

Superresolution microscopy and biochemical approaches identify a novel interaction between MOR and SSTR2 specific to pancreatic ductal adenocarcinoma. Coactivation of the two receptors leads to a distinct signaling pathway, consistent with β-arrestin2 signaling and the increased metastatic potential of pancreatic cancer cells.
Abstract
Pancreatic ductal adenocarcinoma (PDAC), a particularly aggressive malignancy, has been linked to atypical levels, certain mutations, and aberrant signaling of G-protein–coupled receptors (GPCRs). GPCRs have been challenging to target in cancer because they organize into complex networks in tumor cells. To dissect such networks with nanometer-scale precision, here we combine traditional biochemical approaches with superresolution microscopy methods. A novel interaction specific to PDAC is identified between mu opioid receptor (MOR) and somatostatin receptor 2 (SSTR2). Although MOR and SSTR2 did not colocalize in healthy pancreatic cells or matching healthy patient tissues, the pair did significantly colocalize in pancreatic cancer cells, multicellular tumor spheroids, and cancerous patient tissues. Moreover, this association in pancreatic cancer cells correlated with functional cross-talk and increased metastatic potential of cells. Coactivation of MOR and SSTR2 in PDAC cells led to increased expression of mesenchymal markers and decreased expression of an epithelial marker. Together these results suggest that the MOR-SSTR2 heteromer may constitute a novel therapeutic target for PDAC.
INTRODUCTION
In the United States, the fourth-leading cancer-related cause of death is pancreatic ductal adenocarcinoma (PDAC; Howlader et al., 2014). For many PDAC patients, the cancer presents itself at an advanced stage, and only 20% of cases are eligible for surgical resection (Li et al., 2004a). Although conventional therapies (surgery, radiation, chemotherapy) have been improved (Von Hoff et al., 2013) and a new targeted therapy (Moore et al., 2007) has been introduced, the overall survival rate for PDAC patients over the past four decades remains remarkably dismal. Within 5 yr of the diagnosis, the disease claims ∼93% of all patients (Howlader et al., 2014). This high lethality correlates with high rates of metastasis and is partially coupled to limited treatment options (Oberstein and Olive, 2013). It is therefore imperative to identify new therapeutic targets for PDAC.
Toward this goal, we have been studying G-protein–coupled receptors (GPCRs), which play a substantial role in the tumorigenesis of pancreatic cancer. GPCRs activated by chemokines (e.g., from inflammation; Allavena et al., 2008) and neurotransmitters/hormones (from autocrine/paracrine signaling; Heasley, 2001) can lead to pathological signaling (Hanahan and Weinberg, 2011). In addition, GPCRs are often expressed at aberrant levels in PDAC (Koshiba et al., 2000; Balkwill, 2004; Li et al., 2004b; Billadeau et al., 2006; Laklai et al., 2009; Soria and Ben-Baruch, 2009; Call et al., 2013; Shahbaz et al., 2015), and somatic mutations in GPCRs have been found in many tumors, including pancreatic tumors (Kan et al., 2010). Because of this, GPCRs are being targeted in a number of clinical trials for cancer. A Phase 1 trial for pancreatic cancer (NCT02179970) is evaluating a C-X-C motif receptor 4 (CXCR4) ligand; a Phase 1 trial for pancreatic cancer (NCT01385956) is studying a combination of gemcitabine with a somatostatin receptor 2 (SSTR2) ligand; and a Phase 1 trial for advanced malignancies (NCT01015222) is examining a combination treatment involving a mu opioid receptor (MOR) ligand. Despite this significant interest, pharmacological agents that target GPCRs in PDAC are unavailable. The absence of such drugs may be partially attributable to the complexity of GPCR signaling networks.
Although they can organize and function as monomers (Whorton et al., 2007; Kuszak et al., 2009), an increasing body of evidence indicates that GPCRs also interact with each other, either directly or through accessory proteins (Pfeiffer et al., 2002; Gomes et al., 2004, 2013; Wang et al., 2005; Finley et al., 2008; Pello et al., 2008; Kharmate et al., 2013), to form multicomponent functional entities. Compared to the biochemical properties of each GPCR constituent (i.e., protomers), these associated GPCRs exhibit discernibly different biochemical properties and are called heteromers (Ferre et al., 2009). GPCR heteromers are often observed under pathological conditions and appear to be involved in the pathophysiology of a number of diseases, including Parkinson’s disease (Azdad et al., 2009), neuropathic pain (Bushlin et al., 2012), liver fibrosis (Rozenfeld et al., 2011), schizophrenia (Albizu et al., 2011), preeclampsia (AbdAlla et al., 2001), and acromegaly (Grant et al., 2008). Thus many groups have been studying their ability to serve as pharmacological targets. In particular, associated GPCRs have been targeted by heterovalent ligands (Daniels et al., 2005; Jaquet et al., 2005; Yuan et al., 2013) and antibodies (Gupta et al., 2010; Berg et al., 2012); and new methodologies for targeting these entities are emerging (Donaldson et al., 2013; Lewis et al., 2014). Despite the significant success of these approaches, which have been primarily developed for neurological disorders, there are limited examples of targeting GPCR heteromers in cancer (Moreno et al., 2014). Identifying GPCR interactions within the context of human malignancies is challenging, particularly at the molecular level and in the native state (Gomes et al., 2004, 2016; Albizu et al., 2010; Sams et al., 2014; Dudok et al., 2015).
To identify cancer-specific GPCR heteromers, high-resolution information along a large spatial region is needed. Single-molecule pointillistic superresolution microscopy techniques (Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006; Folling et al., 2008; Wombacher et al., 2010), such as direct stochastic optical reconstruction microscopy (dSTORM; Wombacher et al., 2010), are well suited for this purpose (Scarselli et al., 2012; Sams et al., 2014; Tobin et al., 2014; Dudok et al., 2015; Jonas et al., 2015). To dissect the complex arrangement of proteins visualized in this way, we used quantitative analysis of dSTORM data. Applied to superresolution imaging data sets, Voronoï tessellation (Levet et al., 2015) was used to determine receptor organization and cluster sizes, and pair-correlation analysis (Sengupta et al., 2011, 2013) and Monte Carlo simulations were used to precisely interrogate interactions between receptors. This unique approach is quantitative, operates at the single-molecule level, and is well suited for interrogating tight GPCR networks.
Using quantitative superresolution microscopy, we evaluated clustering of GPCRs and colocalization between GPCR pairs in pancreatic environments: healthy cells, cancerous cells, multicellular tumor spheroids (MCTS; a three-dimensional [3D] cell culture system; Sutherland, 1988), and matching tissue samples from three patients. We identified a heteromer between MOR and SSTR2, called MOR-SSTR2, which is specific for PDAC. To the best of our knowledge, this is the first time the association between two GPCRs has been specifically observed in pancreatic malignancy, particularly in a native environment. In addition, it is the first time a quantitative correlation has been obtained on a molecular level between GPCR networks in cultured cells (two-dimensional [2D] and 3D environments) and matching tissue samples. Of importance, we correlated the physical association between MOR and SSTR2 with signaling outcomes. According to our results from biochemical experiments, activation of both MOR and SSTR2 1) correlates with signaling events unique to PDAC cells and 2) leads to increased malignant potency of only cancerous epithelial pancreatic cells. Cumulatively our results suggest that the MOR-SSTR2 heteromer may represent a PDAC-specific therapeutic target.
RESULTS
MOR, SSTR2, and CXCR4 are expressed in both normal and malignant pancreatic cells
MOR, SSTR2, and CXCR4 belong to closely related GPCR families and are expressed at aberrant levels in pancreatic cancer (Reubi et al., 1998; Koshiba et al., 2000; Li et al., 2004b; Chen et al., 2014). For example, in both pancreatic cancer cells and a significant number of primary PDAC tumors (Szende et al., 1990; Li et al., 2004b; Laklai et al., 2009; Shahbaz et al., 2015; Gradiz et al., 2016), SSTR2 has been observed at low but detectable levels. Comparatively high expression levels have been observed for CXCR4 in PDAC (Koshiba et al., 2000; Balkwill, 2004; Billadeau et al., 2006) and for MOR in a number of malignancies (Li et al., 2004b; Zylla et al., 2013; Singleton et al., 2014). Given this landscape of aberrant expression and the fact that all three receptors are known to form heteromers (Pfeiffer et al., 2002; Gomes et al., 2004, 2013; Wang et al., 2005; Finley et al., 2008; Pello et al., 2008; Kharmate et al., 2013), we investigated the expression and organization of MOR, SSTR2, and CXCR4 in PDAC cells. We used real-time PCR (RT-PCR) and Western blot analysis to evaluate the expression levels of the three GPCRs in six cell lines: 1) control CHO-S cells; 2) normal epithelial pancreatic cells; 3) PANC-1 cells, a PDAC cell line with K-Ras mutations (Lieber et al., 1975); 4) BxPC-3 cells, a PDAC cell line with no K-Ras mutations; 5) SU.86.86 cells, a PDAC cell line derived from metastatic liver; and 6) CAPAN-1 cells, a PDAC cell line derived from metastatic liver. According to our results (Figure 1 and Supplemental Figure S13A), which are consistent with previous reports (Koshiba et al., 2000; Li et al., 2004b; Gradiz et al., 2016), SSTR2, CXCR4, and MOR were expressed at higher levels in a pancreatic cancer environment than in normal pancreatic cells. No detectable expression was observed in control CHO-S cells.
FIGURE 1:
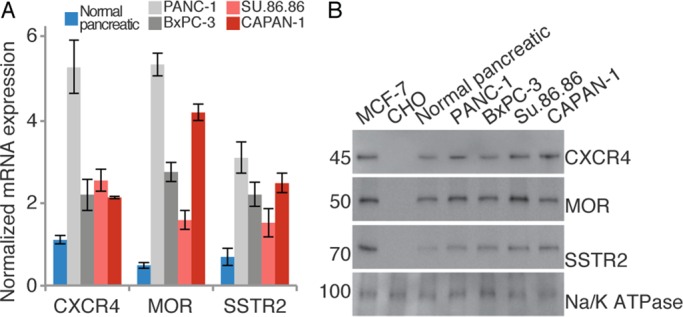
Expression of CXCR4, MOR, and SSTR2 in pancreatic cell lines. (A) Using the mRNA levels of GAPDH in pancreatic cell lines as a point of comparison, we determined the relative mRNA levels of CXCR4, MOR, and SSTR2. Normal epithelial pancreatic cells are shown in blue; primary pancreatic cancer cell lines are shown in gray; metastatic pancreatic cell lines are shown in red. Measurements are from three independent experiments, each done in triplicate. CHO-S cells were used as negative controls and MCF-7 cells as positive controls. Results are expressed as the average with SD. All GPCRs show statistically increased expression in cancerous cell lines compared with normal pancreatic cells (p ≤ 0.02). (B) In the membrane fraction of pancreatic cell lines, the protein levels of CXCR4, MOR, and SSTR2 were determined. CHO-S cells were used as negative controls and MCF-7 cells as positive controls. Loading was validated with Na/K ATPase. Quantitation of GPCR protein levels in different cell lines from three independent experiments is shown in Supplemental Figure S13A. Images were cropped for clarity; full blots are given in Supplemental Figure S13B.
Validation of dSTORM imaging with GPCR-specific antibodies
Having established higher GPCR expression levels in PANC-1 cells compared with normal pancreatic cells, we determined the feasibility of detecting GPCRs using dSTORM. As shown in Supplemental Figure S1, A–C, the plasma membrane organization of MOR, SSTR2, and CXCR4 in pancreatic cells can be observed. The three GPCRs were detected by affinity tagging with specific primary antibodies and fluorescently labeled secondary antibodies, similarly as before (Tobin et al., 2014; Dudok et al., 2015). The specificities of the antibodies for their respective GPCRs were extensively interrogated using both blocking peptides and knockdown cells. Because blocking peptides bind to the antigen-binding region on the antibody, they interrupt antibody–GPCR interactions and are thus useful for isolating nonspecific interactions. As shown in Supplemental Figure S1, A–C (magenta), no appreciable signal was observed when the antibodies were blocked with peptides, which indicates that the antibodies are specific for MOR, SSTR2, and CXCR4. In Supplemental Figure S1D shows an overlay of GPCR sequences with highlighted blocking peptide regions (terminal domains). Antibodies were also validated using MOR-, SSTR2-, and CXCR4-knockdown cells. According to both Western blots and RT-PCR analyses (Supplemental Figure S2, A and B), each GPCR was knocked down in PANC-1 cells with excellent efficiency. Whereas dSTORM imaging with nonsilencing (NS) control cells produced signal comparable to those in wild-type PANC-1 cells, no significant superresolution signal was detected in each knockdown, which confirmed the specificity of the antibodies for their respective GPCR (Supplemental Figure S2C). Therefore antibody labeling and dSTORM detection can be a valid approach for determining the organization of GPCRs in pancreatic cancer.
MOR and SSTR2 show distinct signatures in malignant pancreatic environments
To determine receptor organization and their interactions, we acquired two-color dSTORM images. The far-red photoswitchable organic dye Alexa Fluor 647 and spectrally distinct photoswitchable organic dye Atto 488 were used as reporters. The distribution of MOR and SSTR2 was detected in normal pancreatic cells, malignant PANC-1 cells, and 3D cultures of PANC-1 cells, that is, MCTS (Figure 2, A and B, and Supplemental Figures S3 and S4A). Consistent with our results from Western blot and RT-PCR analyses (Figure 1), dSTORM revealed significantly lower surface density of both MOR and SSTR2 in normal pancreatic cells compared with PANC-1 cells. Although all cells showed receptor clustering, only in PANC-1 cells and PANC-1 MCTS was the overlap between MOR and SSTR2 (dark blue) evident. We next used Voronoï tessellation with the fast, unbiased, and freely available software tool SR-Tesseler (Levet et al., 2015) to compare quantitatively the receptor organization in various cell types. A clustered distribution with an average cluster radius between 35 and 55 nm was observed in all cell types (Supplemental Figure S5). A significantly higher average cluster size was detected for MOR than for SSTR2 in PANC-1 cells and PANC-1 MCTS but not in normal pancreatic cells. Moreover, both MOR and SSTR2 had significantly higher cluster circularity in PANC-1 cells and PANC-1 MCTS than in normal pancreatic cells. These features suggest a differential distribution of MOR and SSTR2 in healthy versus cancerous cells and may reflect their organization into distinct signaling domains in cancerous cells.
FIGURE 2:

Colocalization of MOR and SSTR2. (A) The distribution of SSTR2 (magenta, detected with Atto 488) and MOR (cyan, detected with Alexa Fluor 647) was determined in a region of normal epithelial pancreatic cells. Scale bar, 2 μm. Peak centers are shown. (B) The distribution of SSTR2 (magenta, detected with Atto 488) and MOR (cyan, detected with Alexa Fluor 647) was determined in a region of PANC-1 cells. Scale bar, 2 μm. Overlap is evident in dark blue. Peak centers are shown. (C) Cross-correlation curves with SEM show colocalization between MOR and SSTR2 in both PANC-1 cells (gray diamonds, 41 regions from 22 cells) and PANC-1 MCTS (red triangles, 40 regions from 21 cells). In contrast, colocalization was not observed in normal pancreatic epithelial cells (blue circles, 50 regions from 21 cells). These results represent combined data obtained using two labeling schemes: 1) MOR detected with Alexa Fluor 647 and SSTR2 detected with Atto 488 and, 2) MOR detected with Atto 488 and SSTR2 detected with Alexa Fluor 647. Individual curves are given in Supplemental Figure S6. In all cases, no long-range correlations were observed. (D) Cell lysates from either normal pancreatic epithelial cells or PANC-1 cells were immunoprecipitated with anti-MOR antibody and immunoblotted with anti-SSTR2 antibody. SSTR2 was detected in the MOR immunoprecipitate in the PANC-1 cell line.
In HEK-293 cells (Pfeiffer et al., 2002) and possibly some breast cancer cell lines (Kharmate et al., 2013), MOR and SSTR2 form heterodimers. Heterodimerization of MOR and SSTR2 cross-modulates phosphorylation, internalization, and desensitization (Pfeiffer et al., 2002) and appears to be involved in antiproliferative pathways in breast cancer (Kharmate et al., 2013). To examine quantitatively whether such interactions are present in pancreatic cancer cells and MCTS, we evaluated colocalization between MOR and SSTR2 by PC-dSTORM in 80-μm2 cell regions (Sengupta et al., 2011; Tobin et al., 2014; Stone and Veatch, 2015). According to our results (Figure 2C), colocalization between the receptors does not occur (correlation curve approximately equal to 1) in normal pancreatic cells. In contrast, MOR and SSTR2 colocalize (correlation curve >1) in malignant PANC-1 cells and PANC-1 MCTS.
Colocalization between MOR and SSTR2 was confirmed through multiple control experiments. A combination of Atto 488 and Alexa Fluor 647 has been extensively used without issues for two-color superresolution imaging (Dempsey et al., 2011; Dudok et al., 2015). Consistently, spectral overlap between the two channels was not observed under our acquisition conditions. To investigate whether specific combinations of colors influenced the outcome, first we detected MOR with Atto 488 and SSTR2 with Alexa Fluor 647; next we detected MOR with Alexa Fluor 647 and SSTR2 with Atto 488. As shown in Supplemental Figure S6, the choice of labeling scheme did not influence the correlation curve for normal pancreatic cells, PANC-1 cells, and PANC-1 MCTS. Thus the averaged data from both labeling schemes were combined (Figure 2C and Supplemental Figure S5). In addition to these experimental labeling controls, Monte Carlo simulations were used to examine the effect of antibody detection efficiency on the ability to detect heteromers. As shown in Supplemental Figure S7, an extensive set of conditions was simulated and evaluated. For example, both 20 and 50% detection efficiencies are capable of detecting as little as 5–10% heteromers. As expected, for a simulated system in which there was no colocalization, detection efficiency was not a relevant parameter. Finally, we used coimmunoprecipitation (coIP) experiments to further establish colocalization between MOR and SSTR2 in pancreatic cancer cells: MOR coimmunoprecipitated with SSTR2 only in malignant PANC-1 cells (Figure 2D).
MOR and CXCR4 do not colocalize in pancreatic cells and PANC-1 MCTS
According to reports, CXCR4 is expressed at high levels in PDAC (Billadeau et al., 2006). Consistently, dSTORM revealed a significantly higher surface density of CXCR4 in PANC-1 cells than in normal pancreatic cells. Of interest, quantitative data suggest that CXCR4 makes relatively uniform clusters with a radius of ∼45 nm in both cell lines (Supplemental Figure S5). Colocalization of CXCR4 and MOR was next evaluated by PC-dSTORM on the surface of normal pancreatic cells, PANC-1 cells, and PANC-1 MCTS (Figure 3, A and B, and Supplemental Figure S4C, respectively). As with MOR and SSTR2 (Supplemental Figure 4B), the imaging approach was validated in PANC-1 MCTS using blocking peptide controls to show that the antibodies were specific for MOR and CXCR4 (Supplemental Figure 4D). According to the dSTORM imaging results, the two GPCRs do not colocalize in any of the pancreatic environments. We confirmed this by quantitative correlation analyses (Figure 3C). The correlation curve is approximately equal to 1 in all cases. In addition, coIP experiments (Figure 3D) suggest no association between receptors in both normal pancreatic cells and PANC-1 cells. These findings imply that the density of individual GPCRs is not a sole factor in determining their association.
FIGURE 3:

There is no colocalization between MOR and CXCR4. (A) The distribution of CXCR4 (magenta, detected with Atto 488) and MOR (cyan, detected with Alexa Fluor 647) was determined in a region of normal pancreatic cells. Scale bar, 2 μm. Peak centers are shown. (B) The distribution of CXCR4 (magenta, detected with Atto 488) and MOR (cyan, detected with Alexa Fluor 647) was determined in a region of PANC-1 cells. Scale bar, 2 μm. Peak centers are shown. (C) Cross-correlation curves with SEM show no colocalization between MOR and CXCR4 in PANC-1 cells (gray diamonds, 32 regions from 14 cells), PANC-1 MCTS (red triangles, 20 regions from 12 cells), and normal pancreatic epithelial cells (blue circles, 21 regions from 13 cells). In all cases, no long-range correlations were observed. (D) Cell lysates from either normal pancreatic epithelial cells or PANC-1 cells were immunoprecipitated with anti-MOR antibody and immunoblotted with anti-CXCR4 antibody. CXCR4 was not detected in the MOR immunoprecipitate in either cell line. Quantitation is shown in Supplemental Figure S13D.
dSTORM imaging quantitatively detects GPCR association in patient tissue samples
dSTORM was used to assess colocalization between MOR and SSTR2 in patient tissue samples. Both healthy and malignant samples were obtained from three patients during surgical resection. Via hematoxylin and eosin staining protocols, the samples were confirmed as being from either the primary PDAC tumor or negative surgical margins (representative images are shown in Supplemental Figure S8). For the superresolution experiments, frozen sections of tissues were prepared, labeled, and imaged. Because PDAC tissue contains both epithelial cells and high levels of stroma (Kleeff et al., 2007), keratin 8 and 18 antibodies (specific for epithelial cells; Moll et al., 1982) were used to discriminate between these tissue types. An Alexa Fluor 405–tagged antibody was used to detect the keratins, and Alexa Fluor 488/647–tagged antibodies were used to detect MOR and SSTR2. In each region, fluorescence in the 405 channel was recorded after imaging in the 488/647 channels. Controls demonstrate that the keratin 8 and 18 antibody binds to epithelial PANC-1 cells and that no cross-talk between 405 and 488/647 channels was observed in tissues (Supplemental Figure S9). The distribution of MOR and SSTR2 was determined in representative healthy and cancerous samples (Figure 4). Negligible signal was detected when blocking peptides were used as controls in tissue samples (inset). We analyzed multiple sections/regions from cancer tissues and healthy tissues of three patients. Our tessellation results (Supplemental Figure S5, E–G) suggest that both MOR and SSTR2 have significantly higher average cluster radii in cancer than in healthy tissues (data for three patients were combined). In addition, in both cancer and healthy tissues, MOR formed more circular clusters on average compared with SSTR2 clusters. Of importance, significant overlap (dark blue) between receptors is evident only in cancer samples. We performed PC-dSTORM analysis and give the cross-correlation curves in Figure 4C. Results suggest that MOR and SSTR2 colocalize in cancer tissue, where the extent of cross-correlation was patient dependent, but not in healthy margins. Although this is only a three-patient study, it clearly demonstrates the feasibility of quantitative superresolution imaging of fresh-frozen patient samples and shows promise for future translational investigations.
FIGURE 4:

Colocalization of MOR and SSTR2 in patient tissues. (A) The distribution of SSTR2 (cyan, detected with Alexa Fluor 647) and MOR (magenta, detected with Atto 488) was determined in healthy pancreatic tissue margins. Scale bar, 2 μm. Inset, controls with blocking peptides; scale bar, 5 μm. Peak centers are shown. (B) The distribution of SSTR2 (cyan, detected with Alexa Fluor 647) and MOR (magenta, detected with Atto 488) was determined in matching cancerous pancreatic tissue. Scale bar, 2 μm. Controls with blocking peptides are given in the inset; scale bar, 5 μm. Overlap is evident in dark blue. Peak centers are shown. (C) Cross-correlation with SEM demonstrates colocalization between MOR and SSTR2 in tumor tissue: red triangles (patient 1, N = 9), red circles (patient 2, N = 8) and red diamonds (patient 3, N = 15). Colocalization was not detected in matching healthy tissue: blue triangles (patient 1, N = 7), blue circles (patient 2, N = 8), and blue diamonds (patient 3, N = 14). Only areas positive for keratin 8 and 18 were used for quantification. In all cases, no long-range correlations are observed.
Activation of MOR and SSTR2 with selective agonists leads to receptor internalization
Given that in pancreatic cancer environments, MOR and SSTR2 colocalize on the membrane, we next examined the effect of selective agonists on the cellular localization of these receptors with confocal microscopy. We used a specific MOR agonist, dermorphin (Melchiorri and Negri, 1996), and a specific SSTR2 agonist, L-054,264 (Kailey et al., 2012). Our data (Supplemental Figure S10) demonstrate largely membrane localization of MOR and SSTR2 in steady state, as expected, and their subsequent internalization upon coactivation with selective agonists in PANC-1 cells.
Simultaneous activation of MOR and SSTR2 influences both epidermal growth factor receptor 1 and extracellular signal-regulated kinase 1/2 phosphorylation in PANC-1 cells
To examine signaling pathways upon activation of MOR and/or SSTR2, we performed downstream phenotypic assays measuring extracellular signal-regulated kinase 1/2 (ERK1/2) and epidermal growth factor receptor 1 (EGFR) phosphorylation. Although their signaling pathways are complex, GPCR activation often converges at ERK1/2, which is critical for cellular proliferation, differentiation, and survival (Marshall, 1995; Bonni et al., 1999). The pathways may additionally involve activation of EGFR (Belcheva et al., 2001; Billadeau et al., 2006), which is often overexpressed in PDAC (Korc et al., 1992). To evaluate potential roles played by associated MOR and SSTR2 in PDAC, we activated these two GPCRs in both normal pancreatic cells and cancerous PANC-1 cells. Using a variety of ligands, we examined the phosphorylation status of ERK1/2 and EGFR. Via Western blot analysis, we evaluated three treatments: 1) a specific MOR agonist, dermorphin, 2) a specific SSTR2 agonist, L-054,264, and 3) a combination of these two agonists (Figure 5, A and B). In normal pancreatic cells, EGFR phosphorylation was not detected with any of these treatments. In PANC-1 cells, however, the MOR agonist dermorphin induced significant EGFR phosphorylation, whereas the SSTR2 agonist induced modest EGFR phosphorylation. Of importance, a combination of the two agonists did not induce EGFR phosphorylation in PANC-1 cells. This result is consistent with previous results in breast cancer cells (Kharmate et al., 2013). With regard to ERK1/2, all treatments produced transient phosphorylation of ERK1/2 in both normal and PANC-1 cells, albeit to different extents. Compared to levels observed in normal cells, higher levels of treatment-induced pERK1/2 were observed in PANC-1 cells. Whereas single agonists produced similar kinetic profiles of ERK1/2 phosphorylation in both cell lines with maxima at 3 min, the combination of two agonists yielded slower and more sustained (after 3 min) pERK1/2 activation only in PANC-1 cells.
FIGURE 5:

Combined MOR and SSTR2 agonist treatment leads to a distinct signaling pathway in PANC-1 cells. (A) After treatment of either normal epithelial pancreatic cells or PANC-1 cells with agonists, phosphorylation of ERK1/2 and EGFR in cell lysates was analyzed (Western blot detection). The agonist treatments were 10 nM dermorphin, 10 nM L-054,264, or 10 nM dermorphin with 10 nM L-054,264. Treatment time periods are indicated. Images were cropped for clarity; large regions of representative original images are provided in Supplemental Figure S13E. (B) Image Lab software was used to quantify the amount of ERK1/2 or EGFR phosphorylation in each lane. The data are expressed as a ratio of either pERK1/2 over total ERK1/2 or pEGFR over total EGFR and averaged. Results are normalized (the maximum response for dermorphin in PANC-1 cells is taken as 100%) and presented with SE. Dermorphin treatment is presented in purple; L-054,264 treatment is presented in red; and the combined treatment is presented in blue. *p < 0.01 (obtained using the single-tail t test) between dermorphin activation and either L-054,264 or combined L-054,264 and dermorphin activation. For pEGFR/EGFR, the corresponding p < 0.01 in all cases. (C) Confocal imaging was used to determine pERK1/2 localization in cells. The combined MOR and SSTR2 agonists targeted pERK1/2 to the nucleus in normal pancreatic cells (low levels) and to cytoplasm in PANC-1 cells (high levels). Single agonists targeted pERK1/2 to the nucleus in normal pancreatic cells (low levels) and to the nucleus in PANC-1 cells (high levels). Scale bars, 10 μm. (D) After treatment of PANC-1 cells with agonists (10 nM dermorphin, 10 nM L-054,264, or 10 nM dermorphin with 10 nM L-054,264) for the indicated periods of time, nuclear and cytoplasmic cell fractions were separated, and levels of pERK1/2 and pRSK were observed using Western blots. Large regions of representative original images are provided in Supplemental Figure S13F. (E) After treatment of normal pancreatic and PANC-1 cells with agonists (10 nM dermorphin, 10 nM L-054,264, or 10 nM dermorphin with 10 nM L-054,264) for 24 h, mRNA levels of N-cadherin (light gray), MMP-9 (dark gray), vimentin (medium gray), and E-cadherin (red) were measured and compared with their levels in untreated cells. Measurements from three independent experiments, each done in duplicate. Results are expressed as the average with SD.
Simultaneous activation of MOR and SSTR2 influences the localization of pERK1/2 and pRSK in PANC-1 cells
Using confocal imaging of cells, we next investigated the effects of single and combined agonists on pERK1/2 localization (Figure 5C and Supplemental Figure S11). For all treatments in normal pancreatic cells, low levels of pERK1/2 were detected in the nucleus. In PANC-1 cells with single agonists, high levels of pERK1/2 were detected, again in the nucleus. Of interest, treating PANC-1 cells with the combined agonists produced high levels of pERK1/2 located mostly in the cytoplasm. To determine whether this result was a direct function of engaging MOR and SSTR2 simultaneously, we evaluated the combined treatment in PANC-1–knockdown cells. In both PANC-1/MORsi and PANC-1/SSTR2si, treating cells with the combination of agonists resulted in largely nuclear pERK1/2. In PANC-1 NS control cells, largely cytoplasmic pERK1/2 localization was restored (Supplemental Figure S11C). We next generated a PANC-1/β-arrestin2si cell line and confirmed the knockdown using Western blots (Supplemental Figure 11F). Of importance, in PANC-1/β-arrestin2si cells, treatment with the L-054,264/dermorphin combination resulted in largely nuclear pERK1/2 (Supplemental Figure 11F). To confirm further this differential pERK1/2 localization in PANC-1 cells, we performed subcellular fractionation experiments and confirmed the purity of fractionation using cytoplasmic and nuclear markers (Figure 5D). Treatment with a single agonist quickly led to mostly nuclear pERK1/2, whereas even after prolonged treatment (30 min) with the double-agonist combination, mostly cytoplasmic pERK1/2 was observed (Figure 4D). As a control, we evaluated the localization of pERK1/2 after activating MOR and CXCR4, which do not colocalize according to both dSTORM and coIP. When we treated PANC-1 cells with a selective CXCR4 agonist (CXCL12) or a combination of CXCL12 and dermorphin, we observed high levels of nuclear pERK1/2 (Supplemental Figure S11D). Next we evaluated the functional consequences of the cytoplasmic localization of pERK1/2 by monitoring the phosphorylation status of one of its cytoplasmic substrates, p90 ribosomal S6 kinase (RSK; Yoon and Seger, 2006), an important factor for cell motility, invasion, and metastasis. In PANC-1 cells, RSK was rapidly phosphorylated in different locations, depending on the treatment type. Treatment with individual agonists resulted in nuclear pRSK, whereas treatment with the combination resulted in both cytoplasmic and nuclear pRSK (Figure 5D). Thus, in PANC-1 cells, coactivation of MOR and SSTR2 appears to uniquely orchestrate phosphorylation of ERK1/2, consistent with β-arrestin2 signaling.
Simultaneous activation of MOR and SSTR2 influences the metastatic potential of PDAC cells
Coactivation of associating GPCRs could be physiologically relevant for PDAC. Endogenous ligands are able to continuously activate GPCRs (Heasley, 2001), and MOR agonists such as morphine are often used in PDAC palliative treatment (Mercadante et al., 2010). According to clinical studies in PDAC, a strong correlation may exist between aberrant ERK1/2 activation and a process that is important to initiating metastasis, namely epithelial-to-mesenchymal transition (EMT; Javle et al., 2007). Specifically, patient PDAC cells often undergo a switch from E-cadherin to N-cadherin expression (Nakajima et al., 2004; Javle et al., 2007; Berx and van Roy, 2009) and show an increase in vimentin (Javle et al., 2007; Handra-Luca et al., 2011) and MMP9 (Jones et al., 2004). Considering this association between ERK1/2 status and EMT in PDAC, we examined whether MOR-SSTR2 activation changes the metastatic potential of cells. After PANC-1 cells were treated with dermorphin alone, L-054,264 alone, or a combination of the two, expression levels of EMT markers were measured by RT-PCR and Western blots. Of importance, only the combined MOR and SSTR2 agonist treatment (24 h, Figure 5E) in PANC-1 cells showed features consistent with EMT. Compared to levels found with the single-agonist treatments, mRNA levels observed with the combination treatment significantly increased for vimentin, MMP9, and N-cadherin and decreased for E-cadherin. This effect was not observed in normal pancreatic cells upon all treatments. As a control experiment, the agonist combination treatment was evaluated in PANC-1–knockdown cells. In PANC-1/MORsi and PANC-1/SSTR2si cells, the treatment did not affect mRNA levels of the four EMT markers (Supplemental Figure 12B). Moreover, in PANC-1 cells, neither a combination of MOR/CXCR4 agonists nor an individual CXCR4 agonist affected the mRNA levels of the four EMT markers (Supplemental Figure 12C). In addition to these RT-PCR studies, we also confirmed the selective effect of the combination treatment on EMT markers in PANC-1 cells at the protein level. The combined MOR and SSTR2 agonists increased protein levels of vimentin and N-cadherin and decreased levels of E-cadherin in PANC-1 cells but not in normal pancreatic cells (Supplemental Figure 12D). These results imply that coactivation of associating GPCRs may contribute to the aggressive biology of PDAC.
DISCUSSION
Conventional targets in PDAC have not yielded an effective chemotherapeutic agent for a number of reasons: the disease is usually detected at a late stage, when it is more difficult to treat; it progresses aggressively; and it is difficult to deliver PDAC drugs efficiently (Li et al., 2004a; Oberstein and Olive, 2013). The limitations of current approaches are reflected in PDAC’s dismal 5-yr survival rate of 7% (Howlader et al., 2014). To develop an orthogonal approach capable of complementing the current clinical regimen, we have focused our attention on GPCR heteromers. Because GPCR heteromers are more commonly observed in pathological cells (AbdAlla et al., 2001; Grant et al., 2008; Azdad et al., 2009; Albizu et al., 2011; Rozenfeld et al., 2011; Bushlin et al., 2012), they are ideal targets for developing a selective pharmacological agent and are under scrutiny for a number of neurological disorders. However, in pancreatic cancer, they remain a relatively untapped resource. The focus has been predominantly on monomeric GPCRs such as SSTR2 (Cascinu et al., 1995) and CXCR4 (Singh et al., 2010). Because such receptors exhibit complex signaling patterns and tend to make heteromers under pathological conditions, we have investigated their association in PDAC by coupling classical biochemical experiments with quantitative superresolution imaging techniques.
Using Western blots and RT-PCR (Figure 1), we showed that pancreatic cells express MOR, CXCR4, and SSTR2. We then applied pointillistic superresolution microscopy methods to obtain details about receptor distribution at the single-molecule level. To achieve high spatial resolution, pointillistic techniques such as dSTORM use total internal reflection microscopy illumination and single marker switching to restrict light emission to a single fluorophore in a diffraction-limited spot (Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006; Folling et al., 2008; Wombacher et al., 2010). In this way, a resolution of ∼10–25 nm is obtained and single-molecule sensitivity is achieved along large spatial areas. We further applied a rigorous quantitative analysis: Voronoï tessellation analysis (Levet et al., 2015) was used to determine receptor shape and size, and pair-correlation analysis (Sengupta et al., 2011, 2013) was used to obtain information on the extent of colocalization between receptors. Our results suggest that MOR, SSTR2, and CXCR4 form clusters with an average radius between 35 and 55 nm. This is consistent with the organization of receptors into signaling domains and possible partial association with lipid rafts (Tobin et al., 2014).
PC-dSTORM was used to define the colocalization of CXCR4, MOR, and SSTR2. The effect of cellular environment was examined by using healthy cells, cancerous cells, and the external layer of MCTS. MCTS can recapitulate 1) cellular interactions and tumor heterogeneity (e.g., contain both quiescent and proliferative cells; Sutherland, 1988) and 2) histomorphological, functional, and microenvironmental features of human tumor tissue (Hirschhaeuser et al., 2010). According to our results, MOR and SSTR2 colocalize in only malignant samples (Figure 2). This colocalization largely occurs in clustered regions. Conversely, in all the cases, MOR and CXCR4 did not colocalize (Figure 3). Our data were validated with two important controls: no appreciable signal was detected with 1) blocking peptides (Supplemental Figures S1 and S4, B and D) and 2) stable PANC-1 cell knockdowns of the corresponding receptor (Supplemental Figure S2). In NS control PANC-1 cells, signals for MOR, SSTR2, and CXCR4 were detected at levels and distributions similar to those in wild-type PANC-1 cells. In addition, the choice of label (Alexa Fluor 647 vs. Atto 488) did not produce significant differences in cross-correlation curves (Supplemental Figure S5). As demonstrated by Monte Carlo simulations, antibody detection efficiencies do not significantly influence our conclusions obtained from correlation curves (Supplemental Figure S6).
To confirm the dSTORM-detected GPCR interactions, we performed immunoprecipitation experiments. The interactions observed in the superresolution experiments were similar to those observed through the biochemical approach. In both healthy and malignant pancreatic cells, no interactions were observed between MOR and CXCR4, whereas in malignant PANC-1 cells, SSTR2 coimmunoprecipitates with MOR (Figures 2D and 3D).
To examine the relevance of MOR-SSTR2 colocalization under native conditions, we extended our studies to patient tissue samples. Although superresolution imaging of patient samples is time intensive and technically challenging (relatively high background, limited labeling options, and sample heterogeneity among others), in the present study we addressed this by optimizing both sample preparation and imaging conditions and by sampling multiple sections/regions of tissues while selectively labeling epithelial cells. Specifically, we used typical pathology markers for epithelial tissue, keratin 8 and 18 (Moll et al., 1982). Furthermore, the superresolution data sets from tissue imaging were analyzed quantitatively (Figure 4). According to our results, GPCR organization is significantly different in normal and malignant tissue samples for three patients. The cluster size is larger for both MOR and SSTR2 in cancer tissue than in healthy margins. In addition, whereas they do not colocalize in healthy margins of three patients, MOR-SSTR2 clearly colocalize in cancerous patient tissues. Of importance, this is the first time an association has been described between MOR and SSTR2 in pancreatic cancer.
Given that MOR-SSTR2 is a newly identified associated receptor pair in PDAC, we further investigated the cellular localization of the two receptors upon agonist activation with confocal microscopy. As expected, largely membrane localization of MOR and SSTR2 with appreciable colocalization was seen in the steady state, whereas coactivation with selective agonists leads to receptor internalization.
We further investigated signaling of MOR-SSTR2 in pancreatic environments. GPCR signaling pathways are complex and influenced by both the cellular environment (Schmid and Bohn, 2009) and interactions with other receptors, such as receptor tyrosine kinases (RTKs) and/or other GPCRs (Belcheva et al., 2001; Billadeau et al., 2006; Rozenfeld and Devi, 2007; Kenakin, 2011). Whereas signaling pathways proceed by different G-protein subclasses, β-arrestin2, or RTKs (Belcheva et al., 2001, 2003; Billadeau et al., 2006; Fujioka et al., 2011), they often converge at ERK1/2. Canonical G-protein–mediated signaling leads to rapid and transient phosphorylation of ERK1/2, which is targeted to the nucleus. Similarly, signaling through EGFR can induce fast phosphorylation of ERK1/2, which also leads to nuclear targeting. Conversely, β-arrestin2–mediated signaling produces both slower and more sustained phosphorylation of ERK1/2, which is subsequently targeted to the cytosol and endosomes (Ahn et al., 2004; Gesty-Palmer et al., 2006; Shenoy et al., 2006; Rozenfeld and Devi, 2007; Cervantes et al., 2010). Because a downstream phenotypic assay that quantifies spatiotemporal ERK/1/2 phosphorylation (Ahn et al., 2004) is important for investigating functional cross-talk between associated GPCRs, we studied how activating MOR and/or SSTR2 affects ERK1/2 and EGFR phosphorylation. The three treatments consisted of 1) a specific MOR agonist, dermorphin, 2) a specific SSTR2 agonist, L-054,264, and 3) a combination of the two. In malignant PANC-1 cells, activating MOR and SSTR2 simultaneously with the combination produced unique downstream effects: 1) consistent with previous reports in breast cancer cells (Kharmate et al., 2013), the combination failed to induce EGFR phosphorylation; 2) it produced slower and more sustained pERK1/2 activation; and 3) it predominantly promoted cytoplasmic pERK1/2 and resulted in cytoplasmic pRSK component. This is particularly significantly considering that cytoplasmic pERK has been detected in a large number of patient PDAC samples (Pham et al., 2008; Dutruel et al., 2014). Of importance, mostly nuclear pERK1/2 was observed in MOR-, SSTR2-, and β-arrestin2–knockdown PANC-1 cells upon combination treatment. Based on taking the results together, coactivation of MOR-SSTR2 in PANC-1 cells appears to proceed by a distinct pathway that does not appear to involve EGFR transactivation and is consistent with β-arrestin2 signaling.
Given that PDAC is characterized by frequent metastasis to remote sites (only ∼20% of tumors are discovered in early stages; Li et al., 2004a), we next investigated whether this distinct signaling pathway contributes to the metastatic potential of cells. Expression of mesenchymal proteins in cancer cells is an indicator of aggressive tumor biology for PDAC (Javle et al., 2007) and has been associated with poor patient survival (Javle et al., 2007; Handra-Luca et al., 2011; Jiang et al., 2015), chemoresistance (Arumugam et al., 2009; Wang et al., 2009), and invasiveness (Nakajima et al., 2004; Javle et al., 2007; Zhang et al., 2012). On administration of our three treatments, the expression of EMT markers was quantified. According to our results, coactivation of MOR and SSTR2 in PANC-1 cells leads to increased expression of vimentin, MMP9, and N-cadherin and decreased expression of E-cadherin (Figure 5E and Supplemental Figure S12). This effect was not observed in normal pancreatic cells or PANC-1/MORsi and PANC-1/SSTR2si cells. Thus the coactivation of MOR and SSTR2 appears to be involved in the metastatic potential of PDAC.
Conclusion
By combining quantitative superresolution analyses with biochemical approaches, we identified a molecular signature unique to pancreatic adenocarcinoma. Two G-protein–coupled receptors, MOR and SSTR2, were shown to associate uniquely in PDAC. Moreover, coactivation of MOR and SSTR2 produced a distinct signaling pathway that appears to use β-arrestin2 signaling and increase the metastatic potential of cells. Thus MOR-SSTR2 in PDAC may constitute a novel and specific pharmacological target.
MATERIALS AND METHODS
Coverslip preparation
Number 1.5, 25-mm coverslips (Warner Instruments, Hamden, CT) were cleaned with 1% Hellmanex III (Fisher Scientific, Hampton, NH) for 3 h at 25°C, rinsed with distilled water, and placed in acetone at 70°C for 10 min. This was followed by a secondary cleaning with a mixture of 1:1:5 (vol/vol) of hydrogen peroxide (30%):ammonium hydroxide:water for 1 h at 70°C. Coverslips were rinsed in distilled water and stored in 100% ethanol. Cleaned coverslips were subsequently flame dried and placed in sterile 35-mm tissue culture dishes. For dSTORM microscopy, cells were grown on coverslips coated with fibronectin-like engineered protein (25 μg/ml in phosphate-buffered saline [PBS], pH 7.4; Sigma-Aldrich, St. Louis, MO) as described previously (Tobin et al., 2014). For tissue imaging, coverslips were coated with 0.03% gelatin for 10 min at room temperature; solution was aspirated, and the coverslips were air-dried just before sample addition.
Cell culture
COS-7, HEK293T, PANC-1, BxPC3, SU.86.86, CAPAN-1, and MCF-7 cells (originally obtained from the American Type Culture Collection, Manassas, VA) were cultured in phenol red–free DMEM or RPMI-1640 (only for BxPC3 cells) supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mM l-alanyl- l-glutamine. Human immortalized pancreatic epithelial cells (AddexBio, San Diego, CA) were grown in Keratinocyte SFM medium with defined Keratinocyte SFM supplements (Life Technologies, Carlsbad, CA) as per the supplier’s suggestion and subsequently cultured in phenol red–free Epilife media supplemented with calcium chloride and Epilife supplements (Life Technologies). CHO-S cells (Invitrogen, Carlsbad, CA) were cultured per manufacturer’ specifications in FreeStyle medium.
To obtain a large and reproducible quantity of MCTS, we placed 1000 cells/well into 96-well round-bottom plates coated with PolyHema (Sigma-Aldrich). A small amount (∼1% of total medium) of phenol red–free Matrigel (Corning, Corning, NY) was included in the growth medium. MCTS of consistent size (450 μm) generally formed within 4 d. For superresolution imaging of surface layers, MCTS were placed on fibronectin-like engineered protein coated coverslips for 3 h at 37°C in the cell incubator to attach and were subsequently fixed.
Knockdowns
MOR, SSTR2, CXCR4, and β-arrestin2 knockdowns were performed using GIPZ lentivirals shRNAmir constructs specific for each GPCR (GE Dharmacon, Lafayette, CO); details are provided in the Supplemental Methods. The microRNA is a polycistronic RNA with TurboGFP, allowing visualization of cells that express the short hairpin RNA. The shRNAmir construct was stably integrated in the PANC-1 cells per the manufacturer’s instructions. First, the lentivirus was packaged using the Trans-LentiviralshRNA packaging system. The lentivirus system was then transfected into HEK293T cells (American Type Culture Collection) using calcium phosphate. Packaged lentivirus was concentrated for 48 h after transfection. The supernatant with lentiviral particles containing shRNAmir was directly transduced into PANC-1 cells. Briefly, ∼5 × 104 PANC-1 cells were added to each well in a 24-well plate. A final concentration of 8 μg/ml Polybrene and 500 μl of packaged lentivirus were added to the cells. The cells were incubated for 8 h, and then the medium was changed. After 48 h, DMEM supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mM l-alanyl- l-glutamine supplemented with 2 μg/ml puromycin was added to start selection for stably transduced cells (PANC-1/SSTR2si. PANC-1/MORsi, PANC-1/CXCR4si, and PANC-1/β-arrestin2si). As a control, GIPZ vector containing a nonsilencing control was also transfected (PANC-1 NS).
Antibodies and fluorescent dye conjugation
The following primary antibodies were purchased: anti-MOR (guinea pig polyclonal; Abcam, Cambridge, MA), anti-SSTR2 (rabbit polyclonal; Neuromics, Edina, MN), anti-CXCR4 (mouse monoclonal; Neuromics), and anti–cytokeratin 8 and 18 (mouse monoclonal; Cell Marque/Sigma-Aldrich). Purchased purified secondary antibodies include rabbit anti-guinea pig (polyclonal; Abcam), goat anti-rabbit (polyclonal; Abcam), and goat anti-mouse (polyclonal; EMD Millipore, Temecula, CA). Antibody references are provided in the Supplemental Methods. Secondary antibodies were labeled with Alexa Fluor 405, Alexa Fluor 647 (Life Technologies), or Atto 488 (Sigma-Aldrich) containing an N-hydroxysuccinidimidyl ester (NHS) group for conjugation to proteins. A solution containing a 6–10 M excess of dye dissolved in dimethyl sulfoxide was mixed with a solution of 1 mg/ml secondary antibody in PBS, pH 7.4, with 0.02 M NaHCO3; the resulting solution was allowed to react for 30 min at room temperature. The solution was quenched with 1.5 M hydroxylamine (pH 8.5) for 10 min. Unconjugated dye was removed by passing the solution through a size exclusion chromatography column (Bio-Rad, Hercules, CA). Before the experiment, labeled antibody was passed through a 300-kDa concentrator to remove any potential aggregates. The concentration of labeled secondary antibodies was measured by a NanoDrop 1000 (Thermo) and calculated with respect to the dye’s correction factor. Approximately one dye per antibody was obtained in all cases for Atto 488 and Alexa Fluor 647. Secondary antibodies were used freshly labeled.
Immunocytochemistry
Immunocytochemistry was done according to established protocols. Antibody concentrations and incubation times were individually optimized. Briefly, cells were fixed for 30 min at room temperature with 4% (wt/vol) paraformaldehyde and 0.2% (wt/vol) glutaraldehyde and inactivated with 25 mM glycine for 10 min. After washes in PBS, cells were incubated in permeabilization buffer (PB; 0.1–0.5% Tween-20 and 5% bovine serum albumin [BSA] in PBS) for 20 min. After a wash, cells were incubated for 1 h with 2 μg/ml primary antibody/antibodies. Subsequently cells were extensively washed and incubated with 2 μg/ml labeled secondary antibody/antibodies for 45 min. To avoid interaction between goat anti-rabbit and rabbit anti-guinea pig secondary antibodies, we first incubated cells with secondary anti-rabbit antibody, extensively washed them, and then incubated cells with secondary anti-guinea pig antibody. After PBS wash, cells were postfixed for 10 min with 4% (wt/vol) paraformaldehyde and 0.2% (wt/vol) glutaraldehyde and inactivated with 25 mM glycine for 10 min at room temperature. For control experiments, 5 M excess of blocking peptide was preincubated with specific primary antibody/antibodies for 10 min at room temperature; all other steps were done according to the foregoing protocol. Coverslips were imaged immediately after preparation in Attofluor cell chambers (Life Technologies) in 50 mM Tris (pH 8.0), 10 mM NaCl, and 10% glucose imaging buffer containing mercaptoethylamine (100 mM) and glucose oxidase and catalase (GLOX; 10% vol/vol) as previously described (Dempsey et al., 2011).
Tissue samples and immunohistochemistry
Fresh-frozen tissues in OCT compound (Fisher Scientific) were cut using a Leica CM3050S cryostat; slice thickness was 8 μm. Glandular tissue was sliced to allow imaging of normal/neoplastic glands. Tissue slices were put on clean coverslips coated with 0.03% gelatin, incubated at room temperature for 5 min, and rehydrated with PBS for 10 min. Samples were fixed for 30 min with 4% (wt/vol) paraformaldehyde and 0.2% (wt/vol) glutaraldehyde and inactivated with 25 mM glycine for 10 min at room temperature. Subsequently samples were incubated with antibodies (as described in the immunocytochemistry protocol) to detect MOR and SSTR2. After extensive washes, tissues were incubated with 2 μg/ml cytokeratin antibody 8 and 18 (Cell Marque, Rocklin, CA) in PB buffer for 30 min. Tissues were subsequently washed and incubated with 2 μg/ml Alexa Fluor 405–labeled secondary antibody in PB buffer for 15 min. Tissue samples were extensively washed again and postfixed for 10 min. Samples were immediately imaged on a Nikon N-STORM superresolution microscope. Images were processed with Nikon Elements N-STORM software to identify peaks. Tissue samples were collected under Institutional Review Board No. 06129, and informed consent was obtained from all subjects.
Optical setup and image acquisition
Measurements were performed on a 3D N-STORM superresolution microscope (Nikon, Melville, NY) configured for TIRF. The N-STORM system (Nikon Instruments) consists of a fully automatic Ti-E inverted microscope with piezo stage on a vibration isolation table with a 100×/1.49 numerical aperture TIRF objective (Apo), an N-STORM lens and λ/4 lens, and a Quad cube C-NSTORM (97355; Chroma, Bellows Falls, VT) with filters for 405-, 488-, 561-, and 640-nm light. The microscope is equipped with a Perfect Focus Motor to maintain imaging on the desired focal plane, an MLC-MBP-ND laser launch with 405-, 488-, 561-, and 647-nm lasers (Agilent, Santa Clara, CA), and an electron-multiplying charge-coupled device camera (iXon DU897-ultra; Andor Technology, South Windsor, CT). Data were acquired using NIS Elements 4.3 software with automatic drift correction. Laser powers used to activate and/or image dyes were 120, 60, and 6 mW (measured out of the optical fiber) for 647, 488, and 405 nm, respectively. Two-color imaging was done with sequential activation, switching the laser from 488 to 647 nm for every frame. We acquired 20,000 frames in each channel using an exposure time of 9 ms.
We imaged 2D cultures of cells, outer layer of MCTS, and thin sections of tissue samples. All samples were imaged in TIRF mode, and trans-light was used to observe selected regions. Before microscopy, we marked locations of MCTS and tissue samples on the coverslip with the marker to aid in region selection. For tissue imaging, epithelial cells were identified with quick scanning using a low-power 405-nm laser. We adjusted the TIRF angle and focus to image tissue regions close to the coverslip with appreciable 488-nm and/or 647-nm signal (appearing in the area that matched keratin 8 and 18 signal). For tissues, 405-channel imaging of Alexa Fluor 405 was performed after the two-color dSTORM acquisition.
To select flat membrane segments and avoid artifacts from the 3D topology of the membrane projecting into the 2D imaging plane, we imaged the same segments with a slightly shifted focal plane (minimal signal was observed above membrane segments). In addition, we also examined the photon count distributions for these regions after processing and confirmed that photon counts were in an expected range and did not show bimodal distributions.
Image analysis
Peak localization was performed using NIS Elements 4.3 software. The minimum number of photons was set to 700. Settings for peak localization were as follows: minimum peak width, 200 nm; maximum peak width, 400 nm; maximum axial ratio, 1.3; and maximum displacement, 1 pixel. Peak height thresholds were set to 8000 and 11,000 for Atto 488 and Alexa Fluor 647, respectively. The average localization precision was 17 nm in the 488 channel and 12 nm in the 647 channel, as detected by NIS-Elements. After peak localization, the density of peaks and cross-correlation functions were computed as described previously (Sengupta et al., 2011, 2013). Briefly, for two-color imaging, two separate binary images of cells were computed using peak coordinate centers obtained from NIS-elements. A value of 1 was assigned for pixels with detected peaks, and a value of 0 was assigned elsewhere. Next square regions of interest with a size of 80 μm2 were randomly selected. Cross-correlation was computed using fast Fourier transforms with a previously published algorithm in MATLAB (Natick, MA) (Sengupta et al., 2013). In all cases, error bars represent SEM calculated individually for each data point. All presented images were color inverted for clarity. No other modifications were done. Image tessellation was performed using SR-Tesseler software as previously described (Levet et al., 2015). Brief details are provided in the Supplemental Methods.
Protein extracts and immunoblotting
To prepare protein extracts, cells were pelleted by centrifugation and washed twice in PBS. The pellet was resuspended in lysis buffer (50 mM Tris, pH 8.0, 150 mM sodium chloride, and 1% NP-40 with supplemented Protease and Phosphatase Inhibitor Mini Tablets; ThermoFisher Pierce, Grand Island, NY) and incubated on a rotator for 30 min at 4°C. Subsequently the cells were pelleted by centrifugation at 10,000 rpm for 20 min at 4°C. The protein lysate (supernatant) was used immediately or stored at −80°C. Membrane proteins were isolated using the Mem-Per Plus Membrane protein isolation kit (Thermo Scientific) per the manufacturer’s instructions. Briefly, adherent cells were scraped and centrifuged for 5 min at 300 × g. The cell pellet was washed twice using Cell Wash solution and centrifuged for 5 min at 300 × g. The cell pellet was permeabilized using permeabilization buffer and incubated at 4°C for 10 min on a rotator. The permeabilized cells were then centrifuged at 16,000 × g for 15 min. The supernatant containing the cytosolic proteins was removed and stored at −80°C. The remaining cell pellet was resuspended in solubilization buffer and incubated for 30 min on a rotator at 4°C. After centrifugation at 16,000 × g for 15 min at 4°C, the supernatant containing the fractionated membrane proteins were removed at stored at −80°C. The lysates were subjected to SDS–PAGE’ and Western blotting using standard procedures. Primary antibodies included anti–phospho-ERK1/2 (Thr202/Tyr204; rabbit monoclonal; Cell Signaling), anti-ERK1/2 (rabbit polyclonal; Abcam), anti-EGFR (rabbit monoclonal; Abcam), anti–phospho EGFR (Tyr-1068; rabbit monoclonal; Cell Signaling), anti-MOR (guinea pig polyclonal; Abcam), anti-SSTR2 (rabbit polyclonal; Neuromics), anti-CXCR4 (mouse monoclonal, Neuromics), anti–β-actin (mouse monoclonal; Cell Signaling), and anti–Na/K ATPase (rabbit monoclonal; Cell Signaling). Proteins were detected with Pierce ECL detection reagents (ThermoFisher Pierce). The blots were imaged using the Chemi-Doc Touch Imaging system or film developer.
Epithelial and mesenchymal markers and Western blots
PANC-1 cells were treated with 1) 10 nM dermorphin (Y(D-ALA)FGYPKC; GenScript, Piscataway, NJ) and 2) 10 nM (1R,1′S,3′R/1R,1′R,3′S)-L-054,264 (Tocris) individually and in combination for 24 h. Cells were lysed as described for immunoblotting with EMT marker antibodies (vimentin rabbit monoclonal, E-cadherin rabbit monoclonal, N-cadherin rabbit monoclonal; Epithelial-Mesenchymal Transition Antibody Sample Kit; Cell Signaling) according to the manufacturer’s protocol. Blots were imaged using the Image Lab software (Bio-Rad).
Kinetic study
Cells were treated with 10 nM dermorphin [Y(d-ALA)FGYPKC; GenScript] or 10 nM (1R,1′S,3′R/1R,1′R,3′S)-L-054,264 (Tocris) individually and in combination for the indicated times. Cells were lysed for immunoblotting with phospho-ERK1/2, total ERK1/2, phospho-EGFR, and total EGFR. Blots were imaged on film and quantified using the Image Lab software. ERK1/2 and EGFR phosphorylation at each time point was quantified and normalized by calculating the ratio of pERK1/2 over total ERK1/2 and pEGFR over total EGFR in each lane. The data represent the percentage of maximum response to dermorphin treatment. The p values were obtained using the single-tail t test for the same time point between dermorphin activation and either L-054,264 or combined L-054,264 and dermorphin activation.
Cell fractionation
Nuclear and cytoplasmic extracts were made according to previously published protocols (Smith et al., 2004; Rozenfeld and Devi, 2007). Briefly, PANC-1 cells were treated with 10 nM dermorphin or 10 nM (1R,1′S,3′R/1R,1′R,3′S)-L-054,264 (Tocris, Bristol, UK) individually and in combination for the indicated times. After the treatment, cells were washed in ice-cold PBS three times and scraped into lysis buffer (10 mM Tris, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 1 mM ethylene glycol tetraacetic acid, 1 mM sodium orthovanadate, and a protease phosphatase inhibitor tablet [ThermoFisher Pierce]) and incubated on ice for 10 min. The lysate was then homogenized and centrifuged at 375 × g for 5 min. The pellet consisting of the nuclear fraction was washed five times with lysis buffer containing 0.1% NP-40 to remove any nonnuclear contamination and resuspended in lysis buffer containing NP-40. The soluble fraction was centrifuged twice at 375 × g to remove nuclear contamination and used as the cytosolic fraction. Fractions were then used for immunoblotting for anti–phospho-ERK1/2, anti–phospho-p90RSK (Thr-573; rabbit polyclonal; Cell Signaling, Danvers, MA), and anti–β-actin as a loading control for the cytoplasmic fraction, and Histone H3 antibody (rabbit monoclonal; Cell Signaling) was used as a loading control for the nuclear fraction.
Extraction of RNA, cDNA synthesis, and RT-PCR
RNA was isolated using the RNAeasy Plus Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. For the EMT markers assay, cells were first treated with appropriate agonists where indicated for 24 h, followed by cell lysis and RNA extraction. cDNA was synthesized from 2 μg of RNA using a Bioline cDNA synthesis kit according to the manufacturer’s protocol. The synthesized cDNAs were used as templates for PCRs with primers as described in the Supplemental Methods. RT-PCR was performed using 5× RT Master Mix (Applied Biosystems). The reactions were analyzed on the CFX96 real-time instrument (Bio-Rad). Themocycler conditions were 95°C for 10 min for 1 cycle, 95°C for 15 s, and 60°C for 1 min for 50 cycles. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control. For GPCR assays, COS-7 cells were used as negative and MCF-7 cells as positive controls. For EMT marker assays, fold change was calculated as a ratio of treated to untreated control.
Coimmunoprecipitation
Coimmunoprecipitation was performed using a coimmunoprecipitation kit (ThermoFisher Pierce) per the manufacturer’s protocol. Briefly, MOR antibody was immobilized using a coupling resin. PANC-1 or normal pancreatic epithelial cells were lysed using buffer containing 1% Nonidet P-40, 10% glycerol, 50 mM Tris-Cl, pH 7.4, 300 mM NaCl, 1.5 mM MgCl2, 1 mM CaCl2, and protease inhibitor tablets (ThermoFisher Pierce) as previously described (Rozenfeld and Devi, 2007), precleared, and added to the antibody-coupled resin and incubated with gentle shaking overnight at 4°C. After thorough washes, the protein sample coimmunoprecipitate was eluted and used for Western blotting with SSTR2 or CXCR4 antibodies. Actin was used as a loading control.
Sample preparation for confocal microscopy
For all studies, cells were plated and grown on coverslips in full media.
For internalization studies, PANC-1 cells were treated with 1) 10 nM dermorphin, 2) 10 nM L-054,264, and 3) 10 nM dermorphin plus 10 nM L-054,264 for 30 min. Untreated or agonist(s)-treated cells were then fixed and immunostained for MOR and SSTR2 detection as described. Images were acquired using a Zeiss LSM880 microscope and LD C-Apochromat 40×/1.1 W Corr M27 objective.
For pERK1/2 activation studies, after 24 h, normal pancreatic and/or PANC-1 cells were treated with 1) 10 nM dermorphin, 2) 10 nM L-054,264, 3) 10 nM dermorphin plus 10 nM L-054,264, 4) 100 ng/ml CXCL12, and 5) 10 nM dermorphin plus 100 ng/ml CXCL12 for either 3 or 30 min as indicated. PANC-1 NS, PANC-1/β arreatin2, PANC-1/SSTR2si, and PANC-1/MORsi cells were treated with 10 nM dermorphin with 10 nM L-054,264 for 3 min. After treatment, cells were washed and fixed with 4% (wt/vol) paraformaldehyde and 0.2% (wt/vol) glutaraldehyde for 30 min and inactivated with 25 mM glycine for 10 min. Cells were then washed in PBS and subsequently permeabilized on ice using 0.5% Tween-20 in PBS for 20 min as before (Smith et al., 2004). Cells were washed with PBS and blocked with 5% BSA in PBS for 10 min. Cells were incubated for 1 h at 37°C with 2 μg/ml primary antibody (anti-pERK1/2). Subsequently cells were washed three times with 0.5% Tween-20 in PBS for 5 min each and incubated with 2 μg/ml Alexa Fluor 647–labeled secondary antibody in PB for 45 min. After incubation with secondary antibody, cells were washed three times with 0.5% Tween-20 in PBS. Cells were covered with 4′,6-diamidino-2-phenylindole mounting medium (Vector Labs, Burlingame, CA), mounted on slides, and imaged on a Zeiss LSM700 confocal microscope.
Monte Carlo simulations
We detected on average ∼2000 receptors in normal cells and ∼6000 receptors in PANC-1 cells. Although this is consistent with previously reported values for GPCRs (Jonas et al., 2015), we wanted to investigate whether antibody binding efficiency significantly influences the discovery of heterodimerizaion via cross-correlation curves. We thus simulated superresolution images of receptors with specified dimerization fractions at different receptor densities and antibody binding efficiencies. We assumed detected densities of ∼4 and 10 receptors/μm2, representing lower- and higher-expressing cells on average, respectively. We generated images with random receptor positions and randomly assigned each receptor to either be a monomer (red or green, denoted R or G), homodimer (RR or GG), or heterodimer (RG) in specified ratio. We assumed that receptor sizes were small enough that excluded-area effects were negligible (forbidding two molecules to be less than one receptor diameter apart).
Using those randomly generated images, specified detection efficiency, and the known photophysical properties of the fluorescent labels from experimental data (mean localization precision, average photon count, distribution of photon counts), we generated sets of localizations in the red and green channels, which we translated into images. These localization maps were in the same format as the processed experimental data. Finally, these simulation data were passed to the same analysis software as the experimental data, and the cross-correlation between the red and green channels was computed.
In our simulations, the key variables were as follows:
The number of red and green receptors per square micrometer.
The relative probabilities of the red and green receptors appearing as monomers or as homodimers and heterodimers.
The labeling probabilities for red and green receptors (corresponding to the antibody detection efficiency). We allowed for the possibility of incomplete labeling, so that only a predefined fraction of the receptors was labeled red or green. We did not assume a priori that the labeling probabilities were the same for both receptors.
The mean number of times a receptor turned on and produced a usable image during the experiment.
The mean localization precision and mean photon count for each label (2500 and 1500 for red and green, respectively). The photon counts were assumed to be exponentially distributed, with a minimum threshold of 1000 photons for an image to be analyzed.
After randomly generating receptor positions and randomly picking a subset of the receptors as fluorescently labeled, we randomly generated a number of appearances from a geometric distribution for labeled receptors. We first investigated the effect of the average number of appearances of a fluorophore on the data; we did not observe significant variability for tested conditions. For the majority of the simulations, we thus used an average of four appearances for both channels. We next generated a series of photon counts (from an exponential distribution with specified mean and minimum); we used the photon counts to determine the precision (SD) of each position estimate, based on the assumption that the localization precision is inversely proportional to the square root of the photon count. For each receptor appearance, we generated position estimates. Each estimate was drawn from a Gaussian (normal) distribution with a mean equal to the true position of the receptor and SD equal to the localization precision. These position estimates were then used to construct a high-resolution map of estimated receptor positions. This image map was subsequently analyzed by the same MATLAB program used to compute the cross-correlation function of the experimental data. All image simulations were performed in Python on a laptop computer. A simulation with a 16 × 16–μm field of view, containing ∼5000 receptors, could be conducted and the output rendered into a high-resolution image (125 Mb) in <5 min.
Supplementary Material
Acknowledgments
We thank J. C. Williams, V. Nagarajan, J. Benovic, and A. Schonbrunn for helpful discussion; C. Kowolik for helpful discussion and assistance in designing RT-PCR primers; I. Talisman for editing; E. Cacao for technical help; and B. Armstrong for assistance with confocal imaging. This work was supported by the Beckman Research Institute of the City of Hope (R.J, S.B., O.G., D.L.W., and T.J.-T.), the City of Hope Medical Center (G.S. and P.C.), and the Irell & Manella Graduate School of Biological Sciences at City of Hope (S.J.T.). The Rose Hills Foundation provided for the Eugene and Ruth Roberts Summer Student Academy to M.K. and K.H. Research reported here includes work performed in the Pathology Core and Light Microscopy and Imaging Core, which are supported by the National Cancer Institute of the National Institutes of Health under Award P30CA33572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used:
- CXCR4
C-X-C motif receptor 4
- dSTORM
direct stochastic optical reconstruction microscopy
- EGFR
epidermal growth factor receptor 1
- ERK1/2
extracellular signal-regulated kinase 1/2
- MCTS
multicellular tumor spheroids
- MOR
mu opioid receptor
- PDAC
pancreatic ductal adenocarcinoma
- RTK
receptor tyrosine kinase
- SSTR2
somatostatin receptor 2.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0427) on September 28, 2016.
REFERENCES
- AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med. 2001;7:1003–1009. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, et al. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol. 2010;6:587–594. doi: 10.1038/nchembio.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albizu L, Holloway T, Gonzalez-Maeso J, Sealfon SC. Functional crosstalk and heteromerization of serotonin 5-HT2A and dopamine D2 receptors. Neuropharmacology. 2011;61:770–777. doi: 10.1016/j.neuropharm.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena P, Garlanda C, Borrello MG, Sica A, Mantovani A. Pathways connecting inflammation and cancer. Curr Opin Genet Dev. 2008;18:3–10. doi: 10.1016/j.gde.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azdad K, Gall D, Woods AS, Ledent C, Ferre S, Schiffmann SN. Dopamine D2 and adenosine A2A receptors regulate NMDA-mediated excitation in accumbens neurons through A2A-D2 receptor heteromerization. Neuropsychopharmacology. 2009;34:972–986. doi: 10.1038/npp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Szucs M, Wang DX, Sadee W, Coscia CJ. mu-opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ. mu opioid transactivation and down-regulation of the epidermal growth factor receptor in astrocytes: Implications for mitogen-activated protein kinase signaling. Mol Pharmacol. 2003;64:1391–1401. doi: 10.1124/mol.64.6.1391. [DOI] [PubMed] [Google Scholar]
- Berg KA, Rowan MP, Gupta A, Sanchez TA, Silva M, Gomes I, McGuire BA, Portoghese PS, Hargreaves KM, Devi LA, Clarke WP. Allosteric interactions between delta and kappa opioid receptors in peripheral sensory neurons. Mol Pharmacol. 2012;81:264–272. doi: 10.1124/mol.111.072702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009;1:a003129. doi: 10.1101/cshperspect.a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Billadeau DD, Chatterjee S, Bramati P, Sreekumar R, Shah V, Hedin K, Urrutia R. Characterization of the CXCR4 signaling in pancreatic cancer cells. Int J Gastrointest Cancer. 2006;37:110–119. doi: 10.1007/s12029-007-0011-7. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Bushlin I, Gupta A, Stockton SD, Jr, Miller LK, Devi LA. Dimerization with cannabinoid receptors allosterically modulates delta opioid receptor activity during neuropathic pain. PLoS One. 2012;7:e49789. doi: 10.1371/journal.pone.0049789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call TR, Jedrzkiewicz J, Tripp SR, Wilson AJ, Chortkoff B, Witt BL. Mu opioid receptor expression in various tumor types: creating a mu scoring system. 2013. In: Abstracts and Case Studies from the College of American Pathologists 2013 Annual Meeting, A4225. Available at www.asaabstracts.com/strands/asaabstracts/abstract.htm?year=2013&index=2&absnum=4246.
- Cascinu S, Del Ferro E, Catalano G. A randomised trial of octreotide vs best supportive care only in advanced gastrointestinal cancer patients refractory to chemotherapy. Br J Cancer. 1995;71:97–101. doi: 10.1038/bjc.1995.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes D, Crosby C, Xiang Y. Arrestin orchestrates crosstalk between G protein-coupled receptors to modulate the spatiotemporal activation of ERK MAPK. Circ Res. 2010;106:79–88. doi: 10.1161/CIRCRESAHA.109.198580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fan J, Chen H, Meng Z, Chen Z, Wang P, Liu L. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci Rep. 2014;4:5911. doi: 10.1038/srep05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci USA. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang XW. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JM, Zer C, Avery KN, Bzymek KP, Horne DA, Williams JC. Identification and grafting of a unique peptide-binding site in the Fab framework of monoclonal antibodies. Proc Natl Acad Sci USA. 2013;110:17456–17461. doi: 10.1073/pnas.1307309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudok B, Barna L, Ledri M, Szabo SI, Szabadits E, Pinter B, Woodhams SG, Henstridge CM, Balla GY, Nyilas R, et al. Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat Neurosci. 2015;18:75–86. doi: 10.1038/nn.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutruel C, Bergmann F, Rooman I, Zucknick M, Weichenhan D, Geiselhart L, Kaffenberger T, Rachakonda PS, Bauer A, Giese N, et al. Early epigenetic downregulation of WNK2 kinase during pancreatic ductal adenocarcinoma development. Oncogene. 2014;33:3401–3410. doi: 10.1038/onc.2013.312. [DOI] [PubMed] [Google Scholar]
- Ferre S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, et al. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley MJ, Chen X, Bardi G, Davey P, Geller EB, Zhang L, Adler MW, Rogers TJ. Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the kappa-opioid receptor. J Neuroimmunol. 2008;197:114–123. doi: 10.1016/j.jneuroim.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- Fujioka N, Nguyen J, Chen CS, Li YF, Pasrija T, Niehans G, Johnson KN, Gupta V, Kratzke RA, Gupta K. Morphine-induced epidermal growth factor pathway activation in non-small cell lung cancer. Anesth Analg. 2011;113:1353–1364. doi: 10.1213/ANE.0b013e318232b35a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, Devi LA. G protein-coupled receptor heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Chandrakala MV, Devi LA. Disease-specific heteromerization of G-protein-coupled receptors that target drugs of abuse. Prog Mol Biol Transl Sci. 2013;117:207–265. doi: 10.1016/B978-0-12-386931-9.00009-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA. 2004;101:5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradiz R, Silva HC, Carvalho L, Botelho MF, Mota-Pinto A. MIA PaCa-2 and PANC-1 - pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci Rep. 2016;6:21648. doi: 10.1038/srep21648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant M, Alturaihi H, Jaquet P, Collier B, Kumar U. Cell growth inhibition and functioning of human somatostatin receptor type 2 are modulated by receptor heterodimerization. Mol Endocrinol. 2008;22:2278–2292. doi: 10.1210/me.2007-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E, Lim M, Maillet E, Junek M, Cahill CM, et al. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal. 2010;3:ra54. doi: 10.1126/scisignal.2000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Handra-Luca A, Hong SM, Walter K, Wolfgang C, Hruban R, Goggins M. Tumour epithelial vimentin expression and outcome of pancreatic ductal adenocarcinomas. Br J Cancer. 2011;104:1296–1302. doi: 10.1038/bjc.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasley LE. Autocrine and paracrine signaling through neuropeptide receptors in human cancer. Oncogene. 2001;20:1563–1569. doi: 10.1038/sj.onc.1204183. [DOI] [PubMed] [Google Scholar]
- Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA. Multicellular tumor spheroids: an underestimated tool is catching up again. J Biotechnol. 2010;148:3–15. doi: 10.1016/j.jbiotec.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Howlader N, Noone AM, Krapcho M, Garshell J, Neyman N, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z. SEER Cancer Statistics Review, 1975–2011. Available at http://seer.cancer.gov/archive/csr/1975_2011/ 2014 [Google Scholar]
- Jaquet P, Gunz G, Saveanu A, Dufour H, Taylor J, Dong J, Kim S, Moreau JP, Enjalbert A, Culler MD. Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. Eur J Endocrinol. 2005;153:135–141. doi: 10.1530/eje.1.01950. [DOI] [PubMed] [Google Scholar]
- Javle MM, Gibbs JF, Iwata KK, Pak Y, Rutledge P, Yu J, Black JD, Tan D, Khoury T. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann Surg Oncol. 2007;14:3527–3533. doi: 10.1245/s10434-007-9540-3. [DOI] [PubMed] [Google Scholar]
- Jiang JH, Liu C, Cheng H, Lu Y, Qin Y, Xu YF, Xu J, Long J, Liu L, Ni QX, Yu XJ. Epithelial-mesenchymal transition in pancreatic cancer: Is it a clinically significant factor? Biochim Biophys Acta. 2015;1855:43–49. doi: 10.1016/j.bbcan.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Jonas KC, Fanelli F, Huhtaniemi IT, Hanyaloglu AC. Single molecule analysis of functionally asymmetric GPCR oligomers reveals diverse spatial and structural assemblies. J Biol Chem. 2015;290:3875–3892. doi: 10.1074/jbc.M114.622498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LE, Humphreys MJ, Campbell F, Neoptolemos JP, Boyd MT. Comprehensive analysis of matrix metalloproteinase and tissue inhibitor expression in pancreatic cancer: increased expression of matrix metalloproteinase-7 predicts poor survival. Clin Cancer Res. 2004;10:2832–2845. doi: 10.1158/1078-0432.ccr-1157-03. [DOI] [PubMed] [Google Scholar]
- Kailey B, van de Bunt M, Cheley S, Johnson PR, MacDonald PE, Gloyn AL, Rorsman P, Braun M. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic beta- and alpha-cells. Am J Physiol. Endocrinol Metab. 2012;303:E1107–E1116. doi: 10.1152/ajpendo.00207.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kharmate G, Rajput PS, Lin YC, Kumar U. Inhibition of tumor promoting signals by activation of SSTR2 and opioid receptors in human breast cancer cells. Cancer Cell Int. 2013;13:93. doi: 10.1186/1475-2867-13-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J, Beckhove P, Esposito I, Herzig S, Huber PE, Lohr JM, Friess H. Pancreatic cancer microenvironment. Int J Cancer. 2007;121:699–705. doi: 10.1002/ijc.22871. [DOI] [PubMed] [Google Scholar]
- Korc M, Chandrasekar B, Yamanaka Y, Friess H, Buchier M, Beger HG. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest. 1992;90:1352–1360. doi: 10.1172/JCI116001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Hosotani R, Miyamoto Y, Ida J, Tsuji S, Nakajima S, Kawaguchi M, Kobayashi H, Doi R, Hori T, et al. Expression of stromal cell-derived factor 1 and CXCR4 ligand receptor system in pancreatic cancer: a possible role for tumor progression. Clin Cancer Res. 2000;6:3530–3535. [PubMed] [Google Scholar]
- Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284:26732–26741. doi: 10.1074/jbc.M109.026922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laklai H, Laval S, Dumartin L, Rochaix P, Hagedorn M, Bikfalvi A, Le Guellec S, Delisle MB, Schally AV, Susini C, et al. Thrombospondin-1 is a critical effector of oncosuppressive activity of sst2 somatostatin receptor on pancreatic cancer. Proc Natl Acad Sci USA. 2009;106:17769–17774. doi: 10.1073/pnas.0908674106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levet F, Hosy E, Kechkar A, Butler C, Beghin A, Choquet D, Sibarita JB. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat Methods. 2015;12:1065–1071. doi: 10.1038/nmeth.3579. [DOI] [PubMed] [Google Scholar]
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol. 2014;32:191–198. doi: 10.1038/nbt.2797. [DOI] [PubMed] [Google Scholar]
- Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004a;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- Li M, Li W, Kim HJ, Yao Q, Chen C, Fisher WE. Characterization of somatostatin receptor expression in human pancreatic cancer using real-time RT-PCR. J Surg Res. 2004b;119:130–137. doi: 10.1016/j.jss.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Lieber M, Mazzetta J, Nelson-Rees W, Kaplan M, Todaro G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. International journal of cancer. J Int Cancer. 1975;15:741–747. doi: 10.1002/ijc.2910150505. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Melchiorri P, Negri L. The dermorphin peptide family. Gen Pharmacol. 1996;27:1099–1107. doi: 10.1016/0306-3623(95)02149-3. [DOI] [PubMed] [Google Scholar]
- Mercadante S, Tirelli W, David F, Arcara C, Fulfaro F, Casuccio A, Gebbia V. Morphine versus oxycodone in pancreatic cancer pain: a randomized controlled study. Clin J Pain. 2010;26:794–797. doi: 10.1097/ajp.0b013e3181ecd895. [DOI] [PubMed] [Google Scholar]
- Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- Moreno E, Andradas C, Medrano M, Caffarel MM, Perez-Gomez E, Blasco-Benito S, Gomez-Canas M, Pazos MR, Irving AJ, Lluis C, et al. Targeting CB2-GPR55 receptor heteromers modulates cancer cell signaling. J Biol Chem. 2014;289:21960–21972. doi: 10.1074/jbc.M114.561761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M, Tulachan SS, Ito D, Kami K, Mori T, et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res. 2004;10:4125–4133. doi: 10.1158/1078-0432.CCR-0578-03. [DOI] [PubMed] [Google Scholar]
- Oberstein PE, Olive KP. Pancreatic cancer: why is it so hard to treat? Therap Adv Gastroenterol. 2013;6:321–337. doi: 10.1177/1756283X13478680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pello OM, Martinez-Munoz L, Parrillas V, Serrano A, Rodriguez-Frade JM, Toro MJ, Lucas P, Monterrubio M, Martinez AC, Mellado M. Ligand stabilization of CXCR4/delta-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur J Immunol. 2008;38:537–549. doi: 10.1002/eji.200737630. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Koch T, Schroder H, Laugsch M, Hollt V, Schulz S. Heterodimerization of somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and desensitization. J Biol Chem. 2002;277:19762–19772. doi: 10.1074/jbc.M110373200. [DOI] [PubMed] [Google Scholar]
- Pham NA, Schwock J, Iakovlev V, Pond G, Hedley DW, Tsao MS. Immunohistochemical analysis of changes in signaling pathway activation downstream of growth factor receptors in pancreatic duct cell carcinogenesis. BMC Cancer. 2008;8:43. doi: 10.1186/1471-2407-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubi JC, Waser B, Friess H, Buchler M, Laissue J. Neurotensin receptors: a new marker for human ductal pancreatic adenocarcinoma. Gut. 1998;42:546–550. doi: 10.1136/gut.42.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 2007;21:2455–2465. doi: 10.1096/fj.06-7793com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Gupta A, Gagnidze K, Lim MP, Gomes I, Lee-Ramos D, Nieto N, Devi LA. AT1R-CB(1)R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J. 2011;30:2350–2363. doi: 10.1038/emboj.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang XW. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sams M, Silye R, Gohring J, Muresan L, Schilcher K, Jacak J. Spatial cluster analysis of nanoscopically mapped serotonin receptors for classification of fixed brain tissue. J Biomed Opt. 2014;19:011021. doi: 10.1117/1.JBO.19.1.011021. [DOI] [PubMed] [Google Scholar]
- Scarselli M, Annibale P, Radenovic A. Cell-type-specific β2 adrenergic receptor clusters identified using photo-activated localization microscopy are not lipid raft related, but depend on actin cytoskeleton integrity. J Biol Chem. 2012;287:16768–16780. doi: 10.1074/jbc.M111.329912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Bohn LM. Physiological and pharmacological implications of beta-arrestin regulation. Pharmacol Ther. 2009;121:285–293. doi: 10.1016/j.pharmthera.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Lippincott-Schwartz J. Quantifying spatial organization in point-localization superresolution images using pair correlation analysis. Nat Protoc. 2013;8:345–354. doi: 10.1038/nprot.2013.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbaz M, Ruliang F, Xu Z, Benjia L, Cong W, Zhaobin H, Jun N. mRNA expression of somatostatin receptor subtypes SSTR-2, SSTR-3, and SSTR-5 and its significance in pancreatic cancer. World J Surg Oncol. 2015;13:46. doi: 10.1186/s12957-015-0467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. beta-Arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 2010;103:1671–1679. doi: 10.1038/sj.bjc.6605968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton PA, Mirzapoiazova T, Hasina R, Salgia R, Moss J. Increased mu-opioid receptor expression in metastatic lung cancer. Br J Anaesth. 2014;113(Suppl 1):i103–i108. doi: 10.1093/bja/aeu165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Smedberg JL, Rula ME, Xu XX. Regulation of Ras-MAPK pathway mitogenic activity by restricting nuclear entry of activated MAPK in endoderm differentiation of embryonic carcinoma and stem cells. J Cell Biol. 2004;164:689–699. doi: 10.1083/jcb.200312028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria G, Ben-Baruch A. The CCL5/CCR5 Axis in Cancer. In: Fulton AM, editor. Chemokine Receptors in Cancer. Totowa, NJ: Humana Press; 2009. pp. 109–130. [Google Scholar]
- Stone MB, Veatch SL. Steady-state cross-correlations for live two-colour super-resolution localization data sets. Nat Commun. 2015;6:7347. doi: 10.1038/ncomms8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988;240:177–184. doi: 10.1126/science.2451290. [DOI] [PubMed] [Google Scholar]
- Szende B, Srkalovic G, Schally AV, Lapis K, Groot K. Inhibitory effects of analogs of luteinizing hormone-releasing hormone and somatostatin on pancreatic cancers in hamsters. Events that accompany tumor regression. Cancer. 1990;65:2279–2290. doi: 10.1002/1097-0142(19900515)65:10<2279::aid-cncr2820651020>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Tobin SJ, Cacao EE, Hong DW, Terenius L, Vukojevic V, Jovanovic-Talisman T. Nanoscale effects of ethanol and naltrexone on protein organization in the plasma membrane studied by photoactivated localization microscopy (PALM) PLoS One. 2014;9:e87225. doi: 10.1371/journal.pone.0087225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Sun X, Bohn LM, Sadee W. Opioid receptor homo- and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol Pharmacol. 2005;67:2173–2184. doi: 10.1124/mol.104.010272. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wombacher R, Heidbreder M, van de Linde S, Sheetz MP, Heilemann M, Cornish VW, Sauer M. Live-cell super-resolution imaging with trimethoprim conjugates. Nat Methods. 2010;7:717–719. doi: 10.1038/nmeth.1489. [DOI] [PubMed] [Google Scholar]
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Arnatt CK, El-Hage N, Dever SM, Jacob JC, Selley DE, Hauser KF, Zhang Y. A bivalent ligand targeting the putative mu opioid receptor and chemokine receptor CCR5 heterodimers: binding affinity versus functional activities. Med Chem Commun. 2013;4:847–851. doi: 10.1039/C3MD00080J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wei J, Wang H, Xue X, An Y, Tang D, Yuan Z, Wang F, Wu J, Zhang J, Miao Y. Epithelial mesenchymal transition correlates with CD24+CD44+ and CD133+ cells in pancreatic cancer. Oncol Rep. 2012;27:1599–1605. doi: 10.3892/or.2012.1681. [DOI] [PubMed] [Google Scholar]
- Zylla D, Gourley BL, Vang D, Jackson S, Boatman S, Lindgren B, Kuskowski MA, Le C, Gupta K, Gupta P. Opioid requirement, opioid receptor expression, and clinical outcomes in patients with advanced prostate cancer. Cancer. 2013;119:4103–4110. doi: 10.1002/cncr.28345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.