI and stimulated tyrosine phosphorylation (monitored by anti-phosphotyrosine 4G10), and this serves as a functional readout of the initiation of signaling. Concomitant increases in cross-correlation amplitudes for IgE-FcεRI relative to both Lyn (Figure 1) and stimulated tyrosine phosphorylation (Figure 2) are consistent with previous biochemical studies showing that coupling of Lyn with antigen–cross-linked IgE-FcεRI clusters and consequent receptor phosphorylation is necessary to initiate early stages of signaling (Pribluda et al., 1994; Yamashita et al., 1994). Our previous FLM experiments in live cells showed that antigen-induced IgE-FcεRI clustering sufficient for the initiation of Ca2+ mobilization occurs in the first few minutes after antigen addition (Shelby et al., 2013), and the present two-color FLM experiments demonstrate enhancement of Lyn colocalization during this time frame. Our observation that an order-preferring lipid anchor (PM) but not a disorder-preferring lipid anchor (GG) colocalizes with clustered IgE-FcεRI provides supportive evidence that coupling of Lyn and consequent phosphorylation of FcεRI after antigen stimulation occurs in ordered lipid regions of the plasma membrane.
Cytochalasin and latrunculin enhance colocalization of Lyn, tyrosine phosphorylation, and an order-preferring lipid anchor with IgE-FcεRI
In addition to directly visualizing these early signaling assemblies, our studies were strongly motivated to gain new insight into their regulation by the actin cytoskeleton before and soon after antigen stimulation. We treated cells with cytochalasin D and latrunculin A, which interfere with polymerization of F-actin by different mechanisms: cytochalasin D caps the barbed end of actin filaments (Cooper, 1987), and latrunculin A sequesters monomeric actin (Coué et al., 1987). Both were shown previously to enhance signaling at a point upstream of receptor phosphorylation, although they differ in their specific effects on events leading ultimately to mast cell degranulation (Frigeri and Apgar, 1999; Holowka et al., 2000; Tolarová et al., 2004).
Cross-correlations of IgE-FcεRI with Lyn and phosphotyrosine are both enhanced by cytochalasin and latrunculin. Latrunculin-induced enhancement is more dramatic at early stimulation time points, whereas the effects of cytochalasin are more robust at later times (Figures 3 and 4). Because the total specific activity of plasma membrane Lyn is unaffected by inhibition of actin polymerization (Frigeri and Apgar, 1999; Tolarová et al., 2004; Torigoe et al., 2004), we conclude that actin destabilization–induced enhancement of tyrosine phosphorylation that correlates with IgE-FcεRI/Lyn clusters is a consequence of increased Lyn association. Previous studies reported that latrunculin causes weak phosphorylation of FcεRI β and Syk in the absence of stimulation (Tolarová et al., 2004) and also causes differential depletion of actin from the pool that associates with detergent-insoluble (Lo-like) membranes (Frigeri and Apgar, 1999; Tolarová et al., 2004). These results point to engagement of the actin cytoskeleton with ordered regions of the plasma membrane to restrict spontaneous coupling of FcεRI and Lyn. Cell stimulation mediated by IgE-FcεRI involves de novo actin polymerization (Supplemental Figure S4; Pfeiffer et al., 1985), and some of this cytoskeletal activity acts as a negative feedback inhibitor of signaling that is reduced by latrunculin and cytochalasin treatment (Pierini et al., 1997). This may explain in part why enhancement of phosphotyrosine/IgE-FcεRI colocalization in actin-destabilized cells persists at longer stimulation times than enhancement of Lyn/IgE-FcεRI colocalization (Figures 3 and 4).
Antigen-stimulated increases in cellular phosphotyrosine may include contributions from phosphorylation of clustered FcεRI (β and γ subunits) and other signaling proteins that consequently assemble in proximity to receptor clusters, including Syk and its substrates, as well as other substrates of Lyn (Veatch et al., 2012a). Labeling these with anti-phosphotyrosine 4G10 in our FLM images could account for some of the differences that we observe in the time dependence of phosphotyrosine/IgE-FcεRI colocalization and more transient FcεRI β- and γ-chain phosphorylation detected by Western blot (Pribluda and Metzger, 1992; Xu et al., 1998; Sheets et al., 1999; Holowka et al., 2000). We found that enhancement of tyrosine phosphorylation caused by cytochalasin and latrunculin is preserved in Syk-negative cells (Figure 4B, top), for which downstream signaling is prevented (Zhang et al., 1996). These results indicate that the enhancement we observe in normal RBL-2H3 cells originates largely from increased Lyn-mediated phosphorylation of FcεRI along with any other Lyn substrates that may colocalize with receptor clusters.
We hypothesize that the regulation of Lyn localization by actin is mediated through Lyn’s preferential association with ordered lipid compositions, and this is supported by our finding that the actin cytoskeleton similarly modulates antigen-induced colocalization of IgE-FcεRI with the saturated (PM) lipid anchor (Figure 5). Furthermore, we found that the unsaturated GG lipid anchor associates with antigen-clustered IgE-FcεRI to a much lesser extent, unaffected by the actin-destabilizing drugs (Figure 5). These results are consistent with previous confocal fluorescence imaging experiments, which indicated that enhancement by cytochalasin D of stimulated Lyn/IgE-FcεRI colocalization involves association with ordered lipid compositions (Holowka et al., 2000). We previously showed, through coreconstitution of FcεRI, Lyn, and phosphatases that were artificially targeted to ordered membranes, that association with ordered lipids can protect receptors from dephosphorylation and facilitate functional coupling in the initiation of signaling (Young et al., 2005). Enhanced protection from phosphatases of both FcεRI and Lyn in receptor clusters may be another cause of the increase in signaling responses that we observe as a result of cytochalasin and latrunculin treatment.
Our experiments extend and refine previous observations of regulated segregation of FcεRI and Lyn from electron and fluorescence microscopy (Holowka et al., 2000; Wilson et al., 2000) and from biochemical data (Frigeri and Apgar, 1999; Torigoe et al., 2004). We provide strong evidence that actin stabilizes membrane structures to minimize spontaneous collisions of Lyn with IgE-FcεRI and that antigen-stimulated coupling of Lyn with IgE-FcεRI, and thereby initiation of signaling, is mediated by ordered lipids and regulated by the actin cytoskeleton.
A model for regulation of IgE-FcεRI coupling with Lyn via its PM lipid anchor through constraint of lipid phase–like behavior by cortical actin
We considered our experimental results in terms of Ising-based simulations that model interactions between FcεRI and ordered membrane components such as Lyn and PM and possible modulation by the actin cytoskeleton. Assuming first that ordered membrane components preferentially associate with the actin meshwork, the simulations show that as the dimensions of the corrals increase from smaller (Figure 6A, top) to larger (Figure 6A, bottom), the same clustering of FcεRI increases the cross-correlation amplitudes of FcεRI and PM (Figure 6, B, bottom, and D, blue curve). We interpret this trend as a combination of two effects. Small corral sizes have dimensions close to or smaller than the diameter of the receptor cluster, set at 100 nm. In this regime, the dimensions of the meshwork limit the size of the ordered membrane domain that can form around the receptor cluster. As a result, the cross-correlation depends strongly on corral size. The second effect, which predominates at larger corral dimensions, arises from competition for ordered components between the actin meshwork and order-preferring FcεRI that are clustered in the middle of corrals. This second effect accounts for the continued increase in cross-correlation amplitudes as the actin meshwork becomes increasingly sparse and the effective competition is lessened. This is an example of how the presence of actin can influence the local environment within receptor clusters even when the actin itself resides far from the receptor cluster. This action at a distance occurs because the number of components remains conserved in these simulations.
This interpretation is consistent with the simulated behavior of the system when disordered membrane components preferentially associate with actin (Figure 6, C and D, red curve). Dimensions of the actin meshwork also limit the potential size of the ordered membrane domain colocalizing with the receptor cluster in the regime of relatively small corral sizes. However, when disordered components preferentially associate with actin, the second effect of competition for ordered components is reversed: at increasing larger corral sizes, the cross-correlation amplitudes reach a maximum and then decrease. Thus, in the context of this simple model, preferential association of either ordered or disordered components with the actin meshwork attenuates interactions between FcεRI and ordered membrane components when actin corrals are small relative to the size of receptor clusters. The differences in amplitude values, slopes of the ascending curves, and behavior at large corral sizes for the ordered and disordered cases are related to the asymmetry of the comparison: in both cases, we evaluate accumulation of ordered components at a cluster of order-preferring receptors.
These simulations provide a framework for viewing our experimental FLM results together with measured effects of cytochalasin and latrunculin on the cortical actin meshwork, although other explanations are also possible. Our measurements of the clustered IgE-FcεRI auto-correlation (Supplemental Figure S1) provided a reasonable value for receptor cluster size. Diffusion and EM tomography measurements of other cell lines place corral dimensions in the range of 40–230 nm (Morone et al., 2006; Fujiwara et al., 2016), but we do not know these values for RBL-2H3 cells or the degree to which they are altered by cytochalasin, latrunculin, or antigen stimulation under our conditions. Nonetheless, we observe qualitatively similar trends in our experimental results and in the simulation. Enhanced cross-correlation due to inhibition of actin polymerization occurs in the first few minutes after addition of cross-linking antigen (Figures 3 and 5), with more limited evidence for small enhancement in unstimulated cells (Figure 3C compared with Figure 6A, top).
Our comparative model assumes that formation of corrals by the cortical actin meshwork is mediated by selective association with actin structures of either order- or disorder-preferring components. Although our FLM data do not distinguish definitively between the two possibilities, recent evidence supports the view that order-preferring adapter proteins mediate actin contacts with ordered membrane components in lymphocyte signaling (Viola and Gupta, 2007; Head et al., 2014). One example is Cbp/PAG, which is strongly order preferring, a known interaction partner of Lyn, and coupled to the cytoskeleton via interactions with EBP-50 and ezrin (Hrdinka and Horejsi, 2014). Of note, Cbp/PAG associates with Csk, which inhibits Lyn kinase activity and can attenuate Lyn association with FcεRI when overexpressed (Ohtake et al., 2002). In addition, coupling between ordered lipids and the cytoskeleton may occur through long-chain phosphatidylserine attaching via an adaptor to short actin filaments, which are clustered by myosin activity (Goswami et al., 2008; Raghupathy et al., 2015). It appears that both the relatively stable actin meshwork and dynamic short actin filaments, when pinned to the plasma membrane, can provide the means to regulate lipid phase–like behavior, which surely has functional consequences when the poised resting cell is stimulated and the polymerization state of actin changes.
Concluding remarks
Our experiments using inhibitors of actin polymerization offer a route toward understanding the role of cortical actin in the remodeling that accompanies the early stages of signaling. Some outcomes of the rapid depolymerization of cortical actin that occurs within seconds of antigen addition (Wollman and Meyer, 2012; Wilson et al., 2016; Supplemental Figure S3) may be roughly approximated by latrunculin and cytochalasin treatments. Our data indicate that the actin cytoskeleton regulates FcεRI signaling by controlling the spatial localization of Lyn, and this is exerted through the order-preferring lipid anchorage of Lyn. This view extends the existing body of literature suggesting that the actin cytoskeleton plays a role in organization of the plasma membrane, suppressing FcεRI phosphorylation before antigen engagement, and underscores the importance of lipid-mediated association of Lyn with IgE-FcεRI as part of the initiation of signaling. We hypothesize that actin contact sites with the plasma membrane actively sequester Lyn through mutually preferential interaction with ordered regions of the plasma membrane. In a living system, this would limit spurious inflammatory signaling until IgE-FcεRI clustering becomes sufficient to stabilize ordered lipid regions and compete for order-preferring Lyn while excluding disorder-preferring phosphatases (Field et al., 1995, 1997; Sheets et al., 1999; Young et al., 2003). Signaling steps soon after the initiating event cause actin depolymerization to amplify the response to antigen in the earliest stages, before further cytoskeleton rearrangements modulate the downstream events. The interplay between the actin cytoskeleton and phase-like properties of the plasma membrane provides a highly tunable mechanism for preventing transduction of a subthreshold signal while creating a membrane environment that is highly responsive and primed for activation by regulating colocalization of receptors and signaling molecules.
MATERIALS AND METHODS
Chemicals and reagents
Cell culture reagents, including MEM, trypsin-EDTA, and gentamicin sulfate, were acquired from Life Technologies (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Atlanta, GA). Coverslip dishes were purchased from MatTek (Ashland, MA), and 125-nm Tetraspeck fluorescent beads were purchased from Life Technologies. The amine-reactive fluorophore Dy654 was purchased from Dyomics GmbH (Jena, Germany). Latrunculin A was purchased from both Life Technologies and Sigma-Aldrich (St. Louis, MO), and cytochalasin D was from Sigma-Aldrich. Glutaraldehyde (25% stock) was purchased from Ted Pella (Redding, CA). Paraformaldehyde was purchased from Electron Microscopy Services (Hatfield, PA). Supplies for FLM imaging buffer, including β-mercaptoethanol, glucose oxidase, catalase, and Tris-HCl, were acquired from Sigma-Aldrich.
Multivalent antigen, DNP-BSA with an average of 15 DNP molecules per BSA, was prepared as described previously (Hardy, 1986). Anti-phosphotyrosine clone 4G10 antibody was obtained from Millipore (Billerica, MA), and the Alexa Fluor 488-conjugated anti-mouse IgG2b secondary antibody was purchased from Life Technologies. Mouse monoclonal anti-DNP IgE, prepared as described previously (Gosse et al., 2005), was conjugated to amine-reactive Dy654 according to manufacturer’s instructions and measured to have a dye:protein ratio of 2.7:1. Dy654 IgE displayed normal function in RBL-2H3 cells, that is, capacity to label cells and elicit robust antigen-induced degranulation responses comparable to unlabeled IgE, as measured by a standard degranulation assay (Naal et al., 2004), without causing antigen-independent degranulation.
Fluorescent constructs and variant RBL cell lines
Lyn-mEos3.2 and mEos3.2-LifeAct constructs were prepared through site-directed mutagenesis to incorporate I102N, H158E, and Y189A mutations into mEos2 (Zhang et al., 2012). The use of Lyn-mEos3.2 to study immunoreceptor signaling has been reported previously (Stone and Veatch, 2015). Functionality of fluorescent fusion Lyn constructs, specifically Lyn–enhanced green fluorescent protein (EGFP), has been validated in previous experiments (Larson et al., 2005) through observation of Lyn-EGFP–dependent phosphorylation of FcεRI in a reconstituted system in which Lyn-EGFP, the protein tyrosine phosphatase PTPα, and FcεRI were expressed in CHO cells, which lack endogenous Lyn. PM-mEos3.2 and mEos3.2-GG constructs were prepared through restriction enzyme excision of EGFP from PM-EGFP and EGFP-GG constructs as described previously (Pyenta et al., 2001), followed by insertion and ligation of the mEos3.2 sequence. The Syk-negative RBL TB1A2 (Zhang et al., 1996) cell line was a gift from Reuben Siraganian (National Institutes of Health, Bethesda, MD).
Preparation of fixed RBL-2H3 samples for imaging
Cell culture and transfection.
RBL-2H3 cells and the RBL-derived Syk-negative TB1A2 cells were maintained in culture using RBL medium containing MEM with l-glutamine and phenol red, 20% FBS, and 10 μg/ml gentamicin sulfate at 37°C and 5% CO2 as described previously (Gosse et al., 2005). Cells were harvested using trypsin-EDTA and either sparsely plated onto MatTek coverslip dishes or transiently transfected with Lyn-mEos3.2, mEos3.2-LifeAct, PM-mEos3.2, or mEos3.2-GG fluorescent constructs via electroporation. Approximately 1.0 × 107 cells at a time were transfected with 10–30 μg of DNA plasmid using a Bio-Rad GenePulser Xcell. Cells were resuspended in medium, plated in MatTek dishes, and left in the incubator overnight for recovery and expression of the construct.
Sensitization, stimulation, and fixation.
Cells were sensitized with 3 μg/ml Dy654-labeled IgE in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–buffered RBL medium (80% MEM containing l-glutamine and phenol red, 20% fetal bovine serum, 50 mg/l gentamicin, and 30 mM HEPES, pH 7.4) for 40 min at room temperature. Cells were rinsed with RBL medium and returned to 37°C. For latrunculin A or cytochalasin D treatment, cells were then incubated with 1 μg/ml latrunculin A or cytochalasin D in medium for 5 min. The cells were then stimulated with multivalent antigen (500 ng/ml DNP-BSA in RBL medium with or without 1 μg/ml latrunculin or cytochalasin) for 0, 1, 3, 6, or 12 min at 37ºC. Dishes were rinsed with warm phosphate-buffered saline (PBS) and chemically fixed (4% paraformaldehyde and 0.1% glutaraldehyde in PBS) for 10 min at room temperature. The fix was quenched with blocking buffer (10 mg/ml BSA in PBS) for 10 min. Fixed samples were rinsed three more times in blocking buffer before imaging.
Phosphotyrosine immunolabeling.
Samples stimulated and fixed as described were permeabilized and labeled in a solution of Triton X-100 and anti-phosphotyrosine (4G10) antibody (5 μg/ml 4G10 antibody, 0.1% Triton X-100, and 10 mg/ml BSA in PBS) for 1 h at room temperature. Dishes were rinsed five times with blocking buffer and incubated with A488-conjugated anti-mouse IgG2b (10 μg/ml A488 anti-mouse antibody, 0.1% Triton X-100, and 10 mg/ml BSA in PBS) for 1 h. Dishes were rinsed five times with blocking buffer before imaging.
Superresolution FLM imaging
Imaging setup.
Labeled samples were imaged on an inverted microscope (Leica DM-IRB, Wetzlar, Germany) under illumination through a 1.42 numerical aperture/100× Leica TIRF objective lens. The microscope was equipped with a mercury arc lamp for epifluorescence illumination and 50-mW/405-nm, 100-mW/488-nm, 100-mW/561-nm, and 100-mW/642-nm diode-pumped solid-state lasers (Coherent, Santa Clara, CA) for TIRF imaging. Lasers were outfitted with cleanup filters (Chroma Technology, Burlington, VT), and power was attenuated with neutral density filters (ThorLabs, Newton, NJ). Multi-bandpass excitation, polychroic, and emission filters (ZET405/488/561/640x, ZT405/488/561/640rpc, and ZET405/488/561/640m, respectively; Chroma Technology) allowed for multicolor excitation/emission. The emission path was coupled through a two-channel Optosplit emission splitter (Cairn Optics, Faversham, United Kingdom) for simultaneous recording of either red and far-red (Chroma T650LPXR, ET595/50, and ET655LP) or green and far-red (Chroma T650lpxr, ET525/50m, and ET655LP) emission channels. Images were recorded with an Andor iXon 897 electron-multiplying charge-coupled device (EMCCD) camera (Andor, Belfast, United Kingdom) using a custom image acquisition code written in Matlab (MathWorks, Natick, MA).
Data acquisition.
Fixed cells were imaged in the presence of an oxygen-scavenging and reducing imaging buffer (100 mM Tris, 10 mM NaCl, 1% [wt/wt] glucose, 500 μg/ml glucose-oxidase, 40 μg/ml catalase, and 1% [vol/vol] β-mercaptoethanol at pH 8.5). For Dy654/mEos3.2 two-color imaging, epifluorescence illumination in the green emission channel was used to locate cells moderately expressing mEos3.2 constructs, avoiding highly overexpressing cells. FLM photoswitching data were recorded in far-red and red emission channels for Dy654 and mEos3.2, respectively. Dy654/A488 samples were imaged in far-red and green emission channels. For both Dy654/mEos3.2 and Dy654/A488 imaging, data were acquired at 32 frames/s with an exposure time of 10 ms, with variable EMCCD gain settings and illumination intensity. Data were acquired in a series of 500-frame movies. A given FLM image was reconstructed from at least 15 of these movies, or 7500 individual frames.
Analysis of imaging data
Image reconstruction.
FLM data were analyzed using previously described MATLAB software routines (Veatch et al., 2012b; Shelby et al., 2013). Briefly, single-molecule probe emission was localized in movies of Dy654, mEos3.2, or A488 photoswitching by least-squares fitting of diffraction-limited spots to a 2D Gaussian function. Outliers in spot intensity, aspect ratio, width, and fitting error were culled from the population of localizations.
Registration of the two-color image was achieved by spatially mapping probe localizations in one channel onto the other using a transformation determined empirically by imaging fiducial markers simultaneously in both channels, as described previously (Churchman et al., 2005). An alignment sample consisting of ∼125-nm Tetraspeck fluorescent nanospheres adsorbed onto a coverslip was imaged, and localizations of individual nanospheres in both channels were paired. Paired localizations were used as control points for the built-in Matlab function cp2tform() to infer a 2D “local weighted mean” transformation that spatially mapped one channel onto the other. Registration errors were typically in the range of 12–15 nm.
Corrections due to translational stage drift were calculated at intervals of 250 frames by determining the translational shift that minimizes the cross-correlation of successive 250-frame subimages. After drift correction, images were reconstructed by summing the localizations in each channel, and the localization precision for probes in each channel was calculated from correlation functions as described in Veatch et al. (2012b). Typical localization precision values were ∼20 nm for the Dy654 probe, 23 nm for mEos3.2, and 24 nm for A488. The number of localizations used to reconstruct a given fixed cell image varies based on factors such as labeling density in the case of the 4G10 images or expression levels in the case of mEos3.2 images and differs inherently between probes due to differences in labeling density and probe photophysics. Dy654 FLM images of IgE-FcεRI are generated from ∼300,000–600,000 localizations, mEos3.2 images of Lyn, PM, or LifeAct are generated from 350,000–500,000 localizations, and A488 images of 4G10 are generated from 100,000–700,000 localizations.
Correlation function analysis of FLM images.
Distribution and codistribution of proteins imaged in a FLM experiment are quantified through statistical analysis of the spatial map of localizations using pair auto-correlation and cross-correlation functions, as described in detail in Veatch et al. (2012b) and Shelby et al. (2013).
Briefly, the fast Fourier transform (FFT) is used to calculate the auto-correlation function, g(r), for masked regions of interest within the boundary of individual cells and for each color channel. Cross-correlation functions are calculated from the FFT of masked regions in both color channels. Auto-correlation and cross-correlation functions are normalized by the FFT of the mask and the density of localizations within the mask. Auto-correlation functions for FLM images of Dy654 IgE-FcεRI are corrected for the effects of protein overcounting due to multiple labeling and reversible probe photoswitching as described previously (Veatch et al., 2012b; Shelby et al., 2013). Auto-correlation functions of Lyn-mEos3.2 are not corrected for overcounting due to uncertainty in density of the transfected fluorescent fusion protein on the membrane. Cross-correlation functions generated from two-color images do not contain artifacts from overcounting and are fitted to a single-exponential function:
|
|
We extract A, the cross-correlation amplitude, and ξ from Eq. 1 as fit parameters.
The value of A reported throughout this study is an approximation of the value of the cross-correlation function, c(r), at r = 0. Absolute values of the cross-correlation amplitude are measures of the relative coenrichment of two species with respect to the average density of each species on the membrane.
In addition to overcounting, disproportionate undercounting of tightly clustered molecules is a possible technical issue that could bias Dy654 IgE-FcεRI auto-correlation functions or cross-correlation functions of other probes with Dy654 IgE-FcεRI from stimulated cells. This effect could arise from experimental or postprocessing factors such as self-quenching of Dy654 molecules in close proximity to each other, rejection of blinking events within the same diffraction-limited area during image processing, or any other phenomenon that leads to selective undersampling of densely labeled clusters. To mitigate this effect, we used imaging conditions that limited the average density of blinking events during data collection. If present, undersampling would have the effect of decreasing the amplitude of Dy654 IgE-FcεRI auto-correlations and cross-correlation functions.
A potential concern for the interpretation of two-color cross-correlation functions is possible inclusion of erroneous localizations due to bleedthrough from one color channel into the other, resulting in an artifactual contribution to the cross-correlation function. This artifact has been documented for far-red organic dyes, including the popular stochastic optical reconstruction microscopy probe Alexa Fluor 647 (Stone and Veatch, 2014), and is attributed to a fluorescent impurity. We evaluated bleedthrough from Dy654 into the red emission channel in the context of our imaging setup and analysis methods and found that it contributes negligibly to Lyn/IgE-FcεRI cross-correlation functions and to a slightly larger extent to PM/ IgE-FcεRI and can account for most or all of the observed GG/IgE-FcεRI cross-correlation (Supplemental Figure S2).
2D Ising model simulations
A conserved order parameter 2D Ising model was simulated on a square lattice of 400 × 400 pixels as described previously (Machta et al., 2011) with minor modifications. Briefly, components that prefer Lo or Ld regions are represented as pixels that have value of S = +1 and S = −1, respectively. The vast majority of +1 and −1 pixels represent unspecified membrane components (proteins and lipids). In addition, 100 pixels with values of +1 are classified as receptors (FcεRI), and 400 pixels with values +1 are classified as PM anchor (Lyn). The membrane is also coupled to an actin meshwork generated using a Voroni algorithm, in which the number of uniformly distributed control points was varied across simulations in order to vary the average corral size, which is stated in the figure legends. Components positioned at pixels corresponding to the location of this static meshwork feel an additional field that favors either +1 pixels (Lo-preferring) or −1 pixels (Ld-preferring). The final Hamiltonian is given by
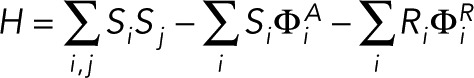 |
The first term sums over the four nearest neighbors ( j ) surrounding the pixel i and applies to all components. The second term describes coupling to the actin meshwork and contributes only for positions i where the mesh is present. At these positions, the actin potential 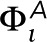 is chosen to be +0.5 for Lo-preferring actin or −0.5 for Ld-preferring actin, which is large enough in magnitude to ensure that most components in contact with the actin mesh have S = +1 or S = −1, respectively. In a third configuration, individual pixels along the mesh are randomly chosen to have
is chosen to be +0.5 for Lo-preferring actin or −0.5 for Ld-preferring actin, which is large enough in magnitude to ensure that most components in contact with the actin mesh have S = +1 or S = −1, respectively. In a third configuration, individual pixels along the mesh are randomly chosen to have 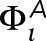 +0.5 or
+0.5 or 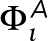 = −0.5, where the populations of +0.5 and −0.5 pixels are equal, so that sum of
= −0.5, where the populations of +0.5 and −0.5 pixels are equal, so that sum of 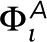 over all pixels is zero. A graphical representation of this potential is shown in Supplemental Figure S8. At positions that are not occupied by the actin mesh, the field
over all pixels is zero. A graphical representation of this potential is shown in Supplemental Figure S8. At positions that are not occupied by the actin mesh, the field 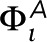 is zero. The third term of the Hamiltonian contributes only when receptors occupy position i, where Ri = 1, otherwise Ri = 0. The receptor field
is zero. The third term of the Hamiltonian contributes only when receptors occupy position i, where Ri = 1, otherwise Ri = 0. The receptor field 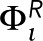 has a Gaussian shape chosen to produce a receptor distribution with a similar shape to those observed in experiment after receptor clustering with antigen (Supplemental Figure S1). This field is placed at a fixed location at the start of the simulation, with its peak centered within an actin corral.
has a Gaussian shape chosen to produce a receptor distribution with a similar shape to those observed in experiment after receptor clustering with antigen (Supplemental Figure S1). This field is placed at a fixed location at the start of the simulation, with its peak centered within an actin corral.
At each update, two random pixels are chosen, the energy cost or gain for exchanging the two pixels is calculated, and the move is either accepted or rejected using a Monte Carlo algorithm that maintains detailed balance. If the resulting configuration is lower or equal in energy, the exchange is always accepted. If the energy is raised, the exchange is accepted stochastically with probability exp(−β∆H), where β is the inverse temperature and ΔH is the change in energy between initial and final states. In this scheme, the critical point occurs at Tc = 2/ln[1 + sqrt(2)]. Simulations were run at T = 1.05Tc and pixel dimensions were chosen to represent a 2 nm × 2 nm patch of membrane, so that the correlation length varies with temperature as observed in experimental observations in isolated plasma membrane vesicles (Veatch et al., 2008). One sweep corresponds to 4002 attempted pixel swaps, so that on average two attempts are made for each pixel. All simulations are run using nonlocal exchanges to decrease equilibration times.
For each condition of actin phase preference (Lo-preferring, Ld-preferring, and random preference), the average density of PM and receptor is calculated by summing matrices representing the positions of these components at 500 simulation snapshots each separated by 100 simulation sweeps. Receptor/PM cross-correlations were also tabulated from receptor and PM configurations taken at 500 simulation snapshots each separated by 100 simulation sweeps. In addition, 12 distinct actin configurations were simulated for each average corral size, totaling 6000 frames used to generate the cross-correlation (c(r)) curves in Figure 6B (left) and Supplemental Figure S9, B and D (left). The same set of 12 actin configurations was used for all three conditions for the actin phase preference. In addition to the raw c(r) curves, we also tabulated curves from simulation snapshots filtered with a Gaussian-shaped point spread function with σ = 24 nm (Supplemental Figure S7 and Figure 6B, right). This is equivalent to convolving the raw c(r) curve with the auto-correlation of the point spread function, gPSF(r) (Veatch et al., 2012b), and allows us to more accurately translate simulation results into the physical context of our experimental data, which were acquired with a localization precision of ∼24 nm.
Supplementary Material
Acknowledgments
We appreciate helpful discussions with Eshan Mitra. We thank Alice Wagenknecht-Wiesner and Joshua Wilson for help with the preparation of mEos3.2 fluorescent constructs. This work was supported by National Institute of General Medicine Grants R01GM117552 and R01GM110052. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used:
- FLM
fluorescence localization microscopy
- GG
lipid anchor consisting of geranylgeranyl modification and polybasic sequence
- PM
palmitoylated, myristoylated lipid anchor of Lyn
- pY
phosphotyrosine.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0425) on September 28, 2016.
REFERENCES
- Andrade DM, Clausen MP, Keller J, Mueller V, Wu C, Bear JE, Hell SW, Lagerholm BC, Eggeling C. Cortical actin networks induce spatio-temporal confinement of phospholipids in the plasma membrane—a minimally invasive investigation by STED-FCS. Sci Rep. 2015;5:11454. doi: 10.1038/srep11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NL, Lidke KA, Pfeiffer JR, Burns AR, Wilson BS, Oliver JM, Lidke DS. Actin restricts FcεRI diffusion and facilitates antigen-induced receptor immobilisation. Nat Cell Biol. 2008;10:955–963. doi: 10.1038/ncb1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Natl Acad Sci USA. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Chichili GR, Cail RC, Rodgers W. Cytoskeletal modulation of lipid interactions regulates Lck kinase activity. J Biol Chem. 2012;287:24186–24194. doi: 10.1074/jbc.M111.320747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichili GR, Rodgers W. Clustering of membrane raft proteins by the actin cytoskeleton. J Biol Chem. 2007;282:36682–36691. doi: 10.1074/jbc.M702959200. [DOI] [PubMed] [Google Scholar]
- Churchman LS, Okten Z, Rock RS, Dawson JF, Spudich JA. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc Natl Acad Sci USA. 2005;102:1419–1423. doi: 10.1073/pnas.0409487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coué M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- Ehrig J, Petrov EP, Schwille P. Near-critical fluctuations and cytoskeleton-assisted phase separation lead to subdiffusion in cell membranes. Biophys J. 2011;100:80–89. doi: 10.1016/j.bpj.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field KA, Holowka D, Baird B. FcεRI-mediated recruitment of p53/56lyn to detergent-resistant membrane domains accompanies cellular signaling. Proc Natl Acad Sci USA. 1995;92:9201–9205. doi: 10.1073/pnas.92.20.9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field KA, Holowka D, Baird B. Compartmentalized activation of the high affinity immunoglobulin E receptor within membrane domains. J Biol Chem. 1997;272:4276–4280. doi: 10.1074/jbc.272.7.4276. [DOI] [PubMed] [Google Scholar]
- Frigeri L, Apgar JR. The role of actin microfilaments in the down-regulation of the degranulation response in RBL-2H3 mast cells. J Immunol. 1999;162:2243–2250. [PubMed] [Google Scholar]
- Fujiwara TK, Iwasawa K, Kalay Z, Tsunoyama TA, Watanabe Y, Umemura YM, Murakoshi H, Suzuki KGN, Nemoto YL, Morone N, et al. Confined diffusion of transmembrane proteins and lipids induced by the same actin meshwork lining the plasma membrane. Mol Biol Cell. 2016;27:1101–1119. doi: 10.1091/mbc.E15-04-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009;228:149–169. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosse JA, Wagenknecht-Wiesner A, Holowka D, Baird B. Transmembrane sequences are determinants of immunoreceptor signaling. J Immunol. 2005;175:2123–2131. doi: 10.4049/jimmunol.175.4.2123. [DOI] [PubMed] [Google Scholar]
- Goswami D, Gowrishankar K, Bilgrami S, Ghosh S, Raghupathy R, Chadda R, Vishwakarma R, Rao M, Mayor S. Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell. 2008;135:1085–1097. doi: 10.1016/j.cell.2008.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR. Handbook of Experimental Immunology. Oxford, UK: Blackwell Scientific; 1986. [Google Scholar]
- Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function. Biochim Biophys Acta. 2014;1838:532–545. doi: 10.1016/j.bbamem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneberg P, Dráberová L, Bambousková M, Pompach P, Dráber P. Down-regulation of protein-tyrosine phosphatases activates an immune receptor in the absence of its translocation into lipid rafts. J Biol Chem. 2010;285:12787–12802. doi: 10.1074/jbc.M109.052555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka D, Baird B. Nanodomains in early and later phases of FcεRI signalling. Essays Biochem. 2015;57:147–163. doi: 10.1042/bse0570147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka D, Calloway N, Cohen R, Gadi D, Lee J, Smith NL, Baird B. Roles for ca(2+) mobilization and its regulation in mast cell functions. Front Immunol. 2012;3:104. doi: 10.3389/fimmu.2012.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka D, Gosse JA, Hammond AT, Han X, Sengupta P, Smith NL, Wagenknecht-Wiesner A, Wu M, Young RM, Baird B. Lipid segregation and IgE receptor signaling: a decade of progress. Biochim Biophys Acta. 2005;1746:252–259. doi: 10.1016/j.bbamcr.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Holowka D, Sheets ED, Baird B. Interactions between FcεRI and lipid raft components are regulated by the actin cytoskeleton. J Cell Sci. 2000;113:1009–1019. doi: 10.1242/jcs.113.6.1009. [DOI] [PubMed] [Google Scholar]
- Holowka D, Sil D, Torigoe C, Baird B. Insights into immunoglobulin E receptor signaling from structurally defined ligands. Immunol Rev. 2007;217:269–279. doi: 10.1111/j.1600-065X.2007.00517.x. [DOI] [PubMed] [Google Scholar]
- Honda Z, Suzuki T, Hirose N, Aihara M, Shimizu T, Nada S, Okada M, Ra C, Morita Y, Ito K. Roles of C-terminal Src kinase in the initiation and the termination of the high affinity IgE receptor-mediated signaling. J Biol Chem. 1997;272:25753–25760. doi: 10.1074/jbc.272.41.25753. [DOI] [PubMed] [Google Scholar]
- Honigmann A, Sadeghi S, Keller J, Hell SW, Eggeling C, Vink R. A lipid bound actin meshwork organizes liquid phase separation in model membranes. Elife. 2014;3:e01671. doi: 10.7554/eLife.01671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdinka M, Horejsi V. PAG—a multipurpose transmembrane adaptor protein. Oncogene. 2014;33:4881–4892. doi: 10.1038/onc.2013.485. [DOI] [PubMed] [Google Scholar]
- Jouvin MH, Adamczewski M, Numerof R, Letourneur O, Vallé A, Kinet JP. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J Biol Chem. 1994;269:5918–5925. [PubMed] [Google Scholar]
- Kraft S, Kinet J-P. New developments in FcεRI regulation, function and inhibition. Nat Rev Immunol. 2007;7:365–378. doi: 10.1038/nri2072. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Fujiwara TK, Morone N, Yoshida KJ, Chadda R, Xie M, Kasai RS, Suzuki KGN. Membrane mechanisms for signal transduction: the coupling of the meso-scale raft domains to membrane-skeleton-induced compartments and dynamic protein complexes. Semin. Cell Dev Biol. 2012;23:126–144. doi: 10.1016/j.semcdb.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Ike H, Nakada C, Murase K, Fujiwara T. Single-molecule tracking of membrane molecules: plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules. Semin Immunol. 2005;17:3–21. doi: 10.1016/j.smim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Larson DR, Gosse JA, Holowka DA, Baird BA, Webb WW. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. J Cell Biol. 2005;171:527–536. doi: 10.1083/jcb.200503110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Levental I. Giant plasma membrane vesicles: models for understanding membrane organization. Curr Top Membr. 2015;75:25–57. doi: 10.1016/bs.ctm.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Machta BB, Papanikolaou S, Sethna JP, Veatch SL. Minimal model of plasma membrane heterogeneity requires coupling cortical actin to criticality. Biophys J. 2011;100:1668–1677. doi: 10.1016/j.bpj.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PK, Feest C, Depoil D, Treanor B, Montaner B, Otipoby KL, Carter R, Justement LB, Bruckbauer A, Batista FD. The actin and tetraspanin networks organize receptor nanoclusters to regulate b cell receptor-mediated signaling. Immunity. 2013;38:461–474. doi: 10.1016/j.immuni.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Morone N, Fujiwara T, Murase K, Kasai RS, Ike H, Yuasa S, Usukura J, Kusumi A. Three-dimensional reconstruction of the membrane skeleton at the plasma membrane interface by electron tomography. J Cell Biol. 2006;174:851–862. doi: 10.1083/jcb.200606007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase K, Fujiwara T, Umemura Y, Suzuki K, Iino R, Yamashita H, Saito M, Murakoshi H, Ritchie K, Kusumi A. Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys J. 2004;86:4075–4093. doi: 10.1529/biophysj.103.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naal RMZG, Tabb J, Holowka D, Baird B. In situ measurement of degranulation as a biosensor based on RBL-2H3 mast cells. Biosens Bioelectron. 2004;20:791–796. doi: 10.1016/j.bios.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Narasimhan V, Holowka D, Baird B. Microfilaments regulate the rate of exocytosis in rat basophilic leukemia cells. Biochem Biophys Res Commun. 1990;171:222–229. doi: 10.1016/0006-291x(90)91380-b. [DOI] [PubMed] [Google Scholar]
- Ohtake H, Ichikawa N, Okada M, Yamashita T. Cutting edge: transmembrane phosphoprotein Csk-binding protein/phosphoprotein associated with glycosphingolipid-enriched microdomains as a negative feedback regulator of mast cell signaling through the FcεRI. J Immunol. 2002;168:2087–2090. doi: 10.4049/jimmunol.168.5.2087. [DOI] [PubMed] [Google Scholar]
- Paolini R, Jouvin MH, Kinet JP. Phosphorylation and dephosphorylation of the high-affinity receptor for immunoglobulin E immediately after receptor engagement and disengagement. Nature. 1991;353:855–858. doi: 10.1038/353855a0. [DOI] [PubMed] [Google Scholar]
- Pfeiffer JR, Seagrave JC, Davis BH, Deanin GG, Oliver JM. Membrane and cytoskeletal changes associated with IgE-mediated serotonin release from rat basophilic leukemia cells. J Cell Biol. 1985;101:2145–2155. doi: 10.1083/jcb.101.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierini L, Harris NT, Holowka D, Baird B. Evidence supporting a role for microfilaments in regulating the coupling between poorly dissociable IgE−FcεRI aggregates and downstream signaling pathways. Biochemistry. 1997;36:7447–7456. doi: 10.1021/bi9629642. [DOI] [PubMed] [Google Scholar]
- Pribluda VS, Metzger H. Transmembrane signaling by the high-affinity IgE receptor on membrane preparations. Proc Natl Acad Sci USA. 1992;89:11446–11450. doi: 10.1073/pnas.89.23.11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribluda VS, Pribluda C, Metzger H. Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc Natl Acad Sci USA. 1994;91:11246–11250. doi: 10.1073/pnas.91.23.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyenta PS, Holowka D, Baird B. Cross-correlation analysis of inner-leaflet-anchored green fluorescent protein co-redistributed with IgE receptors and outer leaflet lipid raft components. Biophys J. 2001;80:2120–2132. doi: 10.1016/S0006-3495(01)76185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyenta PS, Schwille P, Webb WW, Holowka D, Baird B. Lateral diffusion of membrane lipid-anchored probes before and after aggregation of cell surface IgE-receptors†. J Phys Chem A. 2003;107:8310–8318. [Google Scholar]
- Raghupathy R, Anilkumar AA, Polley A, Singh PP, Yadav M, Johnson C, Suryawanshi S, Saikam V, Sawant SD, Panda A, et al. Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell. 2015;161:581–594. doi: 10.1016/j.cell.2015.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Stochastic optical reconstruction microscopy (STORM) provides sub-diffraction-limit image resolution. Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets ED, Holowka D, Baird B. Critical role for cholesterol in lyn-mediated tyrosine phosphorylation of FcεRI and their association with detergent-resistant membranes. J Cell Biol. 1999;145:877–887. doi: 10.1083/jcb.145.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelby SA, Holowka D, Baird B, Veatch SL. Distinct stages of stimulated FcεRI receptor clustering and immobilization are identified through superresolution imaging. Biophys J. 2013;105:2343–2354. doi: 10.1016/j.bpj.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siraganian RP. Mast cell signal transduction from the high-affinity IgE receptor. Curr Opin Immunol. 2003;15:639–646. doi: 10.1016/j.coi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Stone MB, Veatch SL. Far-red organic fluorophores contain a fluorescent impurity. ChemPhysChem. 2014;15:2240–2246. doi: 10.1002/cphc.201402002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MB, Veatch SL. Steady-state cross-correlations for live two-colour super-resolution localization data sets. Nat Commun. 2015;6:7347. doi: 10.1038/ncomms8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolarová H, Dráberová L, Heneberg P, Dráber P. Involvement of filamentous actin in setting the threshold for degranulation in mast cells. Eur J Immunol. 2004;34:1627–1636. doi: 10.1002/eji.200424991. [DOI] [PubMed] [Google Scholar]
- Torigoe C, Song J, Barisas BG, Metzger H. The influence of actin microfilaments on signaling by the receptor with high-affinity for IgE. Mol Immunol. 2004;41:817–829. doi: 10.1016/j.molimm.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Torres AJ, Wu M, Holowka D, Baird B. Nanobiotechnology and cell biology: micro- and nanofabricated surfaces to investigate receptor-mediated signaling. Annu Rev Biophys. 2008;37:265–288. doi: 10.1146/annurev.biophys.36.040306.132651. [DOI] [PubMed] [Google Scholar]
- Treanor B, Depoil D, Gonzalez-Granja A, Barral P, Weber M, Dushek O, Bruckbauer A, Batista FD. The membrane skeleton controls diffusion dynamics and signaling through the B cell receptor. Immunity. 2010;32:187–199. doi: 10.1016/j.immuni.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urata C, Siraganian RP. Pharmacologic modulation of the IgE or Ca2+ ionophore A23187 mediated Ca2+ influx, phospholipase activation, and histamine release in rat basophilic leukemia cells. Int Arch Allergy Appl Immunol. 1985;78:92–100. doi: 10.1159/000233869. [DOI] [PubMed] [Google Scholar]
- Veatch SL, Chiang EN, Sengupta P, Holowka DA, Baird BA. Quantitative nanoscale analysis of IgE-FcεRI clustering and coupling to early signaling proteins. J Phys Chem B. 2012a;116:6923–6935. doi: 10.1021/jp300197p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch SL, Cicuta P, Sengupta P, Honerkamp-Smith A, Holowka D, Baird B. Critical fluctuations in plasma membrane vesicles. ACS Chem Biol. 2008;3:287–293. doi: 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, Baird BA. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One. 2012b;7:e31457. doi: 10.1371/journal.pone.0031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola A, Gupta N. Tether and trap: regulation of membrane-raft dynamics by actin-binding proteins. Nat Rev Immunol. 2007;7:889–896. doi: 10.1038/nri2193. [DOI] [PubMed] [Google Scholar]
- Wilson BS, Pfeiffer JR, Oliver JM. Observing fcεri signaling from the inside of the mast cell membrane. J Cell Biol. 2000;149:1131–1142. doi: 10.1083/jcb.149.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JD, Shelby SA, Holowka D, Baird B. Rab11 regulates the mast cell exocytic response. Traffic. 2016;17:1027–1041. doi: 10.1111/tra.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollman R, Meyer T. Coordinated oscillations in cortical actin and Ca2+ correlate with cycles of vesicle secretion. Nat Cell Biol. 2012;14:1261–1269. doi: 10.1038/ncb2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Holowka D, Craighead HG, Baird B. Visualization of plasma membrane compartmentalization with patterned lipid bilayers. Proc Natl Acad Sci USA. 2004;101:13798–13803. doi: 10.1073/pnas.0403835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Goldstein B, Holowka D, Baird B. Kinetics of multivalent antigen DNP-BSA binding to IgE-FcεRI in relationship to the stimulated tyrosine phosphorylation of FcεRI. J Immunol. 1998;160:3225–3235. [PubMed] [Google Scholar]
- Yamashita T, Mao SY, Metzger H. Aggregation of the high-affinity IgE receptor and enhanced activity of p53/56lyn protein-tyrosine kinase. Proc Natl Acad Sci USA. 1994;91:11251–11255. doi: 10.1073/pnas.91.23.11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Holowka D, Baird B. A lipid raft environment enhances Lyn kinase activity by protecting the active site tyrosine from dephosphorylation. J Biol Chem. 2003;278:20746–20752. doi: 10.1074/jbc.M211402200. [DOI] [PubMed] [Google Scholar]
- Young RM, Zheng X, Holowka D, Baird B. Reconstitution of regulated phosphorylation of FcεRI by a lipid raft-excluded protein-tyrosine phosphatase. J Biol Chem. 2005;280:1230–1235. doi: 10.1074/jbc.M408339200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Berenstein EH, Evans RL, Siraganian RP. Transfection of Syk protein tyrosine kinase reconstitutes high affinity IgE receptor-mediated degranulation in a Syk-negative variant of rat basophilic leukemia RBL-2H3 cells. J Exp Med. 1996;184:71–79. doi: 10.1084/jem.184.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.